

Abstract
Management of neoangiogenesis remains a high-value therapeutic goal. A recently uncovered association between the DNA damage repair pathway and pathological angiogenesis could open previously unexplored possibilities for intervention. An attractive and novel target is the Eyes absent (EYA) tyrosine phosphatase, which plays a critical role in the repair versus apoptosis decision after DNA damage. This study examines the role of EYA in the postnatal development of the retinal vasculature and under conditions of ischemia-reperfusion encountered in proliferative retinopathies. We find that the ability of the EYA proteins to promote endothelial cell (EC) migration contributes to a delay in postnatal development of the retinal vasculature when Eya3 is deleted specifically in ECs. By using genetic and chemical biology tools, we show that EYA contributes to pathological angiogenesis in a model of oxygen-induced retinopathy. Both in vivo and in vitro, loss of EYA tyrosine phosphatase activity leads to defective assembly of γ-H2AX foci and thus to DNA damage repair in ECs under oxidative stress. These data reveal the potential utility of EYA tyrosine phosphatase inhibitors as therapeutic agents in inhibiting pathological neovascularization with a range of clinical applications.
Pathological neovascularization (NV) is associated with common human diseases such as proliferative retinopathies, wet age-related macular degeneration, cancer, atherosclerotic plaque rupture, and rheumatoid arthritis.1 Much evidence points to a role for vascular endothelial growth factor (VEGF) signaling in both pathological and developmental angiogenesis, and drugs targeting the VEGF pathway have been extensively tested as potential anti-angiogenic agents.2, 3 Antibodies to VEGF have achieved clinical success in the treatment of wet age-related macular degeneration, clinically significant macular edema, and retinopathy of prematurity, as well as in improving the odds of progression-free survival in certain cancers. However, intrinsic and induced resistance to anti-VEGF treatment poses limitations to their use in cancer treatment.3, 4 Furthermore, side effects associated with normal angiogenic processes such as wound healing are a concern. It is in these clinical contexts that novel anti-angiogenic agents capable of discriminating between developmental and pathological NV and complementing anti-VEGF treatment would be valuable. Several recent studies have begun to delineate mechanisms that differentiate between developmental and pathological angiogenesis.5, 6, 7, 8, 9 Notably, Okuno et al5 showed that the ataxia telangiectasia mutated (ATM) pathway is activated by tissue hypoxia but is not active during normal angiogenesis, and Economopoulou et al6 report that the H2AX-dependent DNA damage response (DDR) is activated and promotes NV specifically under hypoxic conditions. The Eyes absent (EYA) proteins are protein tyrosine phosphatases (PTPs) that hydrolyze the terminal phosphotyrosine of H2AX, permitting the assembly of the DNA damage response machinery and preventing apoptosis.10 The tyrosine phosphatase activity of EYA is also necessary for endothelial cell (EC) migration and tube formation.11, 12 Taken together, these observations led us to postulate that the EYA proteins could contribute to angiogenesis by multiple mechanisms. They have a specific role in DDR-dependent pathological NV following ischemia-associated DNA damage and an EC migration–associated role in early development of the retinal vasculature. Hence, inhibition of the EYA tyrosine phosphatase activity could be a novel target for development of anti-angiogenic drugs. Here we report on the viability of this new therapeutic mechanism by using both genetic and chemical biology tools. These studies open the door for the development of EYA inhibitors with potential uses in the treatment of a host of vascularization-associated disorders, including cancer and proliferative retinopathies.
Materials and Methods
Antibodies and Reagents
The following antibodies were used in these studies: anti-cleaved caspase-3 (9664S; Cell Signaling Technology, Danvers, MA), anti-CD31 (550274, BD Biosciences, San Jose, CA), isolectin 488 (121411; Life Technologies, Carlsbad CA), bromodeoxyuridine (BrdU) mouse monoclonal Alexa 647 (B35133, Invitrogen, Carlsbad, CA), donkey anti-rabbit Alexa Fluo-594 and goat anti-mouse Alexa Fluo-488 (A21207 and A-11001, Life Technologies, Carlsbad, CA), anti-MDC1 (AB1169; Abcam, Cambridge, MA), anti-γH2AX (MABE205; Millipore, Billerica, MA), VEGF antibody (VG1, Thermo Fisher, Rockford, IL). Human retinal microvascular ECs (HRMECs) were obtained from Cell Systems (Kirkland, WA), and mouse retinal microvascular ECs (MRMECs) were obtained from Cell Biologics (Chicago, IL).
Animals
This study adhered to the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research. C57Bl/6 mice obtained from Jackson Laboratories (Bar Harbor, ME) were bred in the Cincinnati Children's Hospital Medical Center facility (animal protocol 3D09062).
Generation of Eya3flox Allele
We generated a conditional loss-of-function allele, Eya3flox, by conventional gene targeting. We targeted exon 6 for conditional deletion. This design left open the possibility that exons 1 to 5 might produce a functional truncated protein. Incorporation of sequences from the C-terminus of ornithine decarboxylase as a signal for rapid degradation13 was designed to eliminate any expressed Eya3 N-terminal sequence. The allele design incorporated loxP sites into an artificial exon that is initially in reverse orientation. Through two steps of recombination mediated by a pair of wild-type and a pair of variant loxP sequences that are incompatible, the C-terminus of ornithine decarboxylase degradation signal was spliced into the mRNA, simultaneously deleting exon 6. This allele design strategy has been used previously.14 Successful Flp recombination was confirmed by PCR analysis with Eya3fl forward, 5′-CCACTTGGAGTAAGCATCCAGTC-3′ and reverse, 5′-AAGCAAAACGTCCTAGGTGCTC-3′.
Oxygen-Induced Retinopathy Model
The protocol for oxygen-induced retinopathy was one previously described in studies associating DNA damage repair pathway with NV.5, 6 Briefly, neonatal mice and their nursing mothers were kept in room air from postnatal day 0 (P0) to P8. At P8 each pup and its nursing mother were moved to a chamber maintained at 85% oxygen. At P11 the pups and their nursing mothers were returned to room air (surrogate mothers were used if necessary). When appropriate, intravitreal injection of EYA inhibitors [1 μL of either 100 μmol/L stock solution of benzarone (BZ) or 250 μmol/L stock solution of benzbromarone (BBR)] was performed at P12. To examine the effect of genetic deletion of Eya3 in the oxygen-induced retinopathy (OIR) protocol, tamoxifen was administered i.p. at 300 μg per pup daily between P11 and P13 to both Eya3flox/flox, pdgf-iCre, and Eya3flox/flox mice. At various time-points the eyes were enucleated while the pups were maintained under anesthesia. Immunofluorescence imaging and Western blot analysis of ECs demonstrated the effectiveness of this protocol in lowering EYA3 levels in ECs. There was no compensatory up-regulation of other Eyas (Supplemental Figure S1).
Immunostaining of Whole Mount Retinas
Enucleated eyes were fixed for 1 hour in 4% paraformaldehyde/phosphate-buffered saline at room temperature and dissected. Retinas were permeabilized at room temperature for 30 minutes and then incubated with specific antibodies (γ-H2AX, EYA3, or cleaved caspase-3) overnight at 4°C. Antigen was detected with secondary antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 488. To visualize vasculature, retinas were stained with fluoresceinated Griffonia (Bandeiraea) simplicifolia (isolectin B4–Alexa Fluor 594 conjugate, 1:500 dilution; Invitrogen) in phosphate-buffered saline with Tween (PBST). For the BrdU incorporation studies, 10 mg/kg BrdU was administered i.p. at P13. Isolated retinas were treated with 2N HCl for 30 minutes and then extensively washed with PBS, blocked with 10% fetal bovine serum, and incubated with BrdU Alexa Fluor 647 for 1 hour at room temperature. Images were taken at ×400 magnification on a Zeiss microscope (Zeiss, Jena, Germany).
Quantification of NV and VO
Standard published protocols were used to quantitate NV and VO. The number of pixels in the pathological tufts was quantified and compared with the number of pixels in the entire retinal area by a computer-aided method (SWIFT-NV15) that utilizes a series of macros in ImageJ version 1.48 (NIH, Bethesda, MD; http://imagej.nih.gov/ij). Percentage of NV and VO in total retinas was compared between inhibitor- and vehicle-treated eyes, or Eya3VEC-KO and Eya3fl/fl mice. In each case, n was the number of eyes quantified. Each experiment included three independent litters.
RT-PCR Analysis
To determine expression of Eya transcripts in mouse retinal microvascular ECs or retinal ECs from genetically engineered mice, total RNA was extracted from 1 million ECs, and cDNA was synthesized with the Primescript RT reagent kit (Takara Bio, Shiga, Japan). PCR product was analyzed on a 1.5% agarose gel to confirm that amplified products were of the expected sizes. Primers used included EYA1 forward 5′-CATAGCCGACTGAGTGGTAGT-3′ and reverse 5′-GCTCTGTTTTAACTTCGGTGCC-3′; EYA2 forward 5′-CACCGCTGGGCTCTATCAAG-3′ and reverse 5′-GGGGTAGGACGGATAATCCTG-3′; EYA3 forward 5′-CTCAAACCAGGATTATCCCACC-3′ and reverse 5′-CAGCATCACTGTTAGTCTGACC-3′; EYA4 forward 5′-TCCTTGGCCCTGCTAAGAG-3′ and reverse 5′-TGCCTATTTTTGTTGCGCTGT-3′; GAPDH forward 5′-AAGGCCGGGGCCCACTTGAA-3′ and reverse: 5′-CGGCCATCACGCCACAGCTT-3′.
EC Culture, Immunostaining, Transwell Migration, and Proliferation Assays
HRMECs were cultured in Complete Medium (Cell Systems) and used in the first nine passages. ECs from Eya3VEC-KO and control mice were isolated by using magnetic Dynabeads (Life Technologies) coated with anti–PECAM-1 antibody as previously described.16 For analysis of the formation of DNA repair complexes, cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes, and then the coverslips were immunostained for DDR proteins with anti-γH2AX and anti-MDC1 antibodies. Cell proliferation was measured at 72 hours with the WST-8 assay (CCK-8 Kit, Dojindo Molecular Technologies, Rockville, MD) as previously described.12 Proliferation studies were performed in Epithelial Cell Growth Medium 2 (Lonza, Allendale, NJ). Vehicle control contained 0.1% dimethyl sulfoxide, and both EYA inhibitors were dissolved in 0.1% dimethyl sulfoxide. For hypoxia experiments, the cells were maintained at 1% O2 with BioSpherix ProCO2 Model P120 and ProOx Model 110 controllers for C chambers (Biospherix, Parish, NY). Transwell migration experiments were performed as previously described.12
Statistics
Results are presented as the means ± SEM for the in vivo experiments and as means ± SD for the in vitro studies. Statistical analyses were performed with Graphpad PRISM version 5.0 for Mac OSX, (GraphPad Software, La Jolla, CA). A t-test was used when two samples and conditions were compared, and analysis of variance was used for more than two groups. Significance represents P < 0.05.
