

Abstract
Most of polypeptides containing α,β-dehydroamino acids have important biological activity, so exploration of synthetic method has practical significance. In this paper, dipeptides were prepared from l-threonine by protecting of c-terminal allyl acetate, and condensing reaction with a series of N-Boc amino acid. Then, treatment of dipeptides obtained with DMAP, (Boc)2O and tetramethylguanidine in the acetonitrile occured β-elimination reaction to yield stereoselectively dehydrodipeptides. Structures of dehydrodipeptides were confirmed by 1H NMR, 13C NMR and MS. Analysis of 1H NMR, 2D NMR and crystal structure showed that the dehydrodipeptides were Z-configuration.
Graphical abstract.
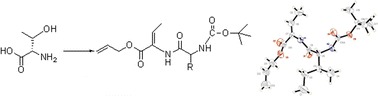
Dehydrodipeptides were prepared from l-threonine. Their structures were confirmed by 1H NMR, 13C NMR and MS
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-016-2005-z) contains supplementary material, which is available to authorized users.
Keywords: l-threonine; α,β-Dehydrobutyrine; Z-isomer; Dipeptide; Synthesis
Background
α,β-Dehydroamino acids, as unnatural amino acids, are found in many natural products including the fungal metabolites, β-lactam antibiotic, sulfide antibiotics, anticarcinoma antibiotics, phytotoxin, the antrimycins, tentoxin, and the phosphatase inhibitors microcystin, nodularin and the synthetic drugs (Gross and Meienhofer 1983; Valentekovich and Schreiber 1995; Botes et al. 1984). In the active peptide, α,β-dehydroamino acids are usually used to fixed peptide main chain and side chain, and make its conformation keeping relatively stable, also can inhibit its biological degradation process. So α,β-dehydroamino acids play an important role in the design and synthesis of biological peptide and the study of its structure–activity relationship (Chang et al. 1998; Rappoport 1994; Harburn et al. 1998; Kohno et al. 1996; Bierbaum 1999).
Many methods available for synthesis of α,β-dehydroamino acids have been reported (Bonauer et al. 2006; Poisel and Schmidt 1976; Kolasa 1983; Maekawa et al. 2004; Schmidt et al. 1992; Trost and Dake 1997; Nagano and Kinoshita 2000; Chen et al. 2005; Goodall and Parsons 1995; Li et al. 1996; Somekh and Shanzer 1983; Miller 1980; Stohimeyer et al. 1999). However, these methods often are low yielding, multistep, require tedious purification steps to remove reagent side products, or incorporate unusual, difficultly obtained amino acid intermediates. Elimination of water from β-hydroxy-α-amino acids is a well-established route to obtain β-dehydroamino acids. This method has been used for the preparation of dehydroalanine and dehydroaminobutanoate from serine and threonine. Shioiri demonstrated Martin’s sulfurane is a mild, neutral and stereospecific dehydrative agent for the dehydrative elimination, which give stereospecific polypeptide with Z-configurational unsaturated amino acid, through removing hydroxyl group of β-hydroxy-α-amino acids in dipeptide or tripeptide (Yokokawa and Shioiri 2002). However, the disadvantage is that the Martin’s sulfurane is expensive. Ferreira reported one important and well-used approach involves the β-elimination reactions of serine and threonine derivatives with Boc-anhydride and 4- (N,N-dimethylamino) pyridine (DMAP) (Ferreira et al. 1999; Ferreira et al. 1998). Furthermore, they use the base N,N,N,N-tetra-methylguanidine (TMG) to induce elimination of the tert-butyl carbonate group from the O- (tert butyl-oxycarbonyl)-β-hydroxyamino acid derivatives, give the corresponding dehydroamino acid derivative. This two-step method can be carried out as a one-pot procedure and is stereoselective, giving only the Z isomer (Ferreira et al. 2007). In this study, We now wish to synthesize a variety of dehydroamino acid derivatives by Ferreira’s synthetic approach and the double bond formed by dehydration reaction is determined Z isomer by NOESY (Shimohigashi et al. 1982; Duhamel et al. 1972) and X-ray crystal diffraction.
Result and discussion
Synthesis
In the synthesis of dehydrodipeptides, first, we obtained dehydroamino acids using N-protected β-hydroxyamino acid esters as raw material. Then, we attempted to obtain dehydrodipeptides by condensation reaction of dehydroamino acids and amino acid. However, owing to the low reactivity of the α-amine group of dehydroamino acids and to the instability of its N-deprotected derivatives, byproducts of reaction are very much, and difficult to separate by this methods. Therefore, this led us to investigate the applicability of Ferreira’s methodology to the dehydration of peptides containing β-hydroxyamino acids as precursors of dehydropeptides. First, dipeptides were prepared from two amino acid, then dehydrodipeptides were obtained by β elimination dehydration of dipeptides.
In order to prepare dipeptides containing l-threonine, the protection of functional groups were very important because of containing three activity groups in l-threonine. Generally, protected groups were selected according to the final products. In this studies, allyl group were used to protect carboxy terminus of amino acid. However, if l-threonine was treated with allyl bromide in dry DMF, besides carboxyl group, the amino group was reacted with allyl bromide. Thus, amino group of l-threonine was protected firstly by Boc group, When N-Boc-protected l-threonine 2 was reacted with allyl bromide in the presence of K2CO3 in dry DMF, it was converted quantitatively into 3 within 12 h. However, in the presence of NaOH, only traces of 3 were detected. This suggests that the by-product may result from a base-induced side-reaction. Thus, the use of an alternative base could possibly reduce this side reaction, so K2CO3 was substituted for NaOH in the reaction of 2 with allyl bromide. Therefore, N-Boc-protected l-threonine 2 were treated with 1.2 equiv. of allyl bromide and K2CO3 in dry DMF to give the corresponding allyl esters of N-protected l-threonine 3.
Dipeptides were readily prepared from 3 by N-deprotection and coupling with the N-protected amino acid using DIPEA/HBTU in higher yield (Scheme 1). Compound 4 was treated with 1.2 molar amounts of N-protected amino acid in the presence of 1.2 molar amounts of HBTU and DIPEA in dichloromethane (DCM) and DMF at room temperature to afford the desired dipeptides 5a-5l in high yield (>90 %).
Scheme 1.
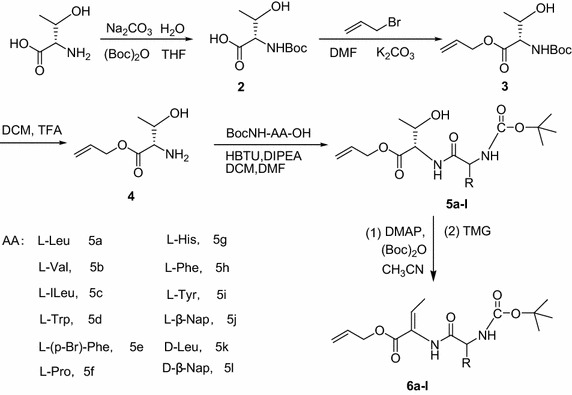
The synthetic route of dehydrodipeptides
Ferