

Abstract
Transgenic mice expressing the tdimer2(12) form of Discosoma red fluorescent protein under control of the proopiomelanocortin gene's regulatory elements are a useful model for studying corticotrophs. Using these mice, we studied the ion channels and mechanisms controlling corticotroph excitability. Corticotrophs were either quiescent or electrically active, with a 22-mV difference in the resting membrane potential (RMP) between the 2 groups. In quiescent cells, CRH depolarized the membrane, leading to initial single spiking and sustained bursting; in active cells, CRH further facilitated or inhibited electrical activity and calcium spiking, depending on the initial activity pattern and CRH concentration. The stimulatory but not inhibitory action of CRH on electrical activity was mimicked by cAMP independently of the presence or absence of arachidonic acid. Removal of bath sodium silenced spiking and hyperpolarized the majority of cells; in contrast, the removal of bath calcium did not affect RMP but reduced CRH-induced depolarization, which abolished bursting electrical activity and decreased the spiking frequency but not the amplitude of single spikes. Corticotrophs with inhibited voltage-gated sodium channels fired calcium-dependent action potentials, whereas cells with inhibited L-type calcium channels fired sodium-dependent spikes; blockade of both channels abolished spiking without affecting the RMP. These results indicate that the background voltage-insensitive sodium conductance influences RMP, the CRH-depolarization current is driven by a cationic conductance, and the interplay between voltage-gated sodium and calcium channels plays a critical role in determining the status and pattern of electrical activity and calcium signaling.
The proopiomelanocortin (POMC) gene is highly expressed in 2 pituitary cell types, corticotrophs and melanotrophs, and transcribed to an approximately 1200-nucleotide mRNA transcript. In mammals, POMC protein is posttranslationally modulated by intracellular proteolytic cleavage in a cell type-specific manner: in corticotrophs, it gives rise to an N-terminal peptide, ACTH 1–39, and β-lipotrophic hormone, whereas in melanotrophs, POMC gives rise to melanocyte-stimulating hormone (α-MSH), γ-lipotrophin, and β-endorphin. Postnatally, melanotrophs are the only secretory cells present in the intermediate lobe and account for more than 95% of the cells found in this lobe. Corticotrophs are derived from the intermediate pituitary but, postnatally, are scattered throughout the anterior lobe (1–3). The main regulation of Pomc expression and ACTH synthesis and release in corticotrophs is mediated by CRH secreted by hypothalamic paraventricular neurons into the hypophysial portal system. CRH binds to Gs-coupled CRH receptors and increases cAMP production and ACTH release. In addition to CRH, the CRH family of peptides and arginine vasopressin (AVP) also directly stimulate ACTH release and act in synergy with CRH to potentiate ACTH release (4). Glucocorticoid receptors, expressed in corticotrophs and CRH neurons, contribute to the negative feedback of glucocorticoids on ACTH secretion (5). This hypothalamic-pituitary-adrenal axis is the major neuroendocrine system that responds to stress.
Corticotrophs are excitable cells and voltage-gated calcium (Cav) influx (VGCI) is physiologically relevant for ACTH release and other cell functions (6). One group reported that rat (7) and mouse (8) corticotrophs are electrically silent, whereas other groups described spontaneous electrical activity in these cells in the same models (9, 10). These cells may exhibit both spontaneous large-amplitude spiking and pseudoplateau bursting (9, 10) and other action potential (AP) waveforms (11). Spontaneous AP firing is associated with fluctuations in intracellular calcium concentration ([Ca2+]i) (12). Corticotrophs express the low threshold T-type and 3 types of high-threshold Cav channels (9), tetrodotoxin (TTX)-sensitive voltage-gated sodium (Nav) channels, and several potassium (K+)-conducting channels, including Ca2+-activated K+ channels (10, 11). Consistent with a hypothesis that corticotrophs fire Ca2+-dependent APs, the L-type Ca2+ channel inhibitor nifedipine abolished their spontaneous and CRH-induced spiking and cytosolic Ca2+ transients (9), whereas TTX did not affect AP firing frequency (10, 11). The ACTH-secreting human pituitary adenoma cells express T- and L-type Cav channels and exhibit TTX-insensitive spontaneous Ca2+ transients (13).
However, the ion channels and mechanisms controlling corticotroph excitability are still not well understood. Different channels may control the resting membrane potential (RMP) in these cells: a background K+ conductance mediated by inwardly rectifying potassium (Kir) channels (10), the basal activities of tandem of pore domains in a weak rectifying K+ channels-related K+ (TREK)-1 channels (8), or the background Na+ (Nab) conductance through unidentified channels (11). CRH depolarizes the RMP within tens of seconds, leading to an increase in the firing rate of spontaneously active cells and causing silent cells to become active (11, 14), but previous researchers concluded that different channels were responsible for CRH's effects. Initially, it was suggested that Kir channel inhibition is partially responsible for the membrane depolarization and enhancement of firing frequency evoked by CRH (10); another report suggested that the inhibition of TREK-1 channels by CRH, which evokes the release of arachidonic acid (AA) from pituitary cells in a cAMP-dependent manner, was responsible for the sustained depolarization (8). Others reported a decrease in the amplitude of CRH-induced depolarization and a significantly delayed depolarization in corticotrophs bathed in Na+-deficient medium, suggesting that Na+ conductance plays a role in this process (11).
In this work, we used transgenic mice expressing the fluorescent protein Discosoma red (DsRed) in POMC-producing melanotrophs and corticotrophs (15). In the initial stage of investigation, we established a protocol to distinguish between these 2 cell types. Next, we characterized the RMP, spontaneous electrical activity, and Ca2+ signaling in corticotrophs. We also studied the effects of different CRH concentrations on electrical activity and Ca2+ signaling, focusing on the role of depolarizing Ca2+, Na+, and cation conductances in CRH-induced depolarization and AP firing.
Materials and Methods
Chemicals
AA, 1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester (BayK 8644), forskolin, nifedipine, 3-isobutyl-l-methylxanthine (IBMX), 2-amino-6-trifluoromethoxybenzothiazole hydrochloride (riluzole), and TTX were purchased from Tocris Bioscience. AVP and CRH were from Bachem, and fura 2-AM, and pluronic F-127 were from Invitrogen. ATP, dibutyryl cAMP (dBcAMP), dopamine, EGTA, γ-aminobutyric acid (GABA), HEPES, N-methyl-D-glucamine (NMDG+), and nystatin were from Sigma.
Animals and procedures
Transgenic mice expressing the dimerized form of DsRed [tdimer2(12)] protein under the control of Pomc-regulatory elements were kindly provided by Dr Malcolm Low (Department of Molecular and Integrative Physiology, University of Michigan Medical School, Ann Arbor, MI). Breeding pairs of congenic C57BL/6J homozygous mice were used to produce male and female mice for pituitary harvest and anterior pituitary cell preparations. Mice were housed in shoebox cages with wood shavings bedding and added nesting material (Nestlers; ParmaServ), 2–4 mice per cage. Cages were kept in ventilated racks in an acclimated room at 21°C–23°C, and a 16-hour light, 8-hour dark cycle. Mice were provided with mouse chow and water at libitum. Euthanasia was performed by decapitation after CO2 sedation, and pituitary glands rapidly dissected and collected in ice-cold Dulbecco's PBS after removal of the neurointermediate lobe. Three to 5 mice were used for each cell preparation; pituitary glands from both sexes were used without regard to sex; in total, 18 preps were performed for electrophysiological and Ca2+ measurement. All animal procedures were approved by the National Institute of Child Health and Human Development Animal Care and Use Committee.
Cell cultures
Anterior pituitary tissue was washed in Dulbecco's PBS containing 0.3% BSA, 2mM glutamine, and MEM vitamins (Invitrogen) and treated with 0.4% trypsin for 15 minutes at 37°C. The cells were mechanically dispersed using a transfer pipette and filtered through a 40-μm nylon mesh to remove tissue fragments. Cells were harvested by centrifugation at 300g for 10 minutes, and the resulting cell pellet was resuspended in medium 199 containing Earle's salts, sodium bicarbonate, 10% heat-inactivated horse serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). For both electrophysiological and Ca2+ measurements, 0.1–0.2 million cells (in a 200-μL drop) were plated onto 25-mm glass coverslips coated with 1% poly-L-lysine and cultured in a humidified 5% CO2 atmosphere at 37°C. All experiments were performed 24–72 hours after dispersion.