Results
Targeted Deletion of Eya3 in ECs Reduces Extension and Branching of the Early Postnatal Retinal Vasculature
The mouse retina is avascular at birth, with a monolayer of vessels extending out from the center to the periphery between birth and P7, providing an accessible and well-characterized system17 to examine the role of EYA in developmental angiogenesis. RT-PCR analysis for Eya1, Eya2, Eya3, and Eya4 in mouse retinal ECs showed a transcript for only Eya3 (Supplemental Figure S1). To investigate the cell-autonomous function of EYA3 in NV, we deleted Eya3 from ECs by using the loxP recombination strategy and the EC-specific tamoxifen-inducible Cre-recombinase Pdgfb-iCreER18 (Eya3VEC-KO). The targeting strategy and the resulting reduction in EYA3 protein levels in endothelial cells is shown in Figure 1. The developing retinal vasculature was examined at P5 and P8 by using flat mounts and staining with isolectin B4.
Figure 1.

Strategy for Eya3 deletion. A: Generation of the Eya3 conditional allele. Orange boxes correspond to exons. Red and yellow arrowheads are LoxP sites. Brown arrowheads are Frt sites used to delete the Neo-positive selectable marker; tk represents the thymidine kinase gene that is selectable against after homologous recombination. B: EYA3 levels are lower in the Eya3VEC-KO endothelial cells. Western blot analysis of lung endothelial cells, and immunostaining of retinal endothelial cells extracted from Eya3fl/fl (CTL) and Eya3VEC-KO mice showing EYA3 (red), and DAPI (blue). CTL, control.
Endothelial deletion of Eya3 caused a significant reduction in the extension of the superficial vascular plexus at P5, as measured by the ratio of the vascular plexus radius to the radius of the total retina (Figure 2, A and B). Branching density was quantitated by counting the number of branch points per unit area of vasculature at the angiogenic front. On average, 24% fewer branch points were observed in the Eya3VEC-KO retinas relative to control littermates (Figure 2, C and D).
Figure 2.

Loss of EYA3 from VECs attenuates development of the superficial vascular plexus in the retina. A: Flat-mounted P5 retina from Eya3VEC-KO conditional mutant mice and control littermates (Eya3fl/fl) labeled with isolectin B4. Deletion of Eya3 was induced by tamoxifen injection between P1 and P3. B: Reduced extension of the vascular plexus as quantitated in both retina of six mice per group. C: Higher magnification images of flat-mounted retina highlight reduced vascular density in Eya3VEC-KO retinas relative to controls. D: Vessel density measured by counting branch points at the advancing vascular front (four fields per retina) shows a significant decrease in branching density upon VEC-specific Eya3 deletion. E: Quantification of BrdU-positive/isolectin B4–positive cells in Eya3VEC-KO and control retina (Eya3fl/fl) at P5. No statistically significant difference is observed. F: Filopodia number was quantified and normalized to the outline of the angiogenic front. Fewer filopodia per millimeter are observed in Eya3VEC-KO retinas. Data presented as means ± SEM (B, D, and F). n = 6 (D); n = 12 (E), 5 (F), control; n = 5 (E), 8 (F), Eya3VEC-KO. ∗∗∗P < 0.001. BrdU, bromodeoxyuridine; CTL, control; P1, postnatal day 1; VECs, vascular endothelial cells.
Both EC proliferation and migration could contribute to the vascular defects observed here. Each of these was examined in retinas of Eya3VEC-KO and control cre-negative littermates at P5. The proliferation of stalk cells plays a key role in the angiogenic process at the vascular front.19 When using bromo-deoxyuridine (BrdU) incorporation by isolectin B4–positive cells at P5 as a measure of EC proliferation, we did not observe a significant difference between retinas from Eya3VEC-KO and their cre-negative littermates (Figure 2E). In retinas from Eya3VEC-KO pups, however, we observed a 27% reduction in the number of filopodia per millimeter of vessel length at the vascular front, suggesting a likely defect in EC migration (Figure 2F).
Targeted Deletion of Eya3 in ECs Attenuates NV in the Oxygen-Induced Retinopathy Model
Despite the early phenotype observed at P5, analysis of the superficial retinal vascular plexus at P8 shows recovery of both extension and branching defects in Eya3VEC-KO animals (Figure 3A), suggesting that lack of EYA3 only delays development of the superficial vascular layer.
Figure 3.

VEC-specific deletion of Eya3 in an OIR model results in reduced neovascularization. A: Flat-mounted P8 retinas from Eya3fl/fl (CTL) and Eya3VEC-KO mice show that development of the vascular plexus in the mutant animals is comparable to that in the controls (tamoxifen injection between P1 and P3). B: Flat-mounted retinas from mice maintained in room air at P8, P11, and P17 compared with mice placed in 85% O2 between P8 and P11 and returned to room air until P17. After hyperoxia treatment, a central zone of vaso-obliteration is evident at P11 and neovascular tufts can be seen at P17. C: P17 retinas of Eya3VEC-KO and control mice subjected to hyperoxia between P8 and P11 with Eya3 deletion induced by tamoxifen injection between P11 and P13. Significantly less neovascularization but comparable vaso-obliteration are seen in Eya3VEC-KO retinas relative to CTL retinas. D: Representative Western blot analysis of extracts from P14 retinas after hyperoxia (left). Quantitation of normalized vascular endothelial growth factor antibody levels from Western blot analyses (three retinas; right). E: Quantification of the percentage of BrdU-positive cells at P14 in OIR retinas. Data are expressed as means ± SEM (D and E). n = 6 retinas in each group (A); n = 17 CTLs, n = 16 Eya3VEC-KO from three independent litters (C); n = 2 CTLs, n = 4 Eya3VEC-KO (E). ∗P < 0.05, ∗∗P < 0.01. CTL, control; OIR, oxygen-induced retinopathy; P8, postnatal day 8; VECs, vascular endothelial cells.
To elucidate the role of EYA in pathological angiogenesis we used a modification of the well-established OIR model20 in which pups are subjected to hyperoxia (75% O2 between P7 and P12) and then returned to room air for 4 to 5 days before analysis. This modified protocol (85% O2 chamber between P8 and P11) was previously described in the study showing that NV requires the DDR-associated minor histone protein H2AX.6 This protocol was validated by the observation of characteristic regression of immature capillaries in the center of the retina leading to a central area of VO upon return to room air (P11-OIR) (Figure 3B). The resulting effective hypoxia leads to EC proliferation and the formation of neovascular tufts (P17-OIR) (Figure 3B), similar to that seen with the 5-day hyperoxia protocol. Maximum severity of NV, the proliferative phase, typically occurs 5 days after return to room air (P16 in this protocol and P17 in the 5-day protocol). Clinically, NV is considered pathological, whereas revascularization is desirable.
Eya3fl/fl (control) and Eya3VEC-KO pups were subjected to hyperoxia from P8 to P11 and injected with tamoxifen from P11 to P13 to induce Eya3 deletion. NV at P17 was quantitated by using an automated protocol.15 Eya3VEC-KO retinas had nearly 40% less NV than did control retinas (Figure 3C). Interestingly, residual VO in the central avascular area was comparable in the control and Eya3VEC-KO retinas (Figure 3C).
The OIR protocol is a VEGF-dependent model in which VEGF levels are suppressed during hyperoxia and elevated upon return to room air. Maximal VEGF levels are typically observed within 3 days after return to room air (P14 in the protocol used here) and remain high until P17. Interestingly, VEGF protein levels were comparable in both the control and Eya3VEC-KO retinas (Figure 3D), although reduced cell proliferation, as measured by BrdU incorporation, was seen in the Eya3VEC-KO retinas at P14 relative to the controls (Figure 3E).
Pharmacological Inhibition of the EYA Tyrosine Phosphatase Activity Attenuates NV in the OIR Model
EYA proteins have multiple and separable biochemical activities (tyrosine phosphatase, threonine phosphatase, and transactivation).21 We hypothesized that only the tyrosine phosphatase activity contributes to pathological angiogenesis through its role in DNA damage repair. To specifically target this enzymatic activity of the EYA proteins, we used a chemical biology approach. We have previously identified and validated EYA tyrosine phosphatase inhibitors that showed in vitro anti-angiogenic activity.11, 12 Two of the best-characterized members of this inhibitor series, BBR and BZ, were used in these studies. These compounds show selectivity toward the EYA family of PTPs, and previous cell-based studies support their ability to target EYA3 in cells.12
At P12 (24 hours after return to room air), each pup subjected to the OIR protocol was injected in one eye with an EYA inhibitor and in the other with a vehicle control. Retinas were dissected and flat-mounted at P17 and stained with isolectin B4 to visualize the vasculature (Figure 4A). There was significant normalization of the vasculature in the eyes treated with either BZ or BBR relative to the vehicle controls (54% and 49% reduction in NV, respectively) (Figure 4, A and B); however, VO was comparable to that in the vehicle-treated eye, suggesting a preferential effect on pathological NV (Figure 4C).
Figure 4.

EYA tyrosine phosphatase inhibition suppresses neovascularization in retinopathy. A: Representative retinal flat mounts at P17 from mice subjected to the OIR protocol and injected at P12 in one eye each with vehicle or EYA inhibitors BZ or BBR. Vessels were visualized with isolectin B4 staining. B: Neovascularization quantitated as percentages of total retinal area. C: Areas of vaso-obliteration quantitated as percentages of total retinal area; nonsignificant. D: Quantification of percentage of BrdU-positive cells at P14 in OIR retinas treated with either vehicle or EYA inhibitors BZ (left) or BBR (right); ∗∗P < 0.01, ∗∗∗P < 0.001. n = 12 BZ, n = 13 BBR and represent three independent litters (B and C); n = 2 BBR, n = 3 BZ (D). BBR, benzbromarone; BrdU, bromodeoxyuridine; BZ, benzarone; OIR, oxygen-induced retinopathy; P17, postnatal day 17.
Increased EC proliferation is known to contribute to NV as well as to vessel tortuosity in the OIR protocol.22 To determine whether EYA inhibitors had an effect on proliferation, we measured incorporation of the nucleotide analog BrdU. Low levels of BrdU incorporation were detected in control vehicle-treated OIR retinas in the proliferative phase (measured here at P14), with a 60% decrease in either the BZ- or BBR-treated retinas (Figure 4D).
EYA-PTP Inhibition or Eya3 Deletion Attenuates the DNA Damage Response in the Oxygen-Induced Retinopathy Model
To establish the kinetics of DNA-damage repair subsequent to ischemic insult induced by the OIR protocol, we monitored γ-H2AX foci in the retina of mice at 48, 72, 96, 120, and 144 hours after return to room air. Widespread γ-H2AX foci containing nuclei were observed within 48 hours and peaked between 120 and 144 hours (P16 to P17); further analyses were conducted at P17 to coincide with evaluation of NV. Although DNA foci were generally more frequent in non-ECs, foci were also observed in ECs at sites of NV (Figure 5A), which is consistent with previous reports that DNA damage repair is coincident with proliferating ECs.6
Figure 5.

