

Abstract
Accumulating evidence suggests that insults occurring during the perinatal period alter the developmental trajectory of the fetus/offspring leading to long-term detrimental outcomes that often culminate in adult pathologies. These perinatal insults include maternal/fetal disease states, nutritional deficits/excess, stress, lifestyle choices, exposure to environmental chemicals, and medical interventions. In addition to reviewing the various insults that contribute to developmental programming and the benefits of animal models in addressing underlying mechanisms, this review focuses on the commonalities in disease outcomes stemming from various insults, the convergence of mechanistic pathways via which various insults can lead to common outcomes, and identifies the knowledge gaps in the field and future directions.
The “fetal origins of adult disease” or “developmental origins of health and disease” hypothesis by Barker (1) has generated immense attention to the concept of developmental programming. Although this phenomenon as it relates to pathogenesis of many adult disorders was proposed in the 1990s (2), the role of programming during development was identified as early as 1873 in imprinting of behavior in birds (3). Additionally, it is long known that developmental exposure to testosterone produced by the fetal testis underlies the programming of male sexual behavior (4–6) and abnormal exposure of the female fetus to testosterone during critical periods of differentiation masculinizes the female brain (7, 8). Although the term “programming” was first introduced by Lucas (9), the developmental origins of health and disease hypothesis (1, 10) gained momentum only after the emergence of epidemiological data from the 1944–1945 Dutch famine cohort showing maternal starvation during gestation correlates with an increased risk for cardiovascular and metabolic diseases in the offspring (11). These observations in conjunction with several subsequent studies (10, 12, 13) demonstrate that the perinatal period, a period in which organogenesis and tissue differentiation occur through a tightly controlled and timed process, is a susceptibility window to the impacts of adverse environment.
Over the years, it has become clear that the mechanisms underlying the developmental origin of adult disease involve reprogramming of the epigenome by environmental factors (14, 15). This is possible during the early life due to the plasticity that allows the developing organism to adopt a phenotype that best suits the environment. As the individual ages, there is loss of plasticity and emergence of a fixed functional capacity. As West-Eberhard describes (16), developmental plasticity is the phenomenon by which a single genotype can give rise to a range of different physiologic or morphologic states in response to different environmental conditions during development. These short-term adaptations made for survival often prove to be detrimental leading to development of diseases. The programming of adult pathology due to perinatal insults is likely the consequence of reduced functional capacity in key organs, a “thrifty” phenotype, where resources are allocated for critical organs at the expense of other organ systems thus increasing vulnerability of these organs to adverse environmental influences in later life (17). In addition, recent findings support a “2-hit” hypothesis to explain the adult onset of diseases (18, 19). This hypothesis suggests that, a genetic susceptibility combined with an insult occurring during perinatal life (“first-hit”) leads to reorganization of the various organ systems, which alone might be insufficient to alter the adult phenotype. However, endocrine imbalances resulting from the perinatal insults and/or adverse stressors/exposures during postnatal life may act as a “second-hit,” which through activational effects might unmask or amplify the underlying defects culminating in disease states (19).
A wide range of gestational events can alter the fetal developmental trajectory (Figure 1). These include maternal nutritional deficit/excess, environmental exposure to endocrine-disrupting chemicals (EDCs), disease states, lifestyle choices, substance abuse, and medical interventions during pregnancy. Although some of these insults lead to alterations that are manifested immediately after birth such as spina bifida associated with folate deficiency or congenital limb malformations due to thalidomide medication for morning sickness (20), a host of adult onset manifestations such as coronary and metabolic diseases may not be apparent until adulthood.
Figure 1.

Impact of perinatal insults in programming adult pathologies in the offspring. Exposure of the fetus/offspring to different insults during critical periods of development may lead to adaptations that prove to be detrimental and associated with adult defects in several organ systems. IUGR, intrauterine growth restriction.
Although a wide variety of perinatal insults during critical windows of differentiation alter the developmental trajectory of the fetus/offspring, there are often many commonalities in the phenotypic outcomes. Most of these insults result in placental alterations, intrauterine growth restriction (IUGR), and catch-up growth culminating in adult diseases. For example, offspring born to women with gestational undernutrition (10, 21) or excess androgen exposure (19, 22, 23) are reported to undergo IUGR, to be born small for their gestational age, and to manifest adult reproductive and metabolic abnormalities. This review addresses the different perinatal insults associated with developmental programming, the commonalities between underlying mechanisms and the resulting adult phenotype, as well as the knowledge gaps in the field.
Perinatal Insults
Undernutrition
Over the last 3 decades, several epidemiological and experimental studies have highlighted the impacts of gestational undernutrition on the offspring's birth size and prevalence of adult metabolic disruptions (24, 25), a matter of considerable importance in developing countries where undernutrition is a major concern. Barker and Osmond (26) were among the first to report the association between gestational undernutrition leading to adverse intrauterine environment and adult cardiometabolic disease in humans. For instance, adverse intrauterine development, as manifested by low birth weight (LBW), was shown to be associated with greater risk of cardiovascular disease, stroke (26), lung disease (27), and polycystic ovary syndrome (PCOS) (28). Importantly, the timing of the prenatal insult was found to play a key role in determining the adult's susceptibility to diseases, emphasizing the importance of critical periods of differentiation of organ systems. Epidemiological data from the Dutch famine demonstrated that nutrient restriction during early gestation was associated with adult hypertension (11), whereas feed restriction during late gestation was linked to increased adiposity, impaired glucose tolerance, and type 2 diabetes (21, 29, 30).
Compensatory growth during early life is also a risk factor for the development of adult diseases in the offspring. It is believed that the degree of mismatch between pre- and postnatal nutrient availability underlies the development of subsequent chronic diseases (1, 10, 31, 32). For instance, rapid compensatory growth was associated with a 6-year reduction in lifespan in boys from the Helsinki Birth cohort (33). Moreover, the combination of small weight at birth and accelerated growth between 3 and 11 years of age was found to predict the differences in the cumulative incidence of coronary heart disease, type 2 diabetes, and hypertension in both men and women from the same cohort (34).
The deleterious impacts of gestational undernutrition on the offspring's health have been experimentally documented in both small and large animal models. In rats, maternal caloric restriction leads to insulin resistance and hypertension in the adult offspring (35–37). In sheep, maternal undernutrition results in decreased adipose tissue depots, glucose intolerance, and hyperinsulinemia in the offspring (37–40). Early to midgestational nutrient restriction in sheep also leads to lower circulating concentrations of progesterone, hyperinsulinemia (41), and marked reduction in fertility in the female offspring (42). Detailed information regarding the effects of maternal undernutrition on the offspring's health can be found in earlier reviews (24, 25).
Overnutrition
Considering the rise in the last decades in the incidence of maternal obesity and associated disorders such as gestational diabetes mellitus (GDM) (43), recent studies have also focused on the effects of maternal overnutrition on the offspring. These studies have demonstrated that maternal overnutrition may result in large for gestational age babies (44) and that high birth weights are also associated with increased risk for the offspring to develop obesity and metabolic alterations during adult life (45–47). These observations in concert with studies of undernutrition indicate that the relationship between birth weight and risk for development of metabolic defects (and other related deficits) is not a simple linear association but rather follows a U-shaped curve (48). Observations from overnutrition studies, as opposed to studies involving undernutrition in underdeveloped countries, are particularly important in developed countries due to the high incidence of obesity (43, 49).
Epidemiological studies have reported a strong association between increased maternal body mass index (50–55) and alterations in glucose-insulin homeostasis in the offspring. Although genetics could play a role in the transmission of these traits, the finding that maternal weight loss after bariatric surgery reduces the risk of obesity and other metabolic disorders in the offspring supports maternal environment as a contributing factor (56). Support for this premise also comes from the finding that maternal weight gain between pregnancies increases the prevalence of obesity in the younger children (57).
Studies of experimental induction of obesity in animal models provide causal evidence in support of detrimental effects of maternal overnutrition on the offspring's health. For instance, offspring of female rats fed a high-fat diet during gestation manifest increased overall adiposity, reduced insulin sensitivity, and hypertension (58, 59). Similarly, increased feed consumption before and during pregnancy in sheep was found to program glucose intolerance in the offspring (60). In nonhuman primates, chronic consumption of a high-fat diet during gestation was found to increase adiposity and lipotoxicity in the liver (61), and increase anxiety in the offspring (62). Interestingly, there are several common traits such as hyperphagia, increased adiposity, reduced insulin sensitivity, and hypertension (47) in the offspring's phenotypic outcomes among the various animal models implicating common mechanistic pathways between the different animal models.
Because the perinatal period is an important window in which adult pathologies may be programmed, intervention strategies adopted before or during gestation or in early postnatal period are likely to prevent the transmission of these traits to the offspring. In sheep, a short period of dietary restriction (1 mo) before pregnancy prevented the negative effects of maternal overnutrition on offspring's adiposity and weight gain (63). Similarly, mild food restriction during the third trimester of gestation in mice prevented the programming effects of a high-fat diet on the offspring's obesity (64). Furthermore, weight loss before gestation in obese women improved the metabolic parameters in their children (56). These observations highlight the importance of dietary and lifestyle interventions in preventing obesity and several associated complications in the subsequent generation. The developmental adaptations in response to gestational overnutrition and potential intervention strategies have been reviewed in more detail elsewhere (47, 48).
Stress
Prenatal stress represents another insult that perturbs the intrauterine environment and developmental trajectory of the fetus. The most common causes of human prenatal stress are major life events like death of a family member, catastrophic devastation resulting from wars, earthquakes, famines, hurricanes or acts of terrorism, hassles of daily life, depression or general anxiety, and pregnancy-specific anxiety (65). Epidemiological studies have shown that maternal bereavement from loss of loved one or maternal depression during pregnancy result in offspring with increased risk of abnormal immune function, obesity, and mental disorders (66, 67). Similar outcomes are reported in offspring of survivors of holocaust, famine, and other natural disasters (68–71). Stress effects on developmental outcomes could be due to direct changes in the hypothalamic-pituitary-adrenal (HPA) axis or indirectly via alterations in nutrient intake. Elevated basal morning cortisol levels observed in individuals exposed to prenatal maternal psychosocial stressors indeed provide support of mediation via programmed alterations of the HPA axis (72, 73).
Various rodent models of perinatal stress such as asphyxia, hypothermia, maternal deprivation, or separation have been used to assess the effects of perinatal stress on the offspring's health. These models have provided experimental evidence that exposure to stressful conditions during prenatal or early life leads to greater adiposity and weight gain with impaired glycemic control in the offspring (74). Other studies have demonstrated that prenatal stress leads to lower birth weight, impaired feedback regulation of the HPA axis, prolonged stress responses, and reduced glucocorticoid and mineralocorticoid receptor expression in the hippocampus (75–77). Similar outcomes in the HPA axis function after prenatal exposure to exogenous glucocorticoids confirm their role in stress-induced programming (78). The consequences of perinatal stress and altered HPA function as it relates to neurologic, immune, and cardiometabolic disorders have been addressed extensively in previous reviews (71, 79–81).
Disease state
The homeostasis of the maternal and fetal endocrine milieus is important to allow normal development and prevent adverse effects arising from early insults. Disease states that perturb hormonal homeostasis are likely to alter the gestational endocrine milieu (82). Disease conditions that are likely to affect the maternal and fetal milieus include PCOS and congenital adrenal hyperplasia (CAH) (83, 84) maternal obesity (52, 55), GDM (85), diabetes (86), preeclampsia (87), and mutations and polymorphisms in genes such as 11β-hydroxysteroid dehydrogenase (HSD11B) (88), sex hormone-binding globulin (SHBG) (89), and aromatase (CYP19) (90). Examples of perturbed hormonal homeostasis in disease states are increased maternal insulin levels in obesity and GDM, increased maternal/fetal androgen levels in PCOS, CAH and mutations associated with SHBG or CYP19, and increase in fetal cortisol levels with HSD11B mutations. In the United States alone, GDM complicates approximately 7% of all pregnancies (85). Perturbed intrauterine environment such as maternal hyperglycaemia in disease states results in macrosomia, impaired glucose tolerance, development of obesity and metabolic disorders in the offspring (85, 86). Preeclampsia complicates approximately 2%–8% of the pregnancies (87) and offspring of preeclamptic pregnancies are born premature, manifest IUGR, develop high blood pressure during childhood and have increased risk of stroke in later life (91).
Similarly, disease conditions that result in elevated androgens during gestation, such as CAH and PCOS, lead to reproductive and metabolic disorders that emerge later in life (19, 92). Homozygous HSD11B mutations in babies lead to increased fetal cortisol levels that have been correlated with lower birth weight (73), a risk factor in the development of cardiometabolic abnormalities. Conditions that lead to adrenal tumors, CAH or mutations in SHBG and CYP19 can result in exposure of developing fetus to elevated androgens, which in turn can lead to development of PCOS in the female offspring (19, 83, 84, 92). Because PCOS affects approximately 6%–15% of the women of reproductive age and these women continue to be hyperandrogenic and hyperinsulinemic during pregnancy, the offspring born to women with PCOS are also at increased risk for development of PCOS (19, 93, 94).
Experimental induction of diabetes in animal models has shown that mild maternal diabetes leads to development of neonatal macrosomia, whereas severe maternal diabetes results in microsomia, alterations in hypothalamic development and compromised insulin-regulatory system (95). As adults, these offspring maintain normal glucose homeostasis during basal conditions but develop diabetes when their glucose metabolism is stressed such as during pregnancy (95). Offspring of some animal models of preeclampsia develop IUGR and manifest altered endothelial function and hypertension during adulthood (91).
Animal models of increased maternal glucocorticoids also present LBW with alterations in the offspring's HPA axis and adverse metabolic and behavioral phenotype during adulthood (75, 76, 78). Similarly, early- to midgestational exposure to excess testosterone can induce phenotypic virilization and masculinization of the brain in female offspring (5, 7, 8) and the adult onset of PCOS-like reproductive and metabolic deficits in several species (rodents, sheep, monkeys) (96, 97). Consequences of disease states such as diabetes, metabolic syndrome and disease models relative to developmental programming of pathologies such as PCOS in offspring have been addressed extensively in other reviews (97–101).
Lifestyle
In addition to maternal nutrition, stress, and disease state directly having a role in developmental programming, the lifestyle led by the pregnant women and imposed on offspring during early life might contribute to disease susceptibility. Lifestyle factors that have a bearing on perinatal programming include dietary choices, physical activity, substance abuse and medical interventions. With the advent of modern technology, physical inactivity is increasing at an alarming rate and is now recognized as the pressing healthcare issue of the 21st century (102). According to the 2014 physical activity council report, about 30% of the Americans report physical inactivity with majority of individuals spending about 70% of their wakeful hours being sedentary (103, 104). Recent studies have found a positive correlation between adult television viewing time and overall sedentary life with increased central adiposity, fasting triglyceride levels and markers of insulin resistance (103, 105). Importantly, physical inactivity during pregnancy has been shown to be associated with obesity in the offspring (106). Being physically active during pregnancy appears to have many benefits that include reduced body mass, reduced risk of impaired glucose tolerance, and development of innate immunity that help prevent adverse metabolic programming in the offspring (107).
Substance abuse with illicit drugs (marijuana, heroin, and cocaine), legal drugs (alcohol and nicotine), misuse of prescription drugs (hallucinogens, inhalants, or psychotherapeutic drugs), or smoking are major prenatal insults, because approximately 5.9%, 8.5%, and 16% of the pregnant women in the United States are current illicit drug users, alcohol users, or cigarette smokers, respectively (data from 2013 Substance Abuse and Mental Health Services Administration and 2012 National Survey on Drug Use and Health) (108). Programming by substance abuse could be facilitated via maternal stress and malnutrition, features common among these individuals. Disruptions in offspring of illicit drug/alcohol/cigarette users include, IUGR, lower birth weight, congenital abnormalities, abnormal brain development, poor performance in many behavioral and cognitive skills, and development of insulin resistance (108–110).
With the advent of assisted reproductive technologies (ARTs), medical interventions during the preimplantation period is another risk factor for adverse developmental programming (111). In the United States alone, there has been a 25% increase in the number of ART cycles over the 10-year period between 2004 and 2013 with approximately 60 000 live births in 2013 (112). Although not a consistent finding, some studies report ART pregnancies to be associated with reduced gestational length and LBW (113, 114). Importantly, embryo manipulations during ARTs appear to result in several epigenetic alterations that might lead to adverse outcomes later in life (111). In addition, medical interventions such as use of antiemetics to treat morning sickness, instrumental vaginal delivery, cesarean section, induction of labor using oxytocin administration and general anesthesia have also been reported to induce epigenetic alterations in the offspring (115). Similarly, inadvertent sex steroid exposure due to mothers continuing to take contraceptive pills or performance-enhancing drugs (116, 117) unaware of their pregnancy can lead to adverse programming. Another aspect to consider is the use of treatments such as metformin to treat PCOS and GDM in pregnant women. Although metformin treatment has not shown adverse outcomes in offspring at birth, the long term effects of such treatment on adult pathology are unknown (118).
Studies in animal models validate the epidemiologic findings in humans. For example, prenatal exposure of rodents to substance abuse drugs leads to reduced birth weight and deficits in several motor and cognitive skills (108, 109), whereas prenatal alcohol exposure leads to alterations in cerebral cortex network, behavior, and insulin resistance in offspring (110, 119). Embryo culture, routinely performed as part of ARTs, has been found to be associated with IUGR and postnatal adiposity (120), risk factors for adult pathologies. The findings that maternal Western-style diet results in offspring that develop nonalcoholic fatty liver and insulin resistance during adulthood (121), and dietary intervention and exercise yield offspring with improved metabolic functions (122) enforce the importance of healthy lifestyle in preventing abnormal programming. Additional details of the role played by lifestyle and benefits achieved by lifestyle interventions can be found in earlier reviews (107, 123, 124).
Environmental exposure to EDCs
Because of the critical role played by hormones, chemicals that mimic hormone actions can disrupt normal endocrine functions and change maternal and fetal endocrine milieus, thus altering the normal trajectory of fetal development and potentially leading to the development of chronic diseases (125, 126). These mimics are called EDCs and as defined by the United States Environmental Protection Agency (127) are “exogenous agents that interfere with synthesis, secretion, transport, metabolism, binding action, or elimination of natural blood-borne hormones that are present in the body and are responsible for homeostasis, reproduction, and developmental process.” EDCs include naturally occurring compounds such as phytoestrogens, genistein and coumestrol or synthetic chemicals used as industrial solvents and lubricants (eg, polychlorinated biphenyls [PCBs], polybrominated biphenyls, and dioxins), plasticizers (eg, bisphenol A), pesticides (eg, dichlorodiphenyltrichloroethane), fungicide (eg, vinclozolin), and pharmaceutical agents (eg, diethylstilbestrol) (128). Because EDCs are ubiquitously present, fetal/offspring exposures during early developmental periods could occur via maternal-fetal transfer and lactation (129, 130). Since mid-20th century, there has been a steady increase in reproductive diseases and a decline in fertility primarily in the developed world, which have been linked to exposure to environmental chemicals, as corroborated by the 20-fold increase in manufacture and use of natural and synthetic chemicals during the same period (131). Epidemiological data support this assertion as demonstrated by neurological, reproductive, and developmental effects in offspring of mothers exposed to mercury poisoning in Japan and PCB in Taiwan (132, 133). Other disruptions found to be associated with developmental exposure to EDCs are obesity, cardiovascular diseases, type 2 diabetes, and several hormone-sensitive cancers (125, 134).
Although epidemiological studies point to associations, the direct link between the disruptions observed and the EDC responsible come from experimental studies in animal models. Causal links between developmental exposures to EDCs, such as phthalates, PCB, bisphenol A, and dioxins, and development of adult disorders, such as male infertility, type 2 diabetes, obesity, cancer, and other metabolic and reproductive dysfunctions, have been reviewed earlier (126, 134–136).
Common Outcomes Programmed by Different Perinatal Insults
Although a variety of insults can alter the developmental trajectory and culminate in adult diseases, there are a number of similarities in the outcomes resulting from the various perinatal insults (Figure 1). For instance, alterations in placental function and IUGR are common consequences of gestational undernutrition (137–139), exposure to steroid excess (140, 141), maternal stress (142–144), and smoking (145), among other insults. Furthermore, metabolic derangements, including altered body weight gain and insulin resistance, appear to be mutual phenotypic traits programmed by different insults in early life. For instance, in female sheep, prenatal exposure to androgen excess (146, 147), gestational undernutrition (39), and overnutrition (60) have all been reported to program insulin resistance in the offspring. Additionally, excessive weight gain postnatally was found to amplify the phenotypic severity in offspring subjected to different prenatal insults, such as gestational obesity (148) and prenatal androgen excess (146). Importantly, the risk of offspring of diet-induced obese mothers to become obese was ameliorated by postnatal cross-fostering to lean mothers (148). The similarity in phenotypic outcomes programmed by different perinatal insults and benefits a given lifestyle intervention can have in overcoming similar pathologic outcomes from different perinatal insults suggest that common mechanisms are likely involved.
Common Mechanisms Underlying Developmental Programming
Depending on the type of perinatal insult, organ system affected, and the developmental window in which the insult occurs, a number of mechanisms have been proposed linking perinatal insults to the development of adult diseases. Nonetheless, as our understanding in this field improves, it has become evident that different insults can lead to fetal/offspring responses that converge on common mechanistic pathways culminating in the development of similar adult outcomes (Figure 2). Metabolic hormones, such as insulin and leptin, as well as steroid hormones such as androgens and estrogens that play key roles in controlling cell proliferation and apoptosis, cell differentiation, neurodevelopment, and energy use among other physiological processes (149–155) are potential targets of perinatal insults. Studies in animal models have demonstrated that different prenatal insults such as maternal undernutrition (82, 156) and overnutrition (157), as well as gestational exposure to testosterone excess (158) result in changes in maternal levels of insulin and/or leptin. Additionally, maternal undernutrition (159) and gestational stress (160, 161) have also been shown to alter the maternal levels of androgens during gestation. These alterations in the metabolic and steroid milieus during early life may be common integrative mechanisms via which perinatal insults program similar adult pathologies later in life. In this regard, a reciprocal interplay between androgens and insulin has been proposed to be involved in the pathogenesis and phenotypic expression of disorders such as PCOS (162–164) and metabolic syndrome (165, 166). Other metabolic factors, such as amino acids (167) and fatty acids (168) have also been implicated in the prenatal programming of metabolic function.
Figure 2.
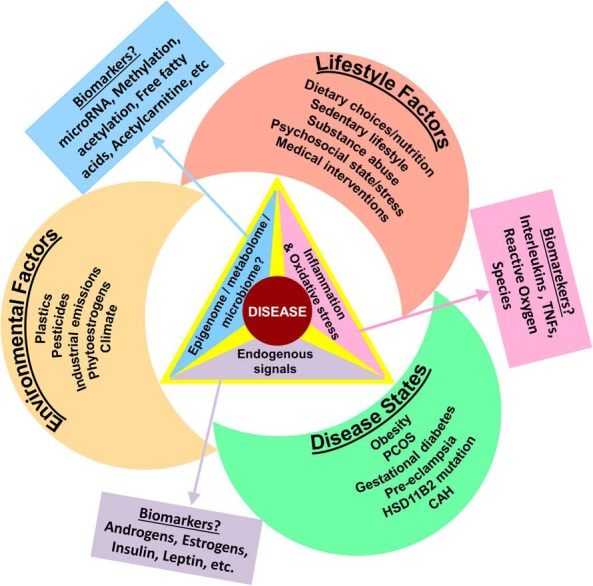
Commonalities in the mechanisms through which different perinatal insults could lead to development of adult disorders. Different perinatal insults (disease states, environment, and lifestyle choices) by altering the epigenome/metabolome/microbiome, inflammatory/oxidative stress mechanisms, or endocrine signals have the potential to alter the fetal developmental trajectory culminating in adult disorders and may also serve as early biomarkers of future disease states.
Another common mechanism believed to be involved in prenatal programming is oxidative stress; a deleterious process that results in damage of cell components, such as proteins, lipids, and DNA (169). Several insults associated with IUGR and development of chronic disease, including gestational hypertension, undernutrition, and smoking have been associated with oxidative stress (169, 170). IUGR has been found to promote low-grade inflammation in the adult offspring, a mechanism linking impaired fetal growth with chronic adult disease (24). For instance, gestational obesity increases the expression of genes associated with oxidative stress and inflammation in the placenta (171) as well as in the skeletal muscle of the developing fetus (172). Similarly, high-fat diet has been shown to increase the expression of enzymes involved in oxidative stress in adult offspring of mice (173). Oxidative stress and inflammation have also been postulated as critical mechanisms underlying several adverse health outcomes associated with prenatal exposure to EDCs (174). The finding that maternal supplementation with antioxidants reduces oxidative stress and prevents adiposity in the offspring of Western diet-fed rats (175), provides support for inflammation and oxidative stress as mediators of adverse metabolic programming in the offspring.
Epigenetic alterations, such as DNA methylation, histone modification, chromatin packing, and microRNA expression, are emerging as key mediators of developmental programming (176, 177). Because epigenetic mechanisms are inherently malleable and can accumulate over time, developmental insults can have profound effect on this process, leading to altered gene expression and development of disease phenotypes (48, 178, 179). For instance, impaired glucose metabolism during pregnancy in women is associated with altered leptin gene DNA methylation and mRNA expression in both the maternal and feto-placental compartments, indicative of increased risk of adult obesity and type 2 diabetes in the offspring (180). Similarly, changes in DNA methylation and histone acetylation in the promoter region of Pdx1, a pancreatic homeobox transcription factor critical for β-cell function and development, have been found to persist throughout development and to be associated with a reduced pancreatic β-cell mass and a prediabetic state in a rat model of IUGR (181). Inappropriate exposure to steroids or EDCs during development has also been observed to promote epigenetic adaptations that likely underlie the development of adult diseases (84, 182, 183). The findings that maternal dietary supplementation with methyl donors, such as folic acid, negated the DNA hypomethylating effect of neonatal exposure to EDCs and prevented EDC-induced changes in the coat color in yellow agouti mice (184) emphasize the role of epigenetics in developmental programming. In conjunction with other recent studies, these data highlight the effects of prenatal insults on genomic plasticity and the potential for maternal dietary supplementation to prevent epigenetic changes.
Importantly, postnatal outcomes resulting from different perinatal insults, such as hormonal imbalances and metabolic disruptions, appear to play an important role in unmasking, maintaining, and/or amplifying the defects programmed in utero. The notion that a combination of prenatal and postnatal insults may be necessary for the development and manifestation of pathology has been proposed for different diseases (19, 185, 186). For instance, observations from the monkey (187) as well as the sheep model (188) of PCOS-like phenotype indicate that postnatal overnutrition exacerbates the reproductive and metabolic alterations programmed by prenatal testosterone excess. Similarly, compensatory growth during early life further increases the risk of chronic diseases in offspring of undernourished mothers. Therefore, it is evident that although prenatal insults can have important organizational effects programming the offspring, postnatal alterations may have activational effects impacting the development, expression and/or severity of disease (19).
Conclusion and Future Directions
Epidemiological data in humans and studies in animal models clearly indicate that a myriad of insults occurring during in utero life may impact on the offspring's long-term health. Nevertheless, the lack of detailed information on the maternal and fetal environments throughout gestation following such insults and the scarcity of longitudinal studies in humans limit our understanding of the susceptibility windows during fetal development as well as the actual impact of such insults on specific organ systems. Additionally, the interactions between prenatal insults and postnatal milieu in the development of adult diseases remain poorly understood. Therefore, animal models of perinatal programming serve as a critical research tool to study the impact of developmental insults on the offspring's health, to unravel the cellular and molecular mechanisms involved in this process, and to test intervention strategies.
An area that requires further investigation is the potential transmission of phenotypic traits to subsequent generations. Studies in animal models have reported that different perinatal insults may lead to epigenetic alterations and adult pathology in multiple generations (182, 189–191). Epidemiological studies in humans also point to multigenerational effects of prenatal exposure to the Dutch famine, with F2 individuals presenting increased neonatal adiposity and poor overall health in later life (192). Going forward, it is important to dissect out the specific contribution of epigenetic inheritance vs the vertical transmission of traits via endocrine imbalances, which may prevail from generation to generation to facilitate the perpetuation of pathologic alterations to subsequent generations. Documentation of the transgenerational epigenetic transmission in the female offspring would require studies of third generation (F3) and beyond (193, 194). Additionally, although this review focuses primarily on maternal exposure to gestational insults, recent experimental and epidemiological observations (195–198) emphasize the need to investigate the role of paternal epigenetic inheritance in the development of adult disease.
An emerging and promising area of research that also merits investigation relative to developmental programming of adult disease is the role of the gut microbiota (199–201). Recent studies point to prevailing microbiome during gestation and postnatal period to have an impact on the developing immune system, a risk factor for development of immune-related pathologies in the offspring (199). Moreover, patterns of microbial colonization appear to influence brain development, infant growth and adiposity (200). Because dietary composition influences the colonization of intestinal bacteria during development (201), strategies to prevent adverse programming should also focus on the impact early nutrition, and use of probiotics/antibiotics have on the microbiome and their relation to developmental programming.
Because perinatal insults may impact multiple physiological systems, the integration of these systems in the development of pathology is an important avenue for future research. For instance, prenatal exposure to androgen excess has been found to impact both the reproductive and the metabolic systems leading to a self-perpetuating cycle with defects at one system having an impact on the other (188). Therefore, interventions targeting at multiple systems may be required to overcome such developmentally programmed disorders. In fact, combined antiandrogen and insulin-sensitizing treatment of young, nonobese women with PCOS has been found to have additive benefits on insulin sensitivity, hyperandrogenemia, and dyslipidemia when compared with monotherapies (202). Importantly, the investigation of potential common mediators affecting the different systems may help identify early biomarkers and therapeutic targets for prevention strategies.
Although many perinatal insults have been identified in the last decades, there is still a long way ahead to identifying all the factors that can adversely impact the offspring's health. Additional epidemiological and animal studies are required to shed more light on this important issue. Furthermore, because the long-term consequences of pharmacological interventions during the preconception period and gestation remain unclear, preventive strategies should focus on promoting healthy lifestyle choices and minimizing unnecessary exposure to potentially harmful agents before and during pregnancy as well as during early life of the child. Postnatal lifestyle interventions represent promising approaches to overcome, or at least alleviate, many deleterious events programmed perinatally.
Acknowledgments
This work was supported by National Institutes of Health Grant P01 HD44232.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ART
- assisted reproductive technology
- CAH
- congenital adrenal hyperplasia
- CYP19
- aromatase
- EDC
- endocrine-disrupting chemical
- GDM
- gestational diabetes mellitus
- HPA
- hypothalamic-pituitary-adrenal
- HSD11B
- 11β-hydroxysteroid dehydrogenase
- IUGR
- intrauterine growth restriction
- LBW
- low birth weight
- PCB
- polychlorinated biphenyl
- PCOS
- polycystic ovary syndrome
- SHBG
- sex hormone-binding globulin.
References
- 1. Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–417. [DOI] [PubMed] [Google Scholar]
- 2. Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gray PH. The descriptive study of imprinting in birds from 1873 to 1953. J Gen Psychol. 1963;68:333–346. [Google Scholar]
- 4. Forest MG. Role of androgens in fetal and pubertal development. Horm Res. 1983;18:69–83. [DOI] [PubMed] [Google Scholar]
- 5. Gorski RA. Sexual dimorphisms of the brain. J Anim Sci. 1985;61(suppl 3):38–61. [DOI] [PubMed] [Google Scholar]
- 6. Jost A. Genetic and hormonal factors in sex differentiation of the brain. Psychoneuroendocrinology. 1983;8:183–193. [DOI] [PubMed] [Google Scholar]
- 7. Phoenix CH, Goy RW, Gerall AA, Young WC. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology. 1959;65:369–382. [DOI] [PubMed] [Google Scholar]
- 8. Wood RI, Foster DL. Sexual differentiation of reproductive neuroendocrine function in sheep. Rev Reprod. 1998;3:130–140. [DOI] [PubMed] [Google Scholar]
- 9. Lucas A. Programming by early nutrition in man. Ciba Found Symp. 1991;156:38–50; discussion 50–35. [PubMed] [Google Scholar]
- 10. Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S–595S. [DOI] [PubMed] [Google Scholar]
- 11. Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. [DOI] [PubMed] [Google Scholar]
- 12. Gluckman PD, Hanson MA. Maternal constraint of fetal growth and its consequences. Semin Fetal Neonatal Med. 2004;9:419–425. [DOI] [PubMed] [Google Scholar]
- 13. Nijland MJ, Ford SP, Nathanielsz PW. Prenatal origins of adult disease. Curr Opin Obstet Gynecol. 2008;20:132–138. [DOI] [PubMed] [Google Scholar]
- 14. Treviño LS, Wang Q, Walker CL. Phosphorylation of epigenetic “readers, writers and erasers”: implications for developmental reprogramming and the epigenetic basis for health and disease. Prog Biophys Mol Biol. 2015;118:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ganu RS, Harris RA, Collins K, Aagaard KM. Maternal diet: a modulator for epigenomic regulation during development in nonhuman primates and humans. Int J Obes Suppl. 2012;2:S14–S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. West-Eberhard MJ. Developmental plasticity and the origin of species differences. Proc Natl Acad Sci USA. 2005;102(suppl 1):6543–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20. [DOI] [PubMed] [Google Scholar]
- 18. Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord. 2007;8:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puttabyatappa M, Cardoso RC, Padmanabhan V. Effect of maternal PCOS and PCOS-like phenotype on the offspring's health [published online ahead of print November 27, 2015]. Mol Cell Endocrinol. doi:10.1016/j.mce.2015.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finnell RH, Waes JG, Eudy JD, Rosenquist TH. Molecular basis of environmentally induced birth defects. Annu Rev Pharmacol Toxicol. 2002;42:181–208. [DOI] [PubMed] [Google Scholar]
- 21. Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185:93–98. [DOI] [PubMed] [Google Scholar]
- 22. Sir-Petermann T, Hitchsfeld C, Maliqueo M, et al. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod. 2005;20:2122–2126. [DOI] [PubMed] [Google Scholar]
- 23. Ibáñez L, Potau N, Francois I, de Zegher F. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83:3558–3562. [DOI] [PubMed] [Google Scholar]
- 24. Lakshmy R. Metabolic syndrome: role of maternal undernutrition and fetal programming. Rev Endocr Metab Disord. 2013;14:229–240. [DOI] [PubMed] [Google Scholar]
- 25. Symonds ME, Sebert SP, Hyatt MA, Budge H. Nutritional programming of the metabolic syndrome. Nat Rev Endocrinol. 2009;5:604–610. [DOI] [PubMed] [Google Scholar]
- 26. Barker D, Osmond C. Low birth weight and hypertension. BMJ. 1988;297:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lal MK, Manktelow BN, Draper ES, Field DJ. Chronic lung disease of prematurity and intrauterine growth retardation: a population-based study. Pediatrics. 2003;111:483–487. [DOI] [PubMed] [Google Scholar]
- 28. Melo AS, Vieira CS, Barbieri MA, et al. High prevalence of polycystic ovary syndrome in women born small for gestational age. Hum Reprod. 2010;25:2124–2131. [DOI] [PubMed] [Google Scholar]
- 29. Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70:811–816. [DOI] [PubMed] [Google Scholar]
- 30. Ravelli AC, van der Meulen JH, Michels R, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–177. [DOI] [PubMed] [Google Scholar]
- 31. Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr Res. 2004;56:311–317. [DOI] [PubMed] [Google Scholar]
- 32. Armitage JA, Taylor PD, Poston L. Experimental models of developmental programming: consequences of exposure to an energy rich diet during development. J Physiol. 2005;565:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. J P Barker D, Kajantie E, Osmond C, Thornburg KL, Eriksson JG. How boys grow determines how long they live. Am J Hum Biol. 2011;23:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–1239. [DOI] [PubMed] [Google Scholar]
- 35. Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50:2279–2286. [DOI] [PubMed] [Google Scholar]
- 36. Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development. 2000;127:4195–4202. [DOI] [PubMed] [Google Scholar]
- 37. Lecoutre S, Breton C. Maternal nutritional manipulations program adipose tissue dysfunction in offspring. Front Physiol. 2015;6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Symonds ME, Mostyn A, Pearce S, Budge H, Stephenson T. Endocrine and nutritional regulation of fetal adipose tissue development. J Endocrinol. 2003;179:293–299. [DOI] [PubMed] [Google Scholar]
- 39. Gardner DS, Tingey K, Van Bon BW, et al. Programming of glucose-insulin metabolism in adult sheep after maternal undernutrition. Am J Physiol Regul Integr Comp Physiol. 2005;289:R947–R954. [DOI] [PubMed] [Google Scholar]
- 40. Todd SE, Oliver MH, Jaquiery AL, Bloomfield FH, Harding JE. Periconceptional undernutrition of ewes impairs glucose tolerance in their adult offspring. Pediatr Res. 2009;65:409–413. [DOI] [PubMed] [Google Scholar]
- 41. Long NM, Tuersunjiang N, George LA, et al. Maternal nutrient restriction in the ewe from early to midgestation programs reduced steroidogenic enzyme expression and tended to reduce progesterone content of corpora lutea, as well as circulating progesterone in nonpregnant aged female offspring. Reprod Biol Endocrinol. 2013;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Long NM, Nijland MJ, Nathanielsz PW, Ford SP. The effect of early to mid-gestational nutrient restriction on female offspring fertility and hypothalamic-pituitary-adrenal axis response to stress. J Anim Sci. 2010;88:2029–2037. [DOI] [PubMed] [Google Scholar]
- 43. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. [DOI] [PubMed] [Google Scholar]
- 44. Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113:1126–1133. [DOI] [PubMed] [Google Scholar]
- 45. Armitage JA, Khan IY, Taylor PD, Nathanielsz PW, Poston L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J Physiol. 2004;561:355–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palatianou ME, Simos YV, Andronikou SK, Kiortsis DN. Long-term metabolic effects of high birth weight: a critical review of the literature. Horm Metab Res. 2014;46:911–920. [DOI] [PubMed] [Google Scholar]
- 47. Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet. 2011;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vickers MH, Sloboda DM. Strategies for reversing the effects of metabolic disorders induced as a consequence of developmental programming. Front Physiol. 2012;3:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fisher SC, Kim SY, Sharma AJ, Rochat R, Morrow B. Is obesity still increasing among pregnant women? Prepregnancy obesity trends in 20 states, 2003–2009. Prev Med. 2013;56:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lake JK, Power C, Cole TJ. Child to adult body mass index in the 1958 British birth cohort: associations with parental obesity. Arch Dis Child. 1997;77:376–381. [DOI] [PubMed] [Google Scholar]
- 51. Laitinen J, Power C, Järvelin MR. Family social class, maternal body mass index, childhood body mass index, and age at menarche as predictors of adult obesity. Am J Clin Nutr. 2001;74:287–294. [DOI] [PubMed] [Google Scholar]
- 52. O'Reilly JR, Reynolds RM. The risk of maternal obesity to the long-term health of the offspring. Clin Endocrinol. 2013;78:9–16. [DOI] [PubMed] [Google Scholar]
- 53. Paliy O, Piyathilake CJ, Kozyrskyj A, Celep G, Marotta F, Rastmanesh R. Excess body weight during pregnancy and offspring obesity: potential mechanisms. Nutrition. 2014;30:245–251. [DOI] [PubMed] [Google Scholar]
- 54. Lau EY, Liu J, Archer E, McDonald SM, Liu J. Maternal weight gain in pregnancy and risk of obesity among offspring: a systematic review. J Obes. 2014;2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lawlor DA, Relton C, Sattar N, Nelson SM. Maternal adiposity–a determinant of perinatal and offspring outcomes? Nat Rev Endocrinol. 2012;8:679–688. [DOI] [PubMed] [Google Scholar]
- 56. Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006;118:e1644–e1649. [DOI] [PubMed] [Google Scholar]
- 57. Villamor E, Cnattingius S. Interpregnancy weight change and risk of adverse pregnancy outcomes: a population-based study. Lancet. 2006;368:1164–1170. [DOI] [PubMed] [Google Scholar]
- 58. Khan IY, Dekou V, Douglas G, et al. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Physiol Regul Integr Comp Physiol. 2005;288:R127–R133. [DOI] [PubMed] [Google Scholar]
- 59. Ainge H, Thompson C, Ozanne SE, Rooney K. A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int J Obes. 2011;35:325–335. [DOI] [PubMed] [Google Scholar]
- 60. Long NM, George LA, Uthlaut AB, et al. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring. J Anim Sci. 2010;88:3546–3553. [DOI] [PubMed] [Google Scholar]