Electrophysiological measurements
Membrane potentials were measured at room temperature in identified corticotrophs using the nystatin-perforated patch-clamp technique. Briefly, cells were continuously perfused with an extracellular solution containing: 150mM NaCl, 3mM KCl, 2mM CaCl2, 1mM MgCl2, 10mM HEPES, and 10mM glucose; pH was adjusted to 7.3 with NaOH. In some experiments, bath CaCl2 was omitted, and 5mM EGTA was added to the bath medium. The extracellular sodium-deficient solution contained: 155mM NMDG+, 3mM KCl, 2mM CaCl2, 1mM MgCl2, 10mM HEPES, and10mM D-glucose; adjusted to pH 7.3 with HCl. The pipette solution contained: 70mM K-aspartate, 70mM KCl, 3mM MgCl2, and 10mM HEPES; pH was adjusted to 7.2 with KOH. Before measurement, Pluronic F-127, a dispersing agent, and then nystatin were added to the intracellular solution from stock solutions (nystatin, 50 mg/mL in dimethyl sulfoxide, always freshly prepared) to obtain a final concentration of 500-μg/mL Pluronic F-127 and 250-μg/mL nystatin. Recordings were performed using an Axopatch 200B amplifier (Axon Instruments). Data were captured and stored using the pClamp 10 software packages in conjunction with a Digidata 1322A A/D converter (Axon Instruments). Solutions were delivered to the recording chamber by a gravity-driven microperfusion system (RSC-200 Rapid Solution Changer; Biologic). Electrophysiological experiments were performed at room temperature (21°C–23°C) during the first 24 hours after cell dispersion.
Intracellular calcium measurements
Calcium studies were performed at room temperature on single isolated cells of 1- to 3-day-old cultures bathed in Krebs-Ringer buffer with 2μM fura-2 AM for 60 minutes. Coverslips with cells were then mounted onto the stage of an Observer-D1 microscope (Carl Zeiss) attached to an ORCA-ER camera (Hamamatsu Photonics) and a Lambda DG-4 wavelength switcher (Sutter). Hardware control and image analysis were performed using Metafluor software (Molecular Devices). Corticotrophs were identified as smaller cells with faint red fluorescence, as described in Results, and were examined under an oil-immersion objective during exposure to alternating 340- and 380-nm light beams and the intensity of light emission at 520 nm was measured. Responses to CRH and/or AVP served as a final set of corticotroph identification criteria.
Results
Identification of melanotrophs
Single-cell electrophysiological and Ca2+ recordings were used to identify POMC-producing melanotrophs and corticotrophs in a mixed population of anterior pituitary cells from transgenic mice expressing the fluorescent protein DsRed under the control of Pomc-regulatory elements (15). We observed 2 subpopulations of DsRed-positive cells: larger-diameter cells with intense fluorescence and smaller-diameter cells that were about 2 orders of magnitude less fluorescent. The larger cells spontaneously fired APs during whole-cell recording via nystatin-perforated patch. Their activity was not affected by application of up to 100nM AVP (Figure 1A) nor application of up to 20nM CRH (Figure 1B). In these cells, dopamine (0.1μM–10μM) hyperpolarized the cell membrane and inhibited AP firing (Figure 1A). Extracellular ATP also inhibited spontaneous electrical activity due to transient hyperpolarization, consistent with the expression of Ca2+-mobilizing purinergic P2Y receptors in these cells (Figure 1B). Replacement of bath Na+ with NMDG+, a large organic cation, also hyperpolarized cells (Figure 1B), indicating that Nab conductance helps establish the RMP. In intact cells loaded with fura 2-AM, spontaneous Ca2+ transients were also not affected by application of AVP or CRH (data not shown). Dopamine, similar to its effects on AP firing, inhibited spontaneous Ca2+ transients (Figure 1C), whereas GABA induced a rise in [Ca2+]i that was abolished by subsequent dopamine application (Figure 1D). These data are in general agreement with the literature (16–18), indicating that the large cells with intense fluorescence, reflecting higher expression of POMC (19), were melanotrophs.
Figure 1.

Electrophysiological properties and calcium signaling in melanotrophs from transgenic POMC-DsRed mice. In pituitary cultures, 2 types of DsRed-positive cells were observed: larger cells displaying intense fluorescence and smaller, less fluorescent cells. Traces shown in this figure are from larger and brighter cells. A and B, Electrophysiological measurements. Spontaneous firing of APs in these cells was not affected by application of AVP but was abolished by the addition of dopamine (DA). These cells were also unresponsive to CRH application, but application of ATP caused a transient hyperpolarization, indicating the endogenous expression of a purinergic Ca2+-mobilizing P2Y receptor. Replacement of bath Na+ with NMDG+ also hyperpolarized cells, indicating the contribution of a Nab conductance to the RMP (B). C and D, Calcium measurements. A substantial fraction of cells exhibited spontaneous fluctuations in [Ca2+]i that were abolished by DA application (C). GABA facilitated a rise in [Ca2+]i, which was abolished by application of DA (D). In this and other figures, gray areas indicate treatment duration and arrows indicate the moment of drug application; drugs were present in bath medium until the end of [Ca2+]i recording. Electrophysiological and Ca2+ data represent example responses from 3–20 and 8–15 similar cells, respectively.
Identification of corticotrophs
The less-fluorescent cells were either quiescent or spontaneously active; the electrically active cells fired single APs, exhibited plateau bursting in which high amplitude APs are superimposed on a periodic depolarized potential, or underwent both single spiking and plateau bursting (Figure 2, A–C). The frequency of spontaneous APs was 1.5 ± 0.5 Hz, the single spike duration at 0 level was 3.9 ± 0.6 milliseconds, and the overshoot of spikes was, on average, 29.8 ± 4.5 mV. Nystatin-perforated whole-cell recordings also revealed that the RMP of the spontaneously active cells was less negative (−53.2 ± 1.7 mV) than that of the quiescent cells (−74.5 ± 2.1 mV) (Figure 2D).
Figure 2.

Spontaneous electrical activity and calcium signaling in corticotrophs from transgenic POMC-DsRed mice. All traces shown are from less-fluorescent cells of smaller diameter. A–D, Electrophysiological measurements. A fraction of these cells exhibited spontaneous firing of APs, 3 representative traces from different cells (A–C). Notice the firing of 2 types of APs, single spikes and plateau bursting, and that the same cell could switch from single spikes to plateau bursting. D, RMP in spontaneously active (A) and quiescent (Q) cells; mean ± SEM values. E–G, Calcium measurements. Changes in the pattern of spontaneous Ca2+ signaling during prolonged recording in an isolated corticotroph (E). Calcium transients were abolished in Ca2+-free medium and reestablished after returning Ca2+ to the medium (F). The percentage of cells exhibiting spontaneous activity increased during 3-day culture (G). D and G, Numbers in parentheses indicate number of cells and cell preparations, respectively.
Basal [Ca2+]i fluctuated in most corticotrophs, whereas the residual cells were quiescent. Two patterns of Ca2+ signaling were observed, prolonged and periodic elevation; the duration, frequencies, and amplitudes of transients varied between cells (data not shown), as well as in 1 cell across time (Figure 2E). Spontaneous Ca2+ signaling was entirely dependent on extracellular Ca2+; all cells were quiescent in Ca2+-deficient medium, and most cells resumed activity once Ca2+ was reintroduced into the extracellular medium (Figure 2F).
Spontaneous firing of AP and calcium transients was observed immediately after cell dispersion and in cells cultured for prolonged periods. Figure 2G shows mean values of percentage of corticotrophs exhibiting spontaneous electrical activity and Ca2+ transients during the first 3 days in culture. These results indicate that spontaneous excitability is intrinsic property of single corticotrophs and that cell excitability increased with increased postdispersion recovery time, which probably reflects recovery or changes in expression of ion channels after enzymatic cell dispersion.
The quiescent cells responded to CRH application with depolarization of the cell membrane (by 27. 2 ± 1.2 mV; n = 33), leading to AP firing. Figure 3A illustrates the relevance of the duration of 1nM CRH application for the initiation of spiking. A short (5–10 s) pulse of CRH initiated depolarization and stimulated short-lasting APs, whereas longer (20–30 s) application of CRH induced prolonged AP firing. In spontaneously active cells, subnanomolar CRH concentrations produced small depolarizations (by 4.7 ± 1.1 mV, n = 3), which increased the frequency of spiking (Figure 3B). Both quiescent and spontaneously active cells responded to AVP (1nM–100nM, applied for 5–10 s) with transient hyperpolarization, followed by sustained depolarization and AP firing (Figure 3, C and D). These data confirmed that the DsRed-positive and smaller diameter cells with moderate fluorescence were corticotrophs, and this subpopulation of cells was used for further studies.
Figure 3.
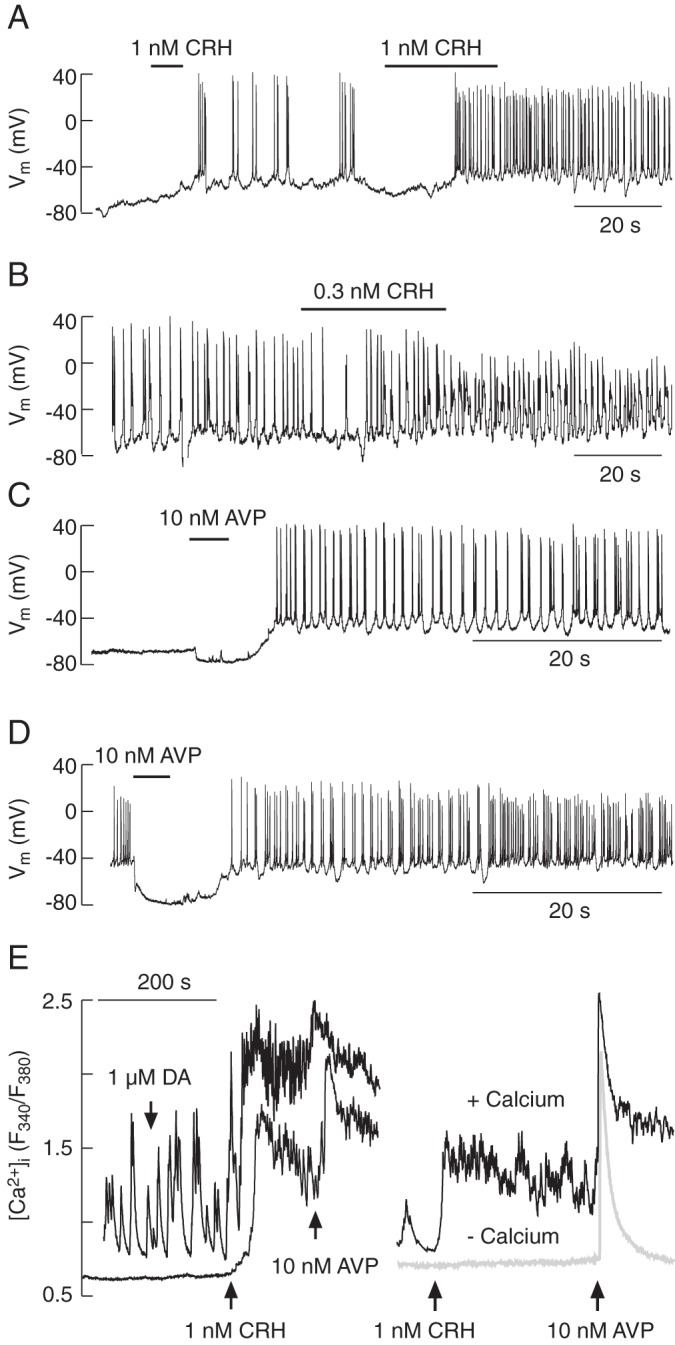
Agonist-induced electrical activity and calcium signaling in corticotrophs from transgenic POMC-DsRed mice. A–D, Electrophysiological measurements. CRH-induced electrical activity in quiescent (A) and spontaneously firing cells (B). Notice the dependence of AP frequency on the duration of CRH application. AVP-induced electrical activity in quiescent (C) and spontaneously firing cells (D). E, Calcium measurements. In both spontaneously active and quiescent cells, CRH increased [Ca2+]i, which was further elevated by application of AVP; DA was ineffective (E, left). CRH-induced rise in [Ca2+]i was only observed in cells bathed in Ca2+-containing medium (E, right). AVP induced the spike elevation in [Ca2+]i in both media, whereas the plateau calcium response was observed only in cells bathed in Ca2+-containing medium (E, right). In this and other figures, horizontal thick lines indicate the durations of treatment.
Similarly to our single-cell electrophysiological recordings, single-cell Ca2+ measurements revealed that CRH (in concentrations up to 1nM) and AVP (in the 1nM–100nM concentration range) increased [Ca2+]i, whereas dopamine application had no effect (Figure 3E, left). The CRH-induced rise in [Ca2+]i was dependent on extracellular Ca2+, as well as AVP-induced sustained response, but not early spike response (Figure 3E, right). These results are consistent with the literature (3), indicating that activation of CRH receptors with physiological agonist concentrations promotes VGCI, whereas AVP promotes Ca2+ mobilization from an intracellular pool and sustained Ca2+ influx. The rest of this study focused on CRH action in both quiescent and spontaneously active corticotrophs.
Concentration-dependent effects of CRH on excitability and calcium signaling
CRH exhibited bidirectional effects on electrical activity and Ca2+ signaling in single corticotrophs, depending on its concentration and activity status of cells. At subnanomolar concentrations, CRH only exhibited stimulatory effect on electrical activity (Figure 4A), whereas at nanomolar concentrations both stimulatory and inhibitory effects were observed (Figure 4, B and C). In quiescent cells, the stimulatory effect was initially manifested by a gradual depolarization; once the CRH-induced depolarization reached the AP threshold, 2 types of electrical activity were observed: single spiking and bursting. In 19 out of 33 quiescent corticotrophs stimulated with 1nM CRH, electrical activity started with single APs (overshoot of +30.8 ± 2.6 mV, duration 4.1 ± 0.5 ms at 0 mV, and frequency of 3.2 ± 0.4 Hz), typically lasting for a period of 5–10 seconds, the time needed to reach the peak in [Ca2+]i response, followed by plateau bursting APs (frequency of bursts = 0.59 ± 0.07 Hz; frequency of spikes within 1 burst = 10.4 ± 1.5 Hz), in which the first spike in the burst displayed overshoot similar to the initially evoked single spikes (Figure 3A). The remaining 14 corticotrophs exhibited either single spiking (n = 6) or bursting (n = 8) activity during the entire response to CRH. When applied during ongoing spontaneous or CRH-evoked activity, transient application of 1nM–100nM CRH induced a transient inhibition of spiking activity followed by increased AP frequency (Figure 4, B and C), but secondary application of high doses of CRH within a short time decreased the frequency of spiking (Figure 4C).
Figure 4.

Bidirectional effects of CRH on electrical activity and calcium signaling. A–C, Electrophysiological measurements. In quiescent cells, 100pM CRH induced gradual depolarization of the plasma membrane resting potential and initiation of electrical activity (A). In spontaneously active cells, 1nM CRH induced a transient cessation of electrical activity, followed by high-frequency AP firing, which persisted for a prolonged period after agonist washout (B). At high (100nM) concentration, CRH inhibited electrical activity and agonist washout was accompanied by recovery of electrical activity; subsequent CRH application reduced the firing frequency (C). D–F, Calcium measurements. 0.1nM CRH application elevated [Ca2+]i in quiescent cells (D). 100nM CRH application inhibited spontaneous calcium transients (E). The stimulatory effect of low (0.1nM) CRH on Ca2+ signaling was also abolished by application of 100nM CRH; Ca2+ influx was restored by application of AVP in the presence of CRH (F).
CRH (100pM) induced sustained increase in [Ca2+]i in both quiescent (Figure 4D) and spontaneously active cells. At higher CRH concentrations (1nM–100nM), prolonged application induced either a sustained inhibition of spontaneous Ca2+ transients (Figure 4E), or stimulation (data not shown). The rise in [Ca2+]i induced by low (0.1nM) CRH was also abolished by high (100nM) CRH concentration in most cells. In the presence of an inhibitory CRH concentration, AVP was able to recover VGCI (Figure 4F). In quiescent cells, 100nM CRH stimulated rise in [Ca2+]i in 2 of 12 cells; the residual cells did not respond, indicating that their silence was further strengthened by pharmacological concentrations of CRH but responded to subsequent application of AVP (data not shown).
The concentration dependence of CRH on Ca2+ signaling and electrical activity is summarized in Figure 5. The data points shown in Figure 5A represent percentage of cells responding to different CRH concentrations with an increase in [Ca2+]i, in both quiescent and spontaneously active cells, or inhibition of spontaneous Ca2+ transients and no effect in quiescent cells. In cells exhibiting stimulatory effect on [Ca2+]i, there was no dependence of the amplitude of Ca2+ signals on CRH concentration (Figure 5B). The latency period was longer at lower CRH concentrations and decreased progressively with increased agonist concentration in both electrophysiological and [Ca2+]i measurements (Figure 5C). The frequency of CRH-induced bursts of APs was also dependent on CRH concentration (Figure 5D) for both the plateau-bursting activity and pseudoplateau bursting activity, in which the periodic depolarization was higher than that of the cells exhibiting plateau-bursting activity and which had superimposed small-amplitude spikes. These results indicate that CRH action is determined by its concentration, the pattern and duration of agonist application, and the activity status of corticotrophs.
Figure 5.

Concentration dependence of CRH on electrical activity and calcium signaling. A, The concentration dependence of CRH on Ca2+ signaling. The data points represent percentage of cells responding with increase in [Ca2+]i in both quiescent and spontaneously active cells (closed circles) or inhibition of spontaneous Ca2+ transients and no effect in quiescent cells (open circles). B, No change in the peak amplitude of Ca2+ transients in response to increasing CRH concentration. C and D, A progressive decrease in the latency (period between CRH application and onset of response) (C) and increase in frequency of bursts (D) with increase in CRH concentration. Numbers in parenthesis indicate number of cells analyzed for each CRH concentration.
Effects of forskolin, dBcAMP, and AA on excitability of corticotrophs
The role of cAMP-protein kinase in stimulatory action of CRH is well established (10, 12, 20). In all quiescent cells tested, forskolin (1μM), an allosteric regulator of adenylyl cyclase capable of stimulating cAMP production in the absence of agonists, mimicked the CRH-induced depolarization of the cell membrane (CRH by 27.2 ± 1.2 mV, n = 33; forskolin by 21.7 ± 4.5 mV, n = 6) and initiated AP firing; forskolin was able to initiate both single spiking (Figure 6A) and bursting or pseudoplateau bursting activity (Figure 6B). dBcAMP (1mM), a cell-permeable cAMP analog, also depolarized the RMP (by 20.9 ± 2.8 mV; n = 3) and stimulated electrical activity in quiescent cells (Figure 6C). Single-cell recording also revealed that changes in electrical activity induced by forskolin were accompanied by rises in [Ca2+]i (Figure 6D); similarly, inhibition of endogenous phosphodiesterase activity by IBMX was accompanied by an elevation in [Ca2+]i (Figure 6E). In spontaneously firing cells, the depolarizing effect of forskolin and dBcAMP was not visible and, in further contrast to CRH, higher doses or repetitive application of forskolin did not inhibit AP firing (data not shown). These results confirm that cAMP mediates CRH's stimulatory effect on electrical activity and associated VGCI, but does not account for CRH-induced inhibition of spontaneous excitability.
Figure 6.
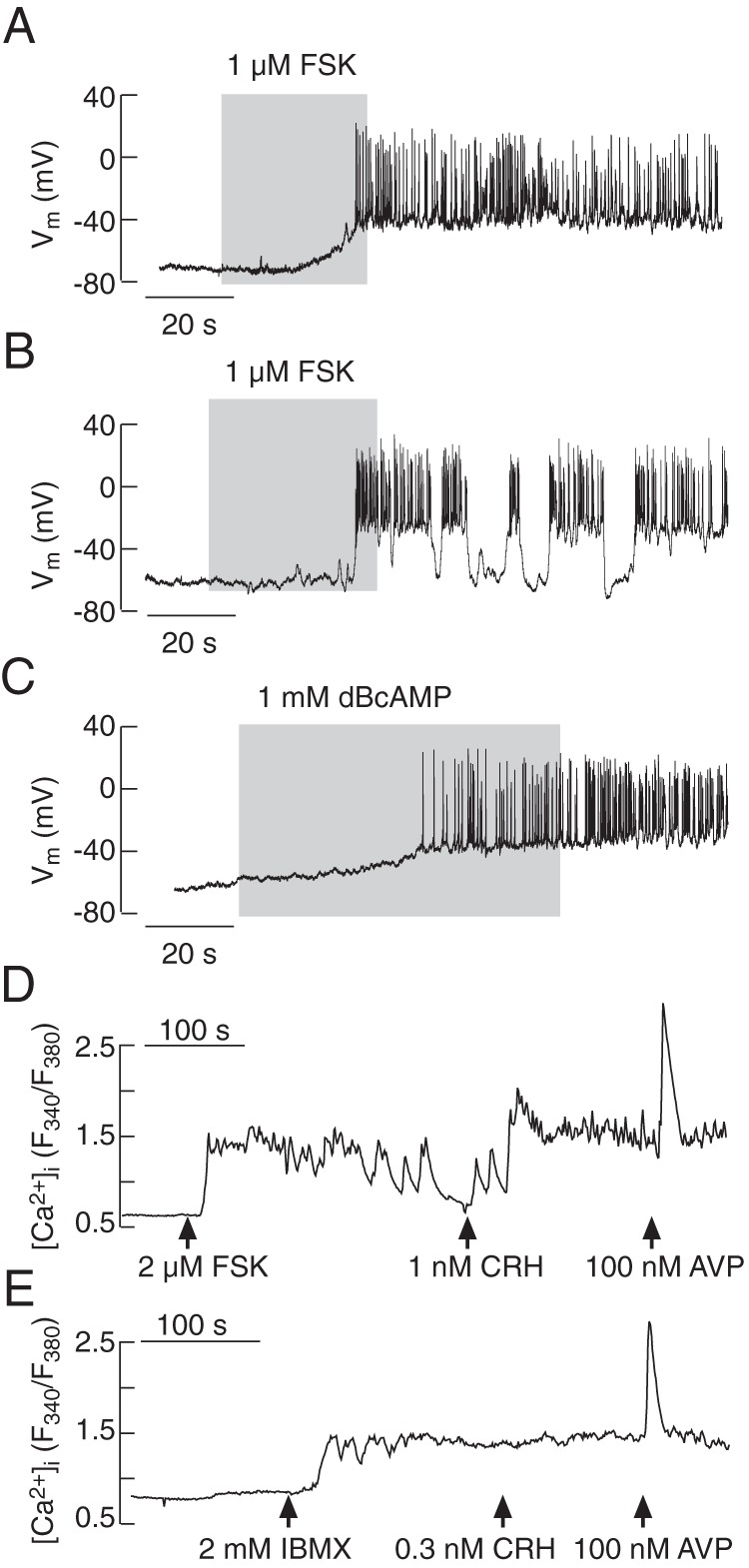
Elevation in cAMP levels mimicked CRH-induced stimulatory effects on electrical activity (A–C) and calcium signaling (D and E). Forskolin (FSK), an allosteric activator of adenylyl cyclase, induced depolarization and stimulated single spiking (A) or pseudoplateau-bursting activity (B). The cell permeable cAMP analog dBcAMP also evoked depolarization and initiated electrical activity (C). Forskolin also stimulated a rise in [Ca2+]i (D), as did IBMX, a common inhibitor of phosphodiesterases (E); in both cases, the subsequent application of AVP stimulated a rise in [Ca2+]i.
It has been reported that AA signaling pathway mediates CRH-induced depolarization of corticotrophs (8); in our experiments, 10μM AA displayed no effect in quiescent cell, but hyperpolarized most spontaneously active cells and CRH-stimulated corticotrophs and inhibited ongoing CRH-induced firing (Figure 7A). CRH in the presence of AA, however, still depolarized the quiescent cells and initiated AP firing (Figure 7B). In the presence of 10μM AA, CRH was also able to elevate [Ca2+]i (Figure 7C). The mean ± SEM values for CRH-induced changes in RMP in the presence and absence of AA as well as effects of AA alone in quiescent and spontaneously active cells are summarized in Figure 7D. These results indicate that AA silences electrical activity in corticotrophs, an action consistent with the TREK-1 or other AA-regulated channel participation in RMP of electrically active cells, but the depolarizing effect of CRH is not affected by AA treatment.
Figure 7.
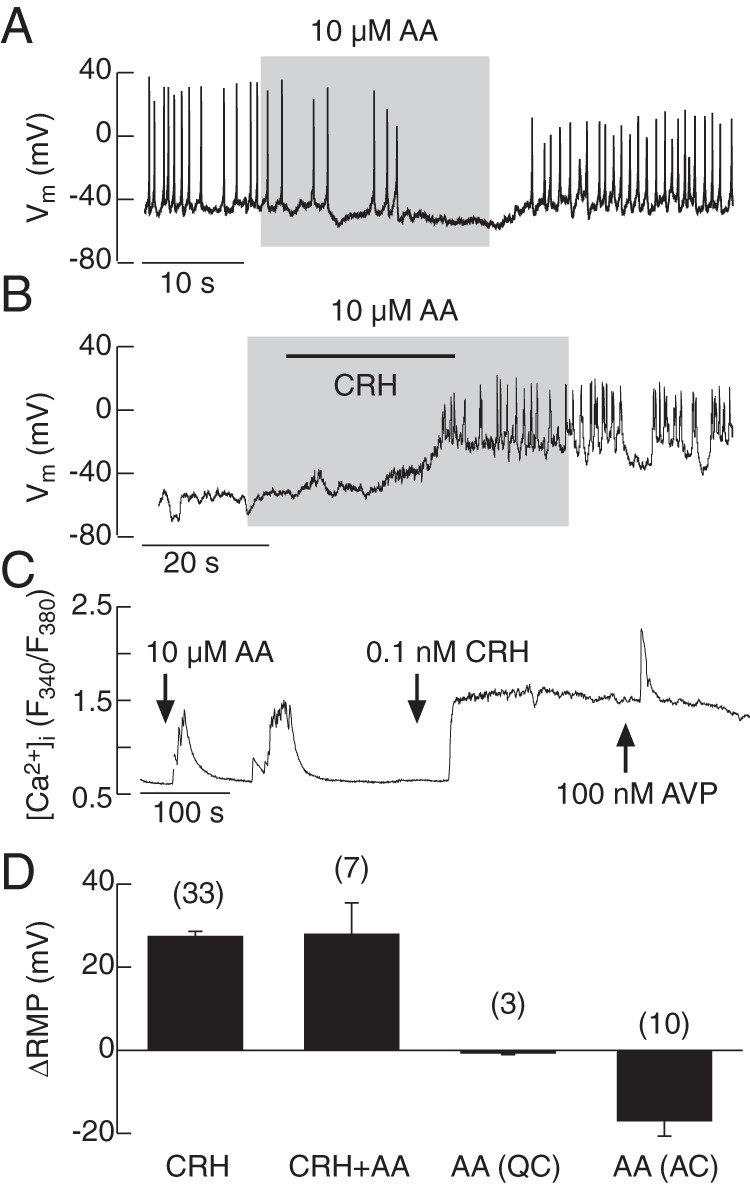
Effects of AA on spontaneous and CRH-induced electrical activity (A and B) and calcium signaling (C). AA induced hyperpolarization and inhibited APs in spontaneously firing corticotrophs (A). In quiescent cells in the presence of AA, CRH was able to depolarize the cell membrane and initiate AP firing (B). CRH facilitated Ca2+ influx in the presence of AA (C). Effects of CRH alone and in the presence of AA as well as AA alone in quiescent cells (QCs) in spontaneously active cells (ACs) on RMP; mean ± SEM values.
The role of Na+ currents in electrical activity
There are discrepancies in the literature whether or not sodium conductance plays a role in RMP and electrical activity of corticotrophs (10, 11, 21–23). In our experiments, removal of bath sodium hyperpolarized the plasma membrane and abolished spontaneous AP firing in most corticotrophs (22 of 37 cells) (Figure 8A), indicating that the Nab conductance contributes to RMP. In cells exhibiting prominent Nab conductances, replacement of bath Na+ with NMDG+ also abolished CRH-induced membrane depolarization and electrical activity in a reversible manner (Figure 8B). Both types of APs, single spiking and bursting, were abolished by the removal of bath Na+; spontaneous [Ca2+]i transients were also abolished by removal of bath Na+ (Figure 8C).
Figure 8.

Dependence of spontaneous and CRH-induced firing of APs and calcium transients on sodium conductance. A–E, The role of the Nab conductance in corticotroph excitability. Substitution of bath Na+ with NMDG+ caused deep hyperpolarization and abolished spontaneous AP firing (A and B) and Ca2+ transients (C) in most corticotrophs. Removal of bath Na+ also abolished CRH-induced electrical activity (B and D). In cells lacking basal Na+ conductance and bathed in Na+-free media, CRH was able to depolarize the plasma membrane and initiate the firing of low amplitude APs; the amplitude and frequency of spiking increased after the return of bath Na+ (E). F–I, The role of Nav channels in corticotroph excitability. Application of riluzole (F) and TTX (G and H), specific blockers of Nav channels, did not abolish spontaneous or CRH-induced firing of APs but reduced the amplitude of spiking. TTX did not alter the pattern of CRH-induced calcium signaling (I).
In the residual corticotrophs (15 of 37), the Nab conductance was not prominent; such cells also responded to CRH application with depolarization and AP firing. A brief removal of bath Na+ abolished spontaneous (data not shown) or ongoing CRH-induced spiking activity (Figure 8D). Furthermore, CRH-induced depolarization observed in cells bathed in Na+-free medium was comparable with that observed in control medium (in controls by 27.2 ± 1.2 mV, n = 33; in Na+-free by 27.4 ± 5.2 mV, n = 6) and was associated with the firing of lower-amplitude APs (Figure 8E). The return of the bath Na+ increased both the frequency and amplitude of spikes (Figure 8E); however, CRH-induced [Ca2+]i increases were always inhibited after the substitution of bath Na+ with NMDG+ (data not shown). These findings demonstrate the relevance of a Nab conductance in RMP but less so in CRH-induced membrane depolarization, which appears to depend on bath Na+ and other cation(s). Furthermore, CRH-triggered APs in cells bathed in NMDG+ medium do not generate global Ca2+ signals.
To examine the contribution of Nav channels in RMP and AP firing, we used the Nav channel blockers riluzole (24) and TTX (25). None of the blockers mimicked the effects of NMDG+ on RMP, nor abolished spontaneous electrical activity, but they did reduce the amplitude of spontaneous and CRH-induced APs. CRH in the presence of riluzole (50μM) was able to depolarize the cell membrane and initiate AP firing; riluzole washout increased the amplitude of CRH-induced APs (Figure 8F). The amplitude of spontaneous APs was reduced during the treatment with TTX (0.5μM) and for a long time after its washout (Figure 8G). TTX also reduced the amplitude of CRH-induced (Figure 8H) and forskolin-induced (data not shown) plateau bursting or pseudoplateau bursting activity. In average, TTX reduced the amplitude of overshoot from +38.5 ± 2.8 to −1.2 ± 3.1 mV (n = 9) and from +36.6 ± 1.6 to +2.5 ± 0.5 mV (n = 5) in CRH-stimulated and spontaneously active corticotrophs, respectively. Thus, all corticotrophs display a TTX- and riluzole-sensitive Nav current that is involved in the high amplitudes of single spikes and burst APs; these channels do not contribute to the control of RMP or CRH-induced slow membrane depolarization. Furthermore, blockade of Nav channels did not obviously affect CRH- or AVP-induced elevations in [Ca2+]i (Figure 8I), indicating that Ca2+-dependent APs are sufficient to drive CRH-induced VGCI.
The role of calcium currents in electrical activity
The expression of Cav channels is well established in corticotrophs but not their roles in RMP and CRH-induced depolarization (7, 9, 13, 26). In contrast to the removal of bath Na+, removal of bath Ca2+ did not hyperpolarize the RMP or abolish spontaneous electrical activity but significantly reduced the frequency of spiking without affecting AP amplitudes (Figure 9A). The CRH-induced electrical activity was also preserved in cells bathed in Ca2+-free medium but with a reduced AP frequency (Figure 9B). Depolarization induced by CRH was significantly reduced but not abolished in the absence of extracellular Ca2+ (control: 27.2 ± 1.7 mV, n = 33; Ca2+ free: 8.5 ± 4.2 mV, n = 4), indicating that Ca2+ and Na+ can substitute for each other in CRH-induced depolarization. In cells bathed in Ca2+-free medium, CRH initiated the firing of single spikes, and the return of bath Ca2+ caused a transition from single spikes to plateau bursting (Figure 9C). Figure 9D summarizes effects of extracellular Ca2+ removal on frequency and amplitude of both spontaneous and CRH-stimulated APs.
Figure 9.
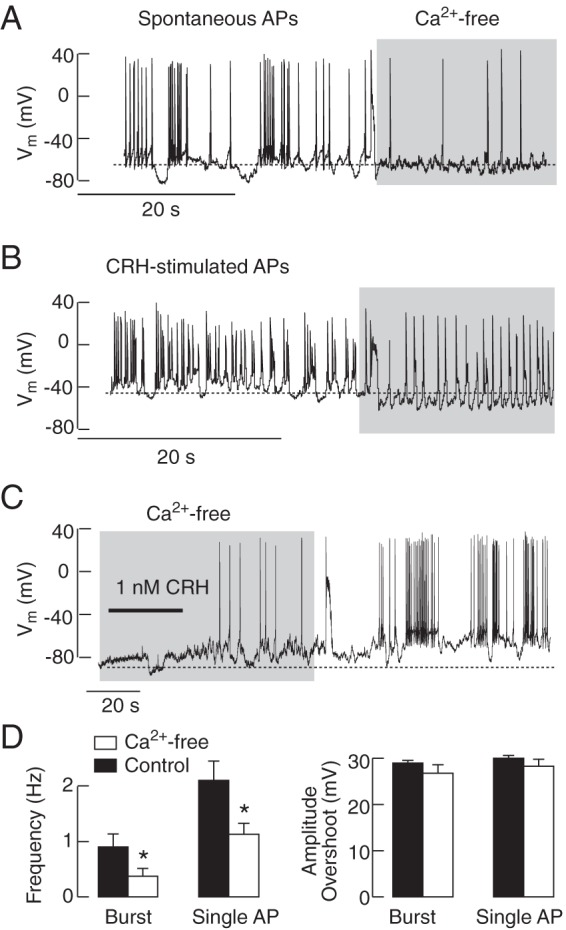
Dependence of spontaneous and CRH-induced electrical activity on Ca2+ influx. Removal of bath Ca2+ did not abolish spontaneous (A) and CRH-induced (B) electrical activity but decreased the frequency of spiking. In cells bathed in Ca2+-deficient medium, CRH was able to depolarize the membrane and initiate spiking (C). Notice the lack of effect of removal of bath Ca2+ on RMP (A) and changes in CRH-induced depolarization (B and C). In summary, removal of bath Ca2+ reduced the frequency of both bursts and single APs (D, left) but did not affect the amplitudes of overshoot (D, right); mean ± SEM values; asterisks indicate P < .05.
To examine the contribution of L-type Cav channels to spontaneous and CRH-induced electrical activity and Ca2+ signaling, we used BayK 8644 (1μM–3μM), and nifedipine (1μM), an activator and blocker of these channels, respectively. Application of BayK 8644 increased the amplitude of [Ca2+]i transients in spontaneously active corticotrophs (Figure 10A) and initiated spiking in quiescent cells (Figure 10B). The stimulatory effect of BayK 8644 was observed in 71% of corticotrophs (Figure 10D, left). In the residual quiescent cells, BayK 8644 was unable to initiate spiking (Figure 10, C and D, left). BayK 8644-stimulated Ca2+ spiking was inhibited in the majority (63%) of cells by 100nM CRH (Figure 10, A and D, right) and facilitated or did not affect the residual cells (Figure 10, B and D, right).
Figure 10.

Role of L-type Cav channels in spontaneous and CRH-induced calcium signaling and electrical activity. A–D, Calcium measurements. In most spontaneously active corticotrophs, 1μM BayK 8644, an L-type Cav channel agonist, increased the amplitude of Ca2+ transients (A) and initiated Ca2+ transients in a fraction of quiescent cells (B) but was ineffective in the residual cells (C). 100nM CRH could inhibit (A) or stimulate (B) Ca2+ influx in cells responding to 1μM BayK 8644 application. D, Percentage of cells responding (+) and not responding (−) to 1μM BayK 8644 application with increase in [Ca2+]i (left) and percentage of cells responding to 100nM CRH application with abolition (−) or no effect/potentiation (+) of BayK 8644-induced Ca2+ transients (right). E–H, Electrophysiological measurements. In a cell firing APs initiated by the previous application of CRH, 1μM BayK 8644, transformed the plateau-bursting type of electrical activity into pseudoplateau bursting, accompanied with transient hyperpolarization in the presence and absence of 1μM TTX (E and F); in the presence of TTX, 1μM BayK 8644 was unable to generate plateau-bursting (E). Treatment with 1μM nifedipine, a blocker of L-type Cav channels, reduced the frequency of firing (G), whereas treatment with 1μM nifedipine and 1μM TTX abolished AP firing (H).
In parallel to single-cell [Ca2+]i measurements, BayK 8644 initiated electrical activity in a fraction of corticotrophs (data not shown) or was without effect (Figure 10E). Furthermore, BayK 8644 transformed single spiking into pseudoplateau bursting activity by potentiation of hyperpolarization between bursts (from −48 ± 2.5 to −76.7 ± 3.4 mV, n = 11) (Figure 10F); the hyperpolarizing effect was also observed in the presence of TTX (Figure 10E). Nifedipine (1μM) reduced the frequency but not the amplitude of spontaneous single spiking (control, 1.93 ± 0.52 Hz; nifedipine treated, 1.31 ± 0.46 Hz) (Figure 10G) and displayed a minor effect on CRH-induced depolarization (14 ± 4.4 mV). Nifedipine also affected the frequency of CRH-induced bursts (control: 0.72 ± 0.1 Hz, nifedipine treated: 0.58 ± 0.09 Hz; n = 5). The combination of TTX and nifedipine completely inhibited CRH-induced spiking; the washout of nifedipine and TTX was accompanied by a slow recovery of spontaneous firing (Figure 10H). These results indicate that L-type Cav channels contribute to the spiking depolarization but not to RMP or CRH-induced cell membrane depolarization.
Discussion
Our investigations indicate that isolated corticotrophs were either silent or spontaneously fired APs that were accompanied by Ca2+ transients. We further show that variances in RMP are determined by the level of Nab conductance and provide an explanation for the differences in firing status. Physiological concentrations of CRH depolarize the cell membrane by facilitating cation influx and initiating spiking in quiescent cells. Depending on the status of Ca2+ entry, cells could fire single spikes or exhibit plateau bursting or pseudobursting; CRH-induced spiking depolarization is mediated by both Nav and Cav channels. At pharmacological concentrations, CRH also inhibits spontaneous or agonist-induced electrical activity and calcium signaling.
Resting membrane potential
Using nystatin-perforated whole-cell recording, we found that RMPs of quiescent corticotrophs were around −75 mV, whereas RMPs of spontaneously active cells were significantly more positive, around −53 mV. Other studies reported similar RMPs for spontaneously active cells, about −54 or −55 mV (perforated whole-cell recording) (11, 27) and −48 mV (standard whole-cell recording) (6) in cultured murine corticotrophs, around −60 mV in rat corticotrophs (10), and between −45 and −55 mV in immortalized AtT-20 corticotrophs (21). In general, RMP is determined by the background depolarizing and hyperpolarizing currents (28). Two hyperpolarizing K+ channels were suggested to contribute to the RMP in corticotrophs: a background K+ conductance mediated by Kir channels (10) and/or TREK-1 channels (8). On the other hand, the voltage-insensitive Nab conductance (see reference 31 below) has been identified as a depolarizing current that contributes to the establishment of the RMP in corticotrophs (11) and other pituitary cell types (29–31). We confirmed here that the inward Nab conductance contributes to the RMP in DsRed mouse corticotrophs and show that the expression of this conductance varies among cells, which consequently determines their electrical status. We also observed that the spontaneous excitability of corticotrophs is low during the first 24 hours in culture and progressively increases with the age of the cultures; this suggests that dispersion of cells affects the expression and/or function of channels that account for the background conductance. We also show that Ca2+ cannot substitute for Na+ as a carrier for these channels, ie, that they are specific for Na+. Although the nature of channels accounting for the Nab conductance remains to be determined, the present experiments with TTX and riluzole clearly eliminated the contribution of Nav channels to this conductance.
CRH-induced depolarizing current
It is well established that CRH induces corticotroph depolarization. Liang et al observed about 15 mV of depolarization after 2 minutes of stimulation with a mixture of CRH and AVP (11). Other studies reported CRH-induced depolarization of about 10 mV (10) or 6 mV (27) for spontaneously active murine corticotrophs and 21 mV (8) for quiescent corticotrophs. Here, we demonstrated that the amplitude of CRH-induced depolarization of corticotrophs is determined by the electrical status of cells; in quiescent cells, CRH depolarized cells by 27 mV, and in spontaneously firing cells, by 5 mV, on average. Consistent with the view that cAMP mimics the action of CRH in corticotroph depolarization and the facilitation of spiking and Ca2+ signaling (7, 8, 12), we show here that elevations in intracellular cAMP by forskolin, dBcAMP, or IBMX depolarize DsRed corticotrophs and facilitate electrical activity and Ca2+ signaling.
It has been postulated that the currents that control the RMP also account for CRH action, ie, that inhibition of outward Kir channels partially contributes to the membrane depolarization and enhancement of firing frequency evoked by CRH (10). Another group suggested that inhibition of TREK-1 channels by CRH accounts for sustained depolarization (8). A decrease in the amplitude of CRH-induced depolarization and a significantly delayed depolarization in corticotrophs bathed in Na+-deficient medium was also reported, suggesting that the Na+ conductance plays a role in this process (11). However, the present study provides 2 lines of evidence arguing against the hypothesis that Nab conductance accounts for the bath Na+ dependence of CRH action. First, the Nab channel is specific for Na+, as discussed above, and thus would not permit other ions as carriers, whereas CRH-induced depolarization in the presence of NMDG+ clearly indicates that other ions substitute for Na+. Second, CRH-induced depolarization was significantly reduced in cells bathed in Ca2+-deficient medium, indicating that VGCI contributes to the CRH-induced depolarization of corticotrophs. Thus, it is reasonable to suggest that the gating of cation-conducting channels is facilitated by CRH. This is in accord with our recent report concerning cAMP-activated nonselective cation conductances in several other secretory pituitary cell types (31). Further studies are needed to identify the channels responsible for this conductance and their expression and function in corticotrophs.
Interplay between Nav and Cav channels in spiking activity
It has been reported that rat (9, 10) and mouse (11) corticotrophs either fire APs spontaneously or were silent (7, 8). AtT-20 immortalized corticotrophs also fire APs spontaneously (21) as do human ACTH-secreting pituitary adenoma cells (13). Our results further support the hypothesis that corticotrophs fire APs under basal conditions. The discrepancy about the electrical status of isolated corticotrophs could reflect the variability in the recording conditions, ie, perforated vs. standard whole-cell recordings, the loss of some intracellular factor(s) that control the background conductance in standard recordings, or the age of pituitary cultures.