γ-H2AX foci formation is attenuated in retinas of mice subjected to the OIR protocol. A: Isolectin B4 (green) and γ-H2AX (red) staining of whole mount retinas (P17) from mice subjected to the OIR protocol and injected intravitreally with either vehicle or the EYA inhibitors BZ or BBR. Nuclei are stained blue with DAPI. White arrowheads highlight γ-H2AX staining in endothelial cells. B: Quantification of nuclei with γ-H2AX foci in retinal flat mounts of OIR mice at P17. One eye of each mouse was injected with vehicle and the other with BZ or BBR. C: Quantification of nuclei with γ-H2AX foci in the retinal flat mounts from Eya3fl/fl (CTL), and Eya3VEC-KO mice subjected to the OIR protocol and with Eya3 deletion induced by tamoxifen injections between P11 and P13. Data are expressed as means ± SEM (B and C). n = 4 retinal segments per retina of three mice per group (B); n = 3 three representative segments from eight retina (C). ∗∗P < 0.01, ∗∗∗P < 0.001. BBR, benzbromarone; BZ, benzarone; CTL, control; γ-H2AX, Ser139-phosphorylated form of the DNA damage sensor H2AX; OIR, oxygen-induced retinopathy.
Genetic and chemical biology tools were used to examine the effect of either Eya3 deletion or EYA-PTP inhibition on the presence of γ-H2AX foci at P17 as a marker of DNA damage repair. Eya3VEC-KO and control mice were subjected to the P8 to P11 OIR protocol, and tamoxifen was administered after return to room air. There was more than a 50% reduction in the number of nuclei with γ-H2AX foci in the retinas of the Eya3VEC-KO mice relative to in the retinas of their cre-negative littermates (Figure 5C). Similarly, retinas treated with the EYA inhibitors BZ or BBR had significantly fewer nuclei with γ-H2AX foci relative to the vehicle-treated controls, which is suggestive of an impaired DNA damage response (Figure 5, A and B).
EYA Inhibition Attenuates EC Migration, Proliferation under Hypoxia, and the Formation of γ-H2AX–MDC1 Foci under Oxidative Stress
To better define the molecular mechanism(s) by which the EYAs might play a role in retinal angiogenesis, we used primary HRMECs. We have previously reported that the EYA-PTP inhibitors BZ and BBR inhibit human umbilical vein EC migration in vitro.12 Similarly, BZ and BBR inhibited Transwell migration of HRMECs in a dose-dependent manner (Figure 6A), confirming a role for the EYA-PTP in the motility of retinal ECs as seen in the P5 retina.
Figure 6.

Lowering EYA3 levels or EYA-PTP inhibition in HRMECs leads to reduced cell motility, proliferation after hypoxia, and an attenuated DNA damage response. A: Transwell migration of HRMECs is inhibited in a dose-dependent manner by the EYA-PTP inhibitors BZ and BBR. B: HRMECs were treated with either vehicle or the EYA inhibitors BZ or BBR and maintained in either hypoxic conditions (1% oxygen) or 21% oxygen for 72 hours. Cell proliferation was measured by an MTT assay. C: Representative immunohistochemical staining for γ-H2AX (green), MDC1 (red), and DAPI (blue) in HRMEC maintained at 0.2% O2 for 12 hours in the presence of either vehicle or the EYA inhibitors BZ and BBR. Quantification of the number of nuclei with γ-H2AX foci in three independent experiments. Data plotted are means ± SD (A, B, and C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. BBR, benzbromarone; BZ, benzarone; CTL, control; HRMEC, human retinal endothelial cell; PTP, protein tyrosine phosphatase.
We next examined the effect of EYA-PTP inhibition on HRMEC proliferation under hypoxic conditions. As expected, HRMECs were more proliferative under hypoxic conditions (1% O2) than in 21% O2. Increased proliferation under hypoxic stress distinguishes ECs from most other cell types that undergo cell cycle arrest and apoptosis in hypoxic conditions because of stalled replication forks. Both BZ and BBR attenuated proliferation in a dose-dependent fashion (Figure 6B). Analogous results were previously reported for human umbilical vein endothelial cells transfected with siRNA targeting H2AX: H2AX was required to maintain proliferation only under hypoxic conditions.6 Because the EYA tyrosine phosphatase targets the C-terminal tyrosine 142 of H2AX, these observations were consistent with a model in which the tyrosine 142–dephosphorylated form of H2AX supports EC proliferation in hypoxia.
ATM/ATR-dependent stabilization of stalled replication forks is generally initiated by formation of γ-H2AX foci.23 In ECs this process is ATR and H2AX dependent, permits survival and proliferation under conditions of pathological hypoxia, and consequently promotes NV.5 Critical for the sustenance of γ-H2AX foci is the recruitment of the mediator MDC1, which will only bind to Y142-dephosphorylated γ-H2AX.24, 25, 26 To determine the effect of EYA inhibition on hypoxia-induced DNA damage repair, HRMECs (with or without EYA inhibitors) were maintained in either 0.2% O2 or 21% O2 for 12 hours. Previous studies have established that treatment with such extreme hypoxia is required for the detection of γ-H2AX in vitro.23 In control cells γ-H2AX foci that colocalize with MDC1 were observed, suggesting the formation of DNA repair complexes. Significantly fewer γ-H2AX foci were present in EYA inhibitor–treated HRMECs relative to control cells (Figure 6C).