- 61. McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sullivan EL, Grayson B, Takahashi D, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010;30:3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rattanatray L, MacLaughlin SM, Kleemann DO, Walker SK, Muhlhausler BS, McMillen IC. Impact of maternal periconceptional overnutrition on fat mass and expression of adipogenic and lipogenic genes in visceral and subcutaneous fat depots in the postnatal lamb. Endocrinology. 2010;151:5195–5205. [DOI] [PubMed] [Google Scholar]
- 64. Giraudo SQ, Della-Fera MA, Proctor L, Wickwire K, Ambati S, Baile CA. Maternal high fat feeding and gestational dietary restriction: effects on offspring body weight, food intake and hypothalamic gene expression over three generations in mice. Pharmacol Biochem Behav. 2010;97:121–129. [DOI] [PubMed] [Google Scholar]
- 65. Dunkel Schetter C. Psychological science on pregnancy: stress processes, biopsychosocial models, and emerging research issues. Annu Rev Psychol. 2011;62:531–558. [DOI] [PubMed] [Google Scholar]
- 66. Li J, Olsen J, Vestergaard M, Obel C, Baker JL, Sorensen TI. Prenatal stress exposure related to maternal bereavement and risk of childhood overweight. PLoS One. 2010;5:e11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dixon B, Peña MM, Taveras EM. Lifecourse approach to racial/ethnic disparities in childhood obesity. Adv Nutr. 2012;3:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Harville E, Xiong X, Buekens P. Disasters and perinatal health: a systematic review. Obstet Gynecol Surv. 2010;65:713–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vaiserman AM. Epigenetic programming by early-life stress: evidence from human populations. Dev Dyn. 2015;244:254–265. [DOI] [PubMed] [Google Scholar]
- 70. Keinan-Boker L, Shasha-Lavsky H, Eilat-Zanani S, Edri-Shur A, Shasha SM. Chronic health conditions in Jewish Holocaust survivors born during World War II. Isr Med Assoc J. 2015;17:206–212. [PubMed] [Google Scholar]
- 71. Entringer S, Buss C, Wadhwa PD. Prenatal stress, development, health and disease risk: a psychobiological perspective-2015 Curt Richter Award Paper. Psychoneuroendocrinology. 2015;62:366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beijers R, Buitelaar JK, de Weerth C. Mechanisms underlying the effects of prenatal psychosocial stress on child outcomes: beyond the HPA axis. Eur Child Adolesc Psychiatry. 2014;23:943–956. [DOI] [PubMed] [Google Scholar]
- 73. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol. 2014;10:391–402. [DOI] [PubMed] [Google Scholar]
- 74. Entringer S. Impact of stress and stress physiology during pregnancy on child metabolic function and obesity risk. Curr Opin Clin Nutr Metab Care. 2013;16:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Weinstock M. Does prenatal stress impair coping and regulation of hypothalamic-pituitary-adrenal axis? Neurosci Biobehav Rev. 1997;21:1–10. [DOI] [PubMed] [Google Scholar]
- 76. Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab. 2002;13:373–380. [DOI] [PubMed] [Google Scholar]
- 77. Huizink AC, Mulder EJ, Buitelaar JK. Prenatal stress and risk for psychopathology: specific effects or induction of general susceptibility? Psychol Bull. 2004;130:115–142. [DOI] [PubMed] [Google Scholar]
- 78. Welberg LA, Seckl JR. Prenatal stress, glucocorticoids and the programming of the brain. J Neuroendocrinol. 2001;13:113–128. [DOI] [PubMed] [Google Scholar]
- 79. Veru F, Laplante DP, Luheshi G, King S. Prenatal maternal stress exposure and immune function in the offspring. Stress. 2014;17:133–148. [DOI] [PubMed] [Google Scholar]
- 80. Bale TL. Lifetime stress experience: transgenerational epigenetics and germ cell programming. Dialogues Clin Neurosci. 2014;16:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Constantinof A, Moisiadis VG, Matthews SG. Programming of stress pathways: a transgenerational perspective [published online ahead of print October 22, 2015]. J Steroid Biochem Mol Biol. doi:10.1016/j.jsbmb.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 82. Fowden AL, Forhead AJ. Endocrine mechanisms of intrauterine programming. Reproduction. 2004;127:515–526. [DOI] [PubMed] [Google Scholar]
- 83. Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–1333. [DOI] [PubMed] [Google Scholar]
- 84. Xita N, Tsatsoulis A. Fetal origins of the metabolic syndrome. Ann NY Acad Sci. 2010;1205:148–155. [DOI] [PubMed] [Google Scholar]
- 85. Baz B, Riveline JP, Gautier JF. ENDOCRINOLOGY OF PREGNANCY: gestational diabetes mellitus: definition, aetiological and clinical aspects. Eur J Endocrinol. 2015;174:R43–R51. [DOI] [PubMed] [Google Scholar]
- 86. Silverman BL, Landsberg L, Metzger BE. Fetal hyperinsulinism in offspring of diabetic mothers. Association with the subsequent development of childhood obesity. Ann NY Acad Sci. 1993;699:36–45. [DOI] [PubMed] [Google Scholar]
- 87. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–644. [DOI] [PubMed] [Google Scholar]
- 88. Bar-Lev A, Annes JP. Genetics of adrenocortical disease: an update. Curr Opin Endocrinol Diabetes Obes. 2012;19:159–167. [DOI] [PubMed] [Google Scholar]
- 89. Chen C, Smothers J, Lange A, Nestler JE, Strauss Iii JF, Wickham Iii EP. Sex hormone-binding globulin genetic variation: associations with type 2 diabetes mellitus and polycystic ovary syndrome. Minerva Endocrinol. 2010;35:271–280. [PMC free article] [PubMed] [Google Scholar]
- 90. Tempfer CB, Schneeberger C, Huber JC. Applications of polymorphisms and pharmacogenomics in obstetrics and gynecology. Pharmacogenomics. 2004;5:57–65. [DOI] [PubMed] [Google Scholar]
- 91. Davis EF, Newton L, Lewandowski AJ, et al. Pre-eclampsia and offspring cardiovascular health: mechanistic insights from experimental studies. Clin Sci (Lond). 2012;123:53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med. 2014;32:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sir-Petermann T, Maliqueo M, Codner E, et al. Early metabolic derangements in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:4637–4642. [DOI] [PubMed] [Google Scholar]
- 95. Aerts L, Van Assche FA. Animal evidence for the transgenerational development of diabetes mellitus. Int J Biochem Cell Biol. 2006;38:894–903. [DOI] [PubMed] [Google Scholar]
- 96. Padmanabhan V, Veiga-Lopez A. Animal models of the polycystic ovary syndrome phenotype. Steroids. 2013;78:734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cardoso RC, Puttabyatappa M, Padmanabhan V. Steroidogenic versus metabolic programming of reproductive neuroendocrine, ovarian and metabolic dysfunctions. Neuroendocrinology. 2015;102:226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol. 2010;299:R711–R722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Poston L. Developmental programming and diabetes - The human experience and insight from animal models. Best Pract Res Clin Endocrinol Metab. 2010;24:541–552. [DOI] [PubMed] [Google Scholar]
- 100. Berends LM, Ozanne SE. Early determinants of type-2 diabetes. Best Pract Res Clin Endocrinol Metab. 2012;26:569–580. [DOI] [PubMed] [Google Scholar]
- 101. Capra L, Tezza G, Mazzei F, Boner AL. The origins of health and disease: the influence of maternal diseases and lifestyle during gestation. Ital J Pediatr. 2013;39:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Davis JC, Verhagen E, Bryan S, et al. 2014 consensus statement from the first Economics of Physical Inactivity Consensus (EPIC) conference (Vancouver). Br J Sports Med. 2014;48:947–951. [DOI] [PubMed] [Google Scholar]
- 103. Owen N, Sparling PB, Healy GN, Dunstan DW, Matthews CE. Sedentary behavior: emerging evidence for a new health risk. Mayo Clin Proc. 2010;85:1138–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Barnes AS. Obesity and sedentary lifestyles: risk for cardiovascular disease in women. Tex Heart Inst J. 2012;39:224–227. [PMC free article] [PubMed] [Google Scholar]
- 105. Thorp AA, Healy GN, Owen N, et al. Deleterious associations of sitting time and television viewing time with cardiometabolic risk biomarkers: Australian Diabetes, Obesity and Lifestyle (AusDiab) study 2004–2005. Diabetes Care. 2010;33:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mourtakos SP, Tambalis KD, Panagiotakos DB, et al. Maternal lifestyle characteristics during pregnancy, and the risk of obesity in the offspring: a study of 5,125 children. BMC Pregnancy Childbirth. 2015;15:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mathias PC, Elmhiri G, de Oliveira JC, et al. Maternal diet, bioactive molecules, and exercising as reprogramming tools of metabolic programming. Eur J Nutr. 2014;53:711–722. [DOI] [PubMed] [Google Scholar]
- 108. Ross EJ, Graham DL, Money KM, Stanwood GD. Developmental consequences of fetal exposure to drugs: what we know and what we still must learn. Neuropsychopharmacology. 2015;40:61–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Slotkin TA. Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates. Toxicol Appl Pharmacol. 2004;198:132–151. [DOI] [PubMed] [Google Scholar]
- 110. Ting JW, Lautt WW. The effect of acute, chronic, and prenatal ethanol exposure on insulin sensitivity. Pharmacol Ther. 2006;111:346–373. [DOI] [PubMed] [Google Scholar]
- 111. De Rycke M, Liebaers I, Van Steirteghem A. Epigenetic risks related to assisted reproductive technologies: risk analysis and epigenetic inheritance. Hum Reprod. 2002;17:2487–2494. [DOI] [PubMed] [Google Scholar]
- 112. National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention. 2013 Assisted Reproductive Technology National Summary Report. 2013. Available at http://www.cdc.gov/art/pdf/2013-report/art_2013_national_summary_report.pdf. Updated October 2015 Accessed January 6, 2016.
- 113. De Geyter C, De Geyter M, Steimann S, Zhang H, Holzgreve W. Comparative birth weights of singletons born after assisted reproduction and natural conception in previously infertile women. Hum Reprod. 2006;21:705–712. [DOI] [PubMed] [Google Scholar]
- 114. Romundstad LB, Romundstad PR, Sunde A, et al. Effects of technology or maternal factors on perinatal outcome after assisted fertilisation: a population-based cohort study. Lancet. 2008;372:737–743. [DOI] [PubMed] [Google Scholar]
- 115. Dahlen HG, Kennedy HP, Anderson CM, et al. The EPIIC hypothesis: intrapartum effects on the neonatal epigenome and consequent health outcomes. Med Hypotheses. 2013;80:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bahrke MS, Yesalis CE, Brower KJ. Anabolic-androgenic steroid abuse and performance-enhancing drugs among adolescents. Child Adolesc Psychiatr Clin N Am. 1998;7:821–838. [PubMed] [Google Scholar]
- 117. Smithells RW. Oral contraceptives and birth defects. Dev Med Child Neurol. 1981;23:369–372. [PubMed] [Google Scholar]
- 118. Barbour LA. Unresolved controversies in gestational diabetes: implications on maternal and infant health. Curr Opin Endocrinol Diabetes Obes. 2014;21:264–270. [DOI] [PubMed] [Google Scholar]
- 119. Schneider ML, Moore CF, Adkins MM. The effects of prenatal alcohol exposure on behavior: rodent and primate studies. Neuropsychol Rev. 2011;21:186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sjöblom C, Roberts CT, Wikland M, Robertson SA. Granulocyte-macrophage colony-stimulating factor alleviates adverse consequences of embryo culture on fetal growth trajectory and placental morphogenesis. Endocrinology. 2005;146:2142–2153. [DOI] [PubMed] [Google Scholar]
- 121. Zambrano E, Nathanielsz PW. Mechanisms by which maternal obesity programs offspring for obesity: evidence from animal studies. Nutr Rev. 2013;71(suppl 1):S42–S54. [DOI] [PubMed] [Google Scholar]
- 122. Nathanielsz PW, Ford SP, Long NM, Vega CC, Reyes-Castro LA, Zambrano E. Interventions to prevent adverse fetal programming due to maternal obesity during pregnancy. Nutr Rev. 2013;71(suppl 1):S78–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Koletzko B, Brands B, Chourdakis M, et al. The Power of Programming and the EarlyNutrition project: opportunities for health promotion by nutrition during the first thousand days of life and beyond. Ann Nutr Metab. 2014;64:187–196. [DOI] [PubMed] [Google Scholar]
- 124. Barua S, Junaid MA. Lifestyle, pregnancy and epigenetic effects. Epigenomics. 2015;7:85–102. [DOI] [PubMed] [Google Scholar]
- 125. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gore AC, Chappell VA, Fenton SE, et al. EDC-2: The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev. 2015:36:E1–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. United States Environmental Protection Agency. Endocrine Disruption. 2016. Available at http://www.epa.gov/endocrine-disruption. Updated December 22, 2015 Accessed January 6, 2016.
- 128. Kabir ER, Rahman MS, Rahman I. A review on endocrine disruptors and their possible impacts on human health. Environ Toxicol Pharmacol. 2015;40:241–258. [DOI] [PubMed] [Google Scholar]
- 129. Masuda Y, Kagawa R, Kuroki H, et al. Transfer of polychlorinated biphenyls from mothers to foetuses and infants. Food Cosmet Toxicol. 1978;16:543–546. [DOI] [PubMed] [Google Scholar]
- 130. Ando M, Saito H, Wakisaka I. Transfer of polychlorinated biphenyls (PCBs) to newborn infants through the placenta and mothers' milk. Arch Environ Contam Toxicol. 1985;14:51–57. [DOI] [PubMed] [Google Scholar]
- 131. Woodruff TJ. Bridging epidemiology and model organisms to increase understanding of endocrine disrupting chemicals and human health effects. J Steroid Biochem Mol Biol. 2011;127:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25:1–24. [DOI] [PubMed] [Google Scholar]
- 133. Guo YL, Lambert GH, Hsu CC, Hsu MM. Yucheng: health effects of prenatal exposure to polychlorinated biphenyls and dibenzofurans. Int Arch Occup Environ Health. 2004;77:153–158. [DOI] [PubMed] [Google Scholar]
- 134. Chevalier N, Fénichel P. Endocrine disruptors: new players in the pathophysiology of type 2 diabetes? Diabetes Metab. 2015;41:107–115. [DOI] [PubMed] [Google Scholar]
- 135. Vom Saal FS, Nagel SC, Coe BL, Angle BM, Taylor JA. The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol Cell Endocrinol. 2012;354:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Peretz J, Vrooman L, Ricke WA, et al. Bisphenol a and reproductive health: update of experimental and human evidence, 2007–2013. Environ Health Perspect. 2014;122:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lesage J, Hahn D, Léonhardt M, Blondeau B, Bréant B, Dupouy JP. Maternal undernutrition during late gestation-induced intrauterine growth restriction in the rat is associated with impaired placental GLUT3 expression, but does not correlate with endogenous corticosterone levels. J Endocrinol. 2002;174:37–43. [DOI] [PubMed] [Google Scholar]
- 138. Belkacemi L, Nelson DM, Desai M, Ross MG. Maternal undernutrition influences placental-fetal development. Biol Reprod. 2010;83:325–331. [DOI] [PubMed] [Google Scholar]
- 139. Tarrade A, Panchenko P, Junien C, Gabory A. Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. J Exp Biol. 2015;218:50–58. [DOI] [PubMed] [Google Scholar]
- 140. Veiga-Lopez A, Steckler TL, Abbott DH, et al. Developmental programming: impact of excess prenatal testosterone on intrauterine fetal endocrine milieu and growth in sheep. Biol Reprod. 2011;84:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Beckett EM, Astapova O, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programing: impact of testosterone on placental differentiation. Reproduction. 2014;148:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Welberg LA, Thrivikraman KV, Plotsky PM. Chronic maternal stress inhibits the capacity to up-regulate placental 11β-hydroxysteroid dehydrogenase type 2 activity. J Endocrinol. 2005;186:R7–R12. [DOI] [PubMed] [Google Scholar]
- 143. Mairesse J, Lesage J, Breton C, et al. Maternal stress alters endocrine function of the feto-placental unit in rats. Am J Physiol Endocrinol Metab. 2007;292:E1526–E1533. [DOI] [PubMed] [Google Scholar]
- 144. Lesage J, Del-Favero F, Leonhardt M, et al. Prenatal stress induces intrauterine growth restriction and programmes glucose intolerance and feeding behaviour disturbances in the aged rat. J Endocrinol. 2004;181:291–296. [DOI] [PubMed] [Google Scholar]
- 145. Vogt Isaksen C. Maternal smoking, intrauterine growth restriction, and placental apoptosis. Pediatr Dev Pathol. 2004;7:433–442. [DOI] [PubMed] [Google Scholar]
- 146. Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cardoso RC, Veiga-Lopez A, Moeller J, et al. Developmental programming: impact of gestational steroid and metabolic milieus on adiposity and insulin sensitivity in prenatal testosterone-treated female sheep. Endocrinology. 2016;157:522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gorski JN, Dunn-Meynell AA, Hartman TG, Levin BE. Postnatal environment overrides genetic and prenatal factors influencing offspring obesity and insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2006;291:R768–R778. [DOI] [PubMed] [Google Scholar]
- 149. Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci. 2004;24:2797–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818–835. [DOI] [PubMed] [Google Scholar]
- 151. Hiort O. The differential role of androgens in early human sex development. BMC Med. 2013;11:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Forhead AJ, Fowden AL. The hungry fetus? Role of leptin as a nutritional signal before birth. J Physiol. 2009;587:1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wood CE. Estrogen in the fetus. In: Advances in Fetal and Neonatal Physiology. San Diego, CA: Springer; 2014:217–228. [DOI] [PubMed] [Google Scholar]
- 154. Bondesson M, Hao R, Lin CY, Williams C, Gustafsson JÅ. Estrogen receptor signaling during vertebrate development. Biochim Biophys Acta. 2015;1849:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fowden AL. The insulin-like growth factors and feto-placental growth. Placenta. 2003;24:803–812. [DOI] [PubMed] [Google Scholar]
- 156. Bispham J, Gopalakrishnan GS, Dandrea J, et al. Maternal endocrine adaptation throughout pregnancy to nutritional manipulation: consequences for maternal plasma leptin and cortisol and the programming of fetal adipose tissue development. Endocrinology. 2003;144:3575–3585. [DOI] [PubMed] [Google Scholar]
- 157. Holemans K, Caluwaerts S, Poston L, Van Assche FA. Diet-induced obesity in the rat: a model for gestational diabetes mellitus. Am J Obstet Gynecol. 2004;190:858–865. [DOI] [PubMed] [Google Scholar]
- 158. Abi Salloum B, Veiga-Lopez A, Abbott DH, Burant CF, Padmanabhan V. Developmental programming: exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology. 2015;156:2323–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Whyte JJ, Alexenko AP, Davis AM, Ellersieck MR, Fountain ED, Rosenfeld CS. Maternal diet composition alters serum steroid and free fatty acid concentrations and vaginal pH in mice. J Endocrinol. 2007;192:75–81. [DOI] [PubMed] [Google Scholar]
- 160. Takahashi LK, Turner JG, Kalin NH. Prolonged stress-induced elevation in plasma corticosterone during pregnancy in the rat: implications for prenatal stress studies. Psychoneuroendocrinology. 1998;23:571–581. [DOI] [PubMed] [Google Scholar]
- 161. Barrett ES, Swan SH. Stress and androgen activity during fetal development. Endocrinology. 2015;156:3435–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pasquali R, Gambineri A. Insulin-sensitizing agents in polycystic ovary syndrome. European J Endocrinol. 2006;154:763–775. [DOI] [PubMed] [Google Scholar]
- 163. Zulian E, Sartorato P, Benedini S, et al. Spironolactone in the treatment of polycystic ovary syndrome: effects on clinical features, insulin sensitivity and lipid profile. J Endocrinol Invest. 2005;28:49–53. [DOI] [PubMed] [Google Scholar]
- 164. Ganie MA, Khurana ML, Nisar S, et al. Improved efficacy of low-dose spironolactone and metformin combination than either drug alone in the management of women with polycystic ovary syndrome (PCOS): a six-month, open-label randomized study. J Clin Endocrinol Metab. 2013;98:3599–3607. [DOI] [PubMed] [Google Scholar]
- 165. Coviello AD, Legro RS, Dunaif A. Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2006;91:492–497. [DOI] [PubMed] [Google Scholar]
- 166. Corbould A. Effects of androgens on insulin action in women: is androgen excess a component of female metabolic syndrome? Diabetes Metab Res Rev. 2008;24:520–532. [DOI] [PubMed] [Google Scholar]
- 167. Ji Y, Wu Z, Dai Z, Sun K, Wang J, Wu G. Nutritional epigenetics with a focus on amino acids: implications for the development and treatment of metabolic syndrome. J Nutr Biochem. 2016;27:1–8. [DOI] [PubMed] [Google Scholar]
- 168. Mennitti LV, Oliveira JL, Morais CA, et al. Type of fatty acids in maternal diets during pregnancy and/or lactation and metabolic consequences of the offspring. J Nutr Biochem. 2015;26:99–111. [DOI] [PubMed] [Google Scholar]
- 169. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. [DOI] [PubMed] [Google Scholar]
- 170. Luo ZC, Fraser WD, Julien P, et al. Tracing the origins of “fetal origins” of adult diseases: programming by oxidative stress? Med Hypotheses. 2006;66:38–44. [DOI] [PubMed] [Google Scholar]
- 171. Radaelli T, Varastehpour A, Catalano P, Hauguel-de Mouzon S. Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes. 2003;52:2951–2958. [DOI] [PubMed] [Google Scholar]
- 172. Yan X, Zhu MJ, Xu W, et al. Up-regulation of Toll-like receptor 4/nuclear factor-κB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology. 2010;151:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. [DOI] [PubMed] [Google Scholar]
- 174. Neier K, Marchlewicz EH, Dolinoy DC, Padmanabhan V. Assessing human health risk to endocrine disrupting chemicals: a focus on prenatal exposures and oxidative stress. Endocr Disruptors. 2015;3:e1069916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sen S, Simmons RA. Maternal antioxidant supplementation prevents adiposity in the offspring of Western diet-fed rats. Diabetes. 2010;59:3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R–10R [DOI] [PubMed] [Google Scholar]
- 177. Lane RH. Fetal programming, epigenetics, and adult onset disease. Clin Perinatol. 2014;41:815–831. [DOI] [PubMed] [Google Scholar]
- 178. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Bouchard L, Thibault S, Guay SP, et al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care. 2010;33:2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118:2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Xu N, Chua AK, Jiang H, Liu NA, Goodarzi MO. Early embryonic androgen exposure induces transgenerational epigenetic and metabolic changes. Mol Endocrinol. 2014;28:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Stel J, Legler J. The role of epigenetics in the latent effects of early life exposure to obesogenic endocrine disrupting chemicals. Endocrinology. 2015;156:3466–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007;104:13056–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Bell MR, Hart BG, Gore AC. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 2. Sex-specific neuromolecular effects in the brain. Mol Cell Endocrinol. 2016;420:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Bayer TA, Falkai P, Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis.” J Psychiatr Res. 1999;33:543–548. [DOI] [PubMed] [Google Scholar]
- 187. Abbott DH, Nicol LE, Levine JE, Xu N, Goodarzi MO, Dumesic DA. Nonhuman primate models of polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013;373:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Crews D, Gore AC, Hsu TS, et al. Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci USA. 2007;104:5942–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Xin F, Susiarjo M, Bartolomei MS. Multigenerational and transgenerational effects of endocrine disrupting chemicals: a role for altered epigenetic regulation? Semin Cell Dev Biol. 2015;43:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Walker DM, Gore AC. Transgenerational neuroendocrine disruption of reproduction. Nat Rev Endocrinol. 2011;7:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115:1243–1249. [DOI] [PubMed] [Google Scholar]
- 193. Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol. 2008;25:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rissman EF, Adli M. Minireview: transgenerational epigenetic inheritance: focus on endocrine disrupting compounds. Endocrinology. 2014;155:2770–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. [DOI] [PubMed] [Google Scholar]
- 196. Curley JP, Mashoodh R, Champagne FA. Epigenetics and the origins of paternal effects. Horm Behav. 2011;59:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Braun K, Champagne FA. Paternal influences on offspring development: behavioural and epigenetic pathways. J Neuroendocrinol. 2014;26:697–706. [DOI] [PubMed] [Google Scholar]
- 198. Pembrey ME. Male-line transgenerational responses in humans. Hum Fertil (Camb). 2010;13:268–271. [DOI] [PubMed] [Google Scholar]
- 199. Kaplan JL, Shi HN, Walker WA. The role of microbes in developmental immunologic programming. Pediatr Res. 2011;69:465–472. [DOI] [PubMed] [Google Scholar]
- 200. Manco M. Gut microbiota and developmental programming of the brain: from evidence in behavioral endophenotypes to novel perspective in obesity. Front Cell Infect Microbiol. 2012;2:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Thompson AL. Developmental origins of obesity: early feeding environments, infant growth, and the intestinal microbiome. Am J Hum Biol. 2012;24:350–360. [DOI] [PubMed] [Google Scholar]
- 202. Ibáñez L, de Zegher F. Low-dose combination of flutamide, metformin and an oral contraceptive for non-obese, young women with polycystic ovary syndrome. Hum Reprod. 2003;18:57–60. [DOI] [PubMed] [Google Scholar]