Corticotrophs express several voltage-gated channels, including Cav channels (9) and TTX-sensitive Nav channels (10). Ritchie's group also observed that these cells exhibit Ca2+-dependent spike depolarization and that nifedipine abolished spontaneous and CRH-induced APs and cytosolic Ca2+ transients, indicating the role of L-type Cav channels in spiking (9), whereas TTX did not affect AP firing (10, 11). The ACTH-secreting human pituitary adenoma cells also express Cav channels and exhibit spontaneous Ca2+ transients that were not abolished by TTX (13). Our [Ca2+]i measurements also show no obvious change in the pattern of spontaneous and CRH-induced Ca2+ signaling after application of TTX; however, we and others (11, 27) found that TTX selectively reduces the amplitude of single APs by abolishing the overshoot. On the other hand, in cells bathed in Ca2+-deficient medium, there was no decrease in the amplitude of APs, whereas simultaneous application of TTX and nifedipine abolished spiking. Thus, in the DsRed corticotrophs, both Nav and Cav channels contribute to spike depolarization; cells with blocked Nav channels fire Ca2+-dependent APs, whereas cells with blocked Cav channels fire Na+-dependent APs. Among secretory anterior pituitary cells from rodents, the contribution of TTX-sensitive Nav channels to spiking depolarization appears to be specific for a subpopulation of corticotrophs (3).
Heterogeneity of spontaneous and CRH-modulated electrical activity
In keeping with previous reports (9, 11, 27), the present study clearly shows that corticotrophs exhibit different patterns of electrical activity. We observed 3 types of electrical activity: single spiking, plateau bursting, and pseudoplateau bursting. Under basal conditions, most of the DsRed corticotrophs fired single spikes, whereas bursting activity dominated during the sustained CRH stimulation. In quiescent cells, CRH initially induced single spikes, followed by spike-bursting activity. Facilitation of VGCI by BayK 8644 regularly caused a transition from plateau bursting to pseudoplateau bursting. Finally, we observed that the blockade of VGCI by removal of bath calcium caused a transition from bursting to single spiking.
The initial work with the corticotrophs suggested that spontaneously active cells fire single APs, CRH causes a transition to bursting activity, and AVP only increases the frequency of single spiking (27). Our data provide further clarification: the transitions “single spiking → plateau bursting → pseudoplateau bursting” and vice versa reflect changes in VGCI, which occurs spontaneously and is facilitated by both CRH (more efficiently) and AVP (less efficiently). In general, these findings are consistent with experimental and theoretical work, indicating a critical role for Ca2+-activated big conductance K+ channels in the bursting activity of pituitary cells (32–34). However, further experimental work is needed to identify channels contributing to different patterns of electrical activity and the role of Ca2+ in this process.
Bidirectional effect of CRH on electrical activity and calcium signaling
The threshold concentration for CRH stimulation of ACTH secretion in cultured rat anterior pituitary cells is about 10pM (35) and reported physiological CRH levels in rat pituitary portal circulation are 100–500 pg/mL, ie, 20pM–100pM (36, 37). These basal concentrations obtained from collections under urethane anesthesia may be overestimated due to surgical stress. Using pentobarbital, an anesthetic known to increase HPA axis activity, basal concentrations levels are about 3-fold higher (38). Similarly, a 5-fold increase has been shown in sheep after endotoxin injection (39). Here, we show that effect of CRH on electrical activity depends on its concentration; at lower concentrations stimulatory effects dominate, whereas at higher concentrations inhibitory effect is more frequently present. Such concentration dependence of CRH action indicates that the status of RMP and resting activity do not account for bidirectional effect of CRH on electrical activity.
Two additional observations further support this conclusion. First, most quiescent cells respond to subnanomolar CRH application with increase in [Ca2+]i, whereas most quiescent cells stayed silent during application of nanomolar CRH concentrations, indicating that the inhibitory pathway was activated. Second, the inhibitory effect of CRH is inconsistent with the role of cAMP in this process because, in cultured pituitary cells, CRH increases cAMP production in a concentration-dependent manner, reaching the peak in production at 100nM CRH (40). Forskolin and dBcAMP mimic the stimulatory action of CRH on adenylyl cyclase and electrical activity, but not the inhibitory action, arguing against the role of the cAMP-dependent kinase signaling pathway in the later process. Similarly, the lack of effect of repeated AVP to inhibit electrical activity (H. Zemkova, G. Aguilera, and S. S. Stojilkovic, unpublished observations) suggests that the phospholipase C-dependent signaling pathway is not involved. The finding that CRH stimulates the phospholipase A signaling pathway and increases in AA production (41, 42), combined with the present demonstration that AA hyperpolarizes corticotrophs, raised the possibility that this signaling pathway accounts for the inhibitory effect of high or repetitive CRH application on electrical activity. AA could influence the gating of other channels (43, 44) in addition to TREK-1, or have nonspecific effects in membrane function. However, our observations that CRH depolarized cells and facilitated VGCI in the presence of high concentrations of exogenous AA argues against this possibility.
Conclusions
The data clearly show that RMP in corticotrophs depends on background voltage-insensitive Na+ conductance, and that CRH has biphasic actions: a depolarizing effect driven by a cationic conductance at basal concentrations in pituitary portal circulation and an inhibitory effect at higher stress CRH concentrations. Although further studies are needed to identify the pathways involved in the inhibitory phase, the silencing effect of high CRH concentrations on electrical activity may contribute to the corticotroph desensitization observed in some conditions associated with elevated CRH output, such as melancholic depression (45). Furthermore, during acute stress responses (46), the effect of AVP to overcome the inhibitory effect of high CRH concentrations could be relevant for maintaining corticotroph responsiveness in the presence of elevated CRH secretion.
Acknowledgments
This work was supported by the Grant Agency of the Czech Republic P304/12/G069, the Ministry of Education, Youth and Sports of CR within the LQ1604 National Sustainability Program II (Project BIOCEV-FAR) and by the project “BIOCEV” (CZ.1.05/1.1.00/02.0109) (to H.Z.), and by a grant from the Intramural Research Program of the National Institute of Child Health and Human Development, National Institute of Health (M.T., M.K., G.A., and S.S.S.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- arachidonic acid
- AP
- action potential
- AVP
- arginine vasopressin
- BayK 8644
- 1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester
- [Ca2+]i
- intracellular calcium concentration
- Cav
- voltage-gated calcium
- dBcAMP
- dibutyryl cAMP
- DsRed
- fluorescent protein Discosoma red
- GABA
- γ-aminobutyric acid
- IBMX
- 3-isobutyl-l-methylxanthine
- Kir
- inwardly rectifying potassium
- Nab
- background Na+
- Nav
- voltage-gated sodium
- NMDG+
- N-methyl-D-glucamine
- POMC
- proopiomelanocortin
- riluzole
- 2-amino-6-trifluoromethoxybenzothiazole hydrochloride
- RMP
- resting membrane potential
- TREK
- tandem of pore domains in a weak rectifying K+ channels-related K+
- TTX
- tetrodotoxin
- VGCI
- Cav influx.