Discussion
The EYA proteins are part of a conserved developmental cassette most often associated with development of the fly eye; loss of eya function in Drosophila leads to severe defects, including loss of the compound eye. However, no ocular phenotype has been reported for any mammalian Eya knockout (there are four mammalian orthologs, Eya1 to Eya4). The Eya3-deficient mouse was reported to have minor functional deficits, including reduced movement and changes in respiratory, heart, and muscle function.27 We have previously reported a role for the EYA-PTP activity in endothelial and cancer cell motility.12, 28 Because EC migration is a critical component of angiogenesis, the role of EYA in the development of the retinal vascular plexus of neonatal mice was examined. Developmental angiogenesis involves, at a minimum, proliferation of endothelial stalk cells, migration of tip cells, tube or lumen formation, and degradation of the extracellular matrix. Analysis of retinal vasculature upon EC-specific Eya3 deletion reveals reduced extension and branching density at P5. The observations that there is little change in EC proliferation upon Eya3 deletion at this stage and that fewer filopodial extensions are seen on tip cells at the vascular front are consistent with a role for EYA3 in EC migration. Reduced filopodia upon Eya3 deletion was also previously observed in breast cancer cells,28 supporting the possibility that the EYA proteins contribute to cell migration by altering the actin cytoskeleton.21, 28
Despite the early phenotype observed in the superficial retinal vasculature, the plexus in Eya3VEC-KO extends to the periphery and is comparable to control retinas by P8. This suggests that loss of Eya3 only delays vascular development, which is commensurate with the observation that EC migration and not EC proliferation is affected by loss of Eya3 in EC. A previous report describing germline deletion of Eya3 noted no defects in eye development or function, although the mutant mice were smaller than the controls.27 No examination of vascular development in vivo has been previously reported. Although the present study focused on the role of endothelial EYA3, it is expressed in other relevant cells, including smooth muscle cells and retinal microglia. The role of nonendothelial EYA3 in angiogenesis is the subject of independent ongoing investigations.
Unlike in the early postnatal days of developmental angiogenesis, in established vascular plexus such as the late postnatal retinal vasculature, little EC proliferation is expected. Under hypoxic stress, as is seen in pathological conditions such as retinopathies and tumors, however, ECs are stimulated to proliferate (Figure 6).6 Conditions of oxidative stress have been linked to DNA damage and repair through various mechanisms: hypoxia can cause low levels of DNA damage subsequent to replicative stress, as well as through direct repression of DNA damage repair genes. Additionally, reactive oxygen species released by the mitochondria of hypoxic cells could cause DNA damage. Survival of ECs would then be contingent on an active DNA damage repair pathway (Figure 7), which in turn has been linked to the ability of the EYA tyrosine phosphatase activity to dephosphorylate the terminal tyrosine of H2AX and thus permit interaction with MDC1 and the assembly of the DNA repair complex.10, 24, 30 γ-H2AX foci can be a measure of either DNA double-strand breaks or stabilized, stalled replication forks.
Figure 7.

Schematic showing the proposed role of the EYA3-PTP in DNA damage repair–linked pathological angiogenesis. Oxidative stress is known to induce DNA damage. DNA damage repair is contingent on the assembly of a multiprotein complex nucleated by γ-H2AX. H2AX is constitutively phosphorylated on its terminal tyrosine.29 The EYA3-PTP can dephosphorylate this tyrosine10 and permit interaction with MDC1 (double-headed arrow), an essential step in the assembly of DNA damage repair foci (represented here by the Mre11-Rad50-Nbs1 MRN complex). Repair of DNA damage permits endothelial cell survival and angiogenesis. γ-H2AX, Ser139-phosphorylated form of the DNA damage sensor H2AX; PTP, protein tyrosine phosphatase.
In the context of the OIR model, hypoxia-induced stalled replication forks are thought to signal an ATR-dependent form of DNA damage response that leads to fork stabilization.5 As a consequence, cell viability is maintained and hypoxia-induced growth factors can promote proliferation and pathological angiogenesis (Figure 7). Such an interaction makes sense in the context of disease or injury in which DNA repair mechanisms need to be up-regulated and angiogenesis needs to be activated. By using both chemical inhibition and genetic methods, we have shown that inhibition of EYA tyrosine phosphatase activity affects the assembly of γ-H2AX foci, recruitment of the DNA damage repair protein MDC1, survival following hypoxic stress, and pathological NV in an animal model of proliferative retinopathy. Although the possibility of off-target effects of small molecules cannot be completely excluded in chemical biology approaches, the similar outcomes obtained with both the genetic and chemical inhibition methods increases the likelihood that EYA-PTP inhibition plays a role in the reduced NV described here.
The OIR protocol appears to induce DNA damage in multiple cell types in the retina, with a relatively low frequency in ECs. This is in agreement with previous reports suggesting that γ-H2AX staining is observed only in proliferating, PCNA-positive cells of the retinal vasculature in a similar experiment.6 Interestingly, the time course of formation and resolution of γ-H2AX foci in the OIR protocol parallels that of NV, reaching a maximum 5 days after return to room air and then declining as NV is resolved. Furthermore, continuous angiogenic factor stimulation has been associated with replication stress and γ-H2AX formation independent of hypoxia.
Although either EYA tyrosine phosphatase inhibition or deletion of Eya3 in ECs significantly reduced the formation of neovascular tufts, we observed little effect on revascularization into the vaso-obliterated central zone. This is reminiscent of the previously reported effect of Atm deletion5 and unlike the suppression of both NV and revascularization seen upon blocking either VEGF or VEGFR2.31 Although ATM appears to play a role in angiogenesis through oxidative defense and independent of the canonical DDR pathway,5 our data support a role for the assembly of the DDR-associated γ-H2AX–MDC1 complex in pathological neoangiogenesis.
The EYA proteins are multidomain and have several separable biochemical and cellular functions.21 The tyrosine phosphatase activity examined here is localized to the conserved C-terminal domain of the proteins.32 Independent of this PTP domain, the N-terminal domain has transactivation and threonine phosphatase activities.33, 34 The tyrosine phosphatase activity not only contributes to EYA's role in DDR but also promotes motility of tumor and ECs.11, 12, 28, 35 Attenuation of EC proliferation by EYA PTP inhibitors under hypoxia demonstrates that this catalytic activity contributes to cell proliferation in DNA damage–inducing conditions. It remains possible that the transactivation activity of the EYAs also plays a role. Indeed, published in vitro studies suggest that the PTP and transactivation activities of EYA are linked in some contexts; the tyrosine phosphatase activity of EYA contributes to transcriptional activation of a SIX1-EYA complex and cell proliferation.36
Conclusions
EYA contributes to angiogenesis by multiple mechanisms. They promote angiogenic sprouting and tube formation, possibly as a result of their role in promoting EC migration. Herein, we show that as participants in the DNA damage response pathway, EYAs also promote survival, proliferation, and NV under hypoxic conditions. Together, these results validate the EYA tyrosine phosphatase as a target for the development of anti-angiogenic agents and suggest the BBR family of compounds as leads for the development of potent and effective antiangiogenics. EYA-targeted therapeutics could be synergistic with VEGF blockade, as well as with genotoxic regimens in cancer treatment. EYA inhibition offers a specific mechanism to inhibit pathological angiogenesis while permitting physiological revascularization. This distinction from VEGF inhibitors may prove to be of significant clinical benefit.
Acknowledgments
Y.W., E.T., and R.N.P. performed the experiments; J.A.S. and R.A.L. designed the allele for conditional deletion of Eya3; L.E.H.S. helped with quantitation of NV and interpretation of OIR results; Y.W., R.N.P., J.A.S., L.E.H.S., and R.A.L. edited the manuscript; and R.S.H. designed the experiments, supervised the research, and wrote the manuscript.
Footnotes
Supported by NIH grants EY022917 and EY014648 (both to R.S.H.).
Disclosures: None declared.
Current address of E.T., Department of Biology, Miami University, Oxford, OH; of J.A.S., Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.10.031.
Supplemental Data
Supplemental Figure S1.
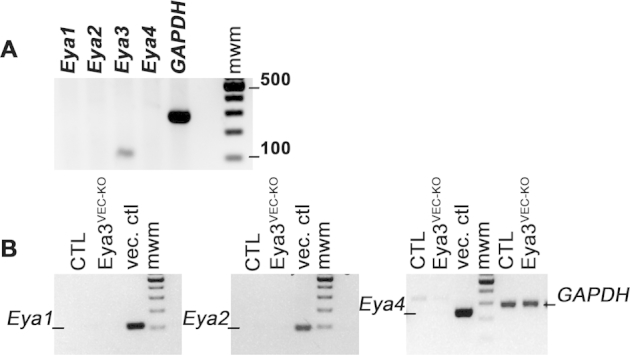
A: RT-PCR analysis on mRNA from mouse retinal endothelial cells. Products after 25 cycles were run on a 1.5% agarose gel. Expected bands are 109 bp for Eya1, 85 bp for Eya2, 107 bp for Eya3, and 237 bp for Eya4. Only Eya3 transcript is detected. A 100-Kb ladder was used for molecular weight markers (mwm). B: RT-PCR analysis on mRNA from endothelial cells isolated from Eya3fl/fl (control) and Eya3VEC-KO mice. No up-regulation of Eya1, Eya2, or Eya4 transcripts is observed.
References
- 1.Chung A.S., Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 2.Campochiaro P.A. Ocular neovascularization. J Mol Med (Berl) 2013;91:311–321. doi: 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welti J., Loges S., Dimmeler S., Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergers G., Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okuno Y., Nakamura-Ishizu A., Otsu K., Suda T., Kubota Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat Med. 2012;18:1208–1216. doi: 10.1038/nm.2846. [DOI] [PubMed] [Google Scholar]
- 6.Economopoulou M., Langer H.F., Celeste A., Orlova V.V., Choi E.Y., Ma M., Vassilopoulos A., Callen E., Deng C., Bassing C.H., Boehm M., Nussenzweig A., Chavakis T. Histone H2AX is integral to hypoxia-driven neovascularization. Nat Med. 2009;15:553–558. doi: 10.1038/nm.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alitalo K. A radical view of pathological vasculature. Cell Metab. 2012;16:287–288. doi: 10.1016/j.cmet.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Rankin E.B., Giaccia A.J., Hammond E.M. Bringing H2AX into the angiogenesis family. Cancer Cell. 2009;15:459–461. doi: 10.1016/j.ccr.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen J., Frye M., Lee B.L., Reinardy J.L., McClung J.M., Ding K., Kojima M., Xia H., Seidel C., Lima e Silva R., Dong A., Hackett S.F., Wang J., Howard B.W., Vestweber D., Kontos C.D., Peters K.G., Campochiaro P.A. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. 2014;124:4564–4576. doi: 10.1172/JCI74527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook P.J., Ju B.G., Telese F., Wang X., Glass C.K., Rosenfeld M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey R.N., Wang T.S., Tadjuidje E., McDonald M.G., Rettie A.E., Hegde R.S. Structure-activity relationships of benzbromarone metabolites and derivatives as EYA inhibitory anti-angiogenic agents. PLoS One. 2013;8:e84582. doi: 10.1371/journal.pone.0084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tadjuidje E., Wang T.S., Pandey R.N., Sumanas S., Lang R.A., Hegde R.S. The EYA tyrosine phosphatase activity is pro-angiogenic and is inhibited by benzbromarone. PLoS One. 2012;7:e34806. doi: 10.1371/journal.pone.0034806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuzawa S., Cuddy M., Fukushima T., Reed J.C. Method for targeting protein destruction by using a ubiquitin-independent, proteasome-mediated degradation pathway. Proc Natl Acad Sci U S A. 2005;102:14982–14987. doi: 10.1073/pnas.0507512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnutgen F., Doerflinger N., Calleja C., Wendling O., Chambon P., Ghyselinck N.B. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol. 2003;21:562–565. doi: 10.1038/nbt811. [DOI] [PubMed] [Google Scholar]
- 15.Stahl A., Connor K.M., Sapieha P., Willett K.L., Krah N.M., Dennison R.J., Chen J., Guerin K.I., Smith L.E. Computer-aided quantification of retinal neovascularization. Angiogenesis. 2009;12:297–301. doi: 10.1007/s10456-009-9155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su X., Sorenson C.M., Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 17.Stahl A., Connor K.M., Sapieha P., Chen J., Dennison R.J., Krah N.M., Seaward M.R., Willett K.L., Aderman C.M., Guerin K.I., Hua J., Lofqvist C., Hellstrom A., Smith L.E. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–2826. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K., Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 19.Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith L.E., Wesolowski E., McLellan A., Kostyk S.K., D'Amato R., Sullivan R., D'Amore P.A. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 21.Tadjuidje E., Hegde R.S. The eyes absent proteins in development and disease. Cell Mol Life Sci. 2013;70:1897–1913. doi: 10.1007/s00018-012-1144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guaiquil V.H., Hewing N.J., Chiang M.F., Rosenblatt M.I., Chan R.V., Blobel C.P. A murine model for retinopathy of prematurity identifies endothelial cell proliferation as a potential mechanism for plus disease. Invest Ophthalmol Vis Sci. 2013;54:5294–5302. doi: 10.1167/iovs.12-11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bencokova Z., Kaufmann M.R., Pires I.M., Lecane P.S., Giaccia A.J., Hammond E.M. ATM activation and signaling under hypoxic conditions. Mol Cell Biol. 2009;29:526–537. doi: 10.1128/MCB.01301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M.S., Edwards R.A., Thede G.L., Glover J.N. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005;280:32053–32056. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 25.Lukas C., Melander F., Stucki M., Falck J., Bekker-Jensen S., Goldberg M., Lerenthal Y., Jackson S.P., Bartek J., Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004;23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stucki M., Clapperton J.A., Mohammad D., Yaffe M.B., Smerdon S.J., Jackson S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 27.Soker T., Dalke C., Puk O., Floss T., Becker L., Bolle I., Favor J., Hans W., Holter S.M., Horsch M., Kallnik M., Kling E., Moerth C., Schrewe A., Stigloher C., Topp S., Gailus-Durner V., Naton B., Beckers J., Fuchs H., Ivandic B., Klopstock T., Schulz H., Wolf E., Wurst W., Bally-Cuif L., de Angelis M.H., Graw J. Pleiotropic effects in Eya3 knockout mice. BMC Dev Biol. 2008;8:118. doi: 10.1186/1471-213X-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandey R.N., Rani R., Yeo E.J., Spencer M., Hu S., Lang R.A., Hegde R.S. The eyes absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene. 2010;29:3715–3722. doi: 10.1038/onc.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao A., Li H., Shechter D., Ahn S.H., Fabrizio L.A., Erdjument-Bromage H., Ishibe-Murakami S., Wang B., Tempst P., Hofmann K., Patel D.J., Elledge S.J., Allis C.D. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnan N., Jeong D.G., Jung S.K., Ryu S.E., Xiao A., Allis C.D., Kim S.J., Tonks N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009;284:16066–16070. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukushima Y., Okada M., Kataoka H., Hirashima M., Yoshida Y., Mann F., Gomi F., Nishida K., Nishikawa S., Uemura A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest. 2011;121:1974–1985. doi: 10.1172/JCI44900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rayapureddi J.P., Kattamuri C., Steinmetz B.D., Frankfort B.J., Ostrin E.J., Mardon G., Hegde R.S. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426:295–298. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda K., Watanabe Y., Ohto H., Kawakami K. Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol Cell Biol. 2002;22:6759–6766. doi: 10.1128/MCB.22.19.6759-6766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okabe Y., Sano T., Nagata S. Regulation of the innate immune response by threonine-phosphatase of eyes absent. Nature. 2009;460:520–524. doi: 10.1038/nature08138. [DOI] [PubMed] [Google Scholar]
- 35.Miller S.J., Lan Z.D., Hardiman A., Wu J., Kordich J.J., Patmore D.M., Hegde R.S., Cripe T.P., Cancelas J.A., Collins M.H., Ratner N. Inhibition of eyes absent homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29:368–379. doi: 10.1038/onc.2009.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., Oghi K.A., Zhang J., Krones A., Bush K.T., Glass C.K., Nigam S.K., Aggarwal A.K., Maas R., Rose D.W., Rosenfeld M.G. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]