References
- 1. Raffin-Sanson ML, de Keyzer Y, Bertagna X. Proopiomelanocortin, a polypeptide precursor with multiple functions: from physiology to pathological conditions. Eur J Endocrinol. 2003;149(2):79–90. [DOI] [PubMed] [Google Scholar]
- 2. Jenks BG. Regulation of proopiomelanocortin gene expression: an overview of the signaling cascades, transcription factors, and responsive elements involved. Ann NY Acad Sci. 2009;1163:17–30. [DOI] [PubMed] [Google Scholar]
- 3. Stojilkovic SS, Tabak J, Bertram R. Ion channels and signaling in the pituitary gland. Endocr Rev. 2010;31(6):845–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aguilera G, Nikodemova M, Wynn PC, Catt KJ. Corticotropin releasing hormone receptors: two decades later. Peptides. 2004;25(3):319–329. [DOI] [PubMed] [Google Scholar]
- 5. McNicol AM, Carbajo-Perez E. Aspects of anterior pituitary growth, with special reference to corticotrophs. Pituitary. 1999;1(3–4):257–268. [DOI] [PubMed] [Google Scholar]
- 6. Tse A, Lee AK, Tse FW. Ca2+ signaling and exocytosis in pituitary corticotropes. Cell Calcium. 2012;51(3–4):253–259. [DOI] [PubMed] [Google Scholar]
- 7. Lee AK, Tse A. Mechanism underlying corticotropin-releasing hormone (CRH) triggered cytosolic Ca2+ rise in identified rat corticotrophs. J Physiol. 1997;504(pt 2):367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee AK, Smart JL, Rubinstein M, Low MJ, Tse A. Reciprocal regulation of TREK-1 channels by arachidonic acid and CRH in mouse corticotropes. Endocrinology. 2011;152(5):1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuryshev YA, Childs GV, Ritchie AK. Corticotropin-releasing hormone stimulates Ca2+ entry through L- and P-type Ca2+ channels in rat corticotropes. Endocrinology. 1996;137(6):2269–2277. [DOI] [PubMed] [Google Scholar]
- 10. Kuryshev YA, Haak L, Childs GV, Ritchie AK. Corticotropin releasing hormone inhibits an inwardly rectifying potassium current in rat corticotropes. J Physiol. 1997;502(pt 2):265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang Z, Chen L, McClafferty H, et al. Control of hypothalamic-pituitary-adrenal stress axis activity by the intermediate conductance calcium-activated potassium channel, SK4. J Physiol. 2011;589(pt 24):5965–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuryshev YA, Childs GV, Ritchie AK. Corticotropin-releasing hormone stimulation of Ca2+ entry in corticotropes is partially dependent on protein kinase A. Endocrinology. 1995;136(9):3925–3935. [DOI] [PubMed] [Google Scholar]
- 13. Guérineau N, Corcuff JB, Tabarin A, Mollard P. Spontaneous and corticotropin-releasing factor-induced cytosolic calcium transients in corticotrophs. Endocrinology. 1991;129(1):409–420. [DOI] [PubMed] [Google Scholar]
- 14. Ritchie AK, Kuryshev YA, Childs GV. Corticotropin-releasing hormone and calcium signaling in corticotropes. Trends Endocrinol Metab. 1996;7(10):365–369. [DOI] [PubMed] [Google Scholar]
- 15. Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29(43):13684–13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shibuya I, Kongsamut S, Douglas WW. Effectiveness of GABAB antagonists in inhibiting baclofen-induced reductions in cytosolic free Ca concentration in isolated melanotrophs of rat. Br J Pharmacol. 1992;105(4):893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Douglas WW, Shibuya I. Calcium signals in melanotrophs and their relation to autonomous secretion and its modification by inhibitory and stimulatory ligands. Ann NY Acad Sci. 1993;680:229–245. [DOI] [PubMed] [Google Scholar]
- 18. Shibuya I, Douglas WW. Spontaneous cytosolic calcium pulsing detected in Xenopus melanotrophs: modulation by secreto-inhibitory and stimulant ligands. Endocrinology. 1993;132(5):2166–2175. [DOI] [PubMed] [Google Scholar]
- 19. Stefaneanu L, Kovacs K, Horvath E, Lloyd RV. In situ hybridization study of pro-opiomelanocortin (POMC) gene expression in human pituitary corticotrophs and their adenomas. Virchows Arch A Pathol Anat Histopathol. 1991;419(2):107–113. [DOI] [PubMed] [Google Scholar]
- 20. Lee AK, Tse FW, Tse A. Arginine vasopressin potentiates the stimulatory action of CRH on pituitary corticotropes via a protein kinase C-dependent reduction of the background TREK-1 current. Endocrinology. 2015;156(10):3661–3672. [DOI] [PubMed] [Google Scholar]
- 21. Surprenant A. Correlation between electrical activity and ACTH/β-endorphin secretion in mouse pituitary tumor cells. J Cell Biol. 1982;95(2 pt 1):559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takano K, Takei T, Teramoto A, Yamashita N. GHRH activates a nonselective cation current in human GH-secreting adenoma cells. Am J Physiol. 1996;270(6 pt 1):E1050–E1057. [DOI] [PubMed] [Google Scholar]
- 23. Mollard P, Vacher P, Guerin J, Rogawski MA, Dufy B. Electrical properties of cultured human adrenocorticotropin-secreting adenoma cells: effects of high K+, corticotropin-releasing factor, and angiotensin II. Endocrinology. 1987;121(1):395–405. [DOI] [PubMed] [Google Scholar]
- 24. Narahashi T, Huang CS, Song JH, Yeh JZ. Ion channels as targets for neuroprotective agents. Ann NY Acad Sci. 1997;825:380–388. [DOI] [PubMed] [Google Scholar]
- 25. Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57(4):397–409. [DOI] [PubMed] [Google Scholar]
- 26. Kuryshev YA, Childs GV, Ritchie AK. Three high threshold calcium channel subtypes in rat corticotropes. Endocrinology. 1995;136(9):3916–3924. [DOI] [PubMed] [Google Scholar]
- 27. Duncan PJ, engül S, Tabak J, Ruth P, Bertram R, Shipston MJ. Large conductance Ca2-activated K (BK) channels promote secretagogue-induced transition from spiking to bursting in murine anterior pituitary corticotrophs. J Physiol. 2015;593(5):1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hille B. Ion Channels of Excitable Cells. Sunderland, MA: Sinauer Associates, Inc; 2001. [Google Scholar]
- 29. Simasko SM. A background sodium conductance is necessary for spontaneous depolarizations in rat pituitary cell line GH3. Am J Physiol. 1994;266(3 pt 1):C709–C719. [DOI] [PubMed] [Google Scholar]
- 30. Sankaranarayanan S, Simasko SM. A role for a background sodium current in spontaneous action potentials and secretion from rat lactotrophs. Am J Physiol. 1996;271(6 pt 1):C1927–C1934. [DOI] [PubMed] [Google Scholar]
- 31. Tomic M, Kucka M, Kretschmannova K, Li S, Nesterova M, Stratakis CA, Stojilkovic SS. Role of nonselective cation channels in spontaneous and protein kinase A-stimulated calcium signaling in pituitary cells. Am J Physiol Endocrinol Metab. 2011;301(2):E370–E379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Goor F, Li YX, Stojilkovic SS. Paradoxical role of large-conductance calcium-activated K+ (BK) channels in controlling action potential-driven Ca2+ entry in anterior pituitary cells. J Neurosci. 2001;21(16):5902–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsaneva-Atanasova K, Sherman A, van Goor F, Stojilkovic SS. Mechanism of spontaneous and receptor-controlled electrical activity in pituitary somatotrophs: experiments and theory. J Neurophysiol. 2007;98(1):131–144. [DOI] [PubMed] [Google Scholar]
- 34. Tabak J, Tomaiuolo M, Gonzalez-Iglesias AE, Milescu LS, Bertram R. Fast-activating voltage- and calcium-dependent potassium (BK) conductance promotes bursting in pituitary cells: a dynamic clamp study. J Neurosci. 2011;31(46):16855–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deng Q, Riquelme D, Trinh L, et al. Rapid glucocorticoid feedback inhibition of ACTH secretion involves ligand-dependent membrane association of glucocorticoid receptors. Endocrinology. 2015;156(9):3215–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fink G, Robinson IC, Tannahill LA. Effects of adrenalectomy and glucocorticoids on the peptides CRF-41, AVP and oxytocin in rat hypophysial portal blood. J Physiol. 1988;401:329–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Makara GB, Sutton S, Otto S, Plotsky PM. Marked changes of arginine vasopressin, oxytocin, and corticotropin-releasing hormone in hypophysial portal plasma after pituitary stalk damage in the rat. Endocrinology. 1995;136(5):1864–1868. [DOI] [PubMed] [Google Scholar]
- 38. Sheward WJ, Fink G. Effects of corticosterone on the secretion of corticotrophin-releasing factor, arginine vasopressin and oxytocin into hypophysial portal blood in long-term hypophysectomized rats. J Endocrinol. 1991;129(1):91–98. [DOI] [PubMed] [Google Scholar]
- 39. Battaglia DF, Brown ME, Krasa HB, Thrun LA, Viguié C, Karsch FJ. Systemic challenge with endotoxin stimulates corticotropin-releasing hormone and arginine vasopressin secretion into hypophyseal portal blood: coincidence with gonadotropin-releasing hormone suppression. Endocrinology. 1998;139(10):4175–4181. [DOI] [PubMed] [Google Scholar]
- 40. Kostic TS, Tomic M, Andric SA, Stojilkovic SS. Calcium-independent and cAMP-dependent modulation of soluble guanylyl cyclase activity by G protein-coupled receptors in pituitary cells. J Biol Chem. 2002;277(19):16412–16418. [DOI] [PubMed] [Google Scholar]
- 41. Won JG, Orth DN. Role of lipoxygenase metabolites of arachidonic acid in the regulation of adrenocorticotropin secretion by perifused rat anterior pituitary cells. Endocrinology. 1994;135(4):1496–1503. [DOI] [PubMed] [Google Scholar]
- 42. Abou-Samra AB, Catt KJ, Aguilera G. Role of arachidonic acid in the regulation of adrenocorticotropin release from rat anterior pituitary cell cultures. Endocrinology. 1986;119(4):1427–1431. [DOI] [PubMed] [Google Scholar]
- 43. Luo J, Hu H. Thermally activated TRPV3 channels. Curr Top Membr. 2014;74:325–364. [DOI] [PubMed] [Google Scholar]
- 44. Dahl G, Muller KJ. Innexin and pannexin channels and their signaling. FEBS Lett. 2014;588(8):1396–1402. [DOI] [PubMed] [Google Scholar]
- 45. Makino S, Hashimoto K, Gold PW. Multiple feedback mechanisms activating corticotropin-releasing hormone system in the brain during stress. Pharmacol Biochem Behav. 2002;73(1):147–158. [DOI] [PubMed] [Google Scholar]
- 46. Aguilera G, Liu Y. The molecular physiology of CRH neurons. Front Neuroendocrinol. 2012;33(1):67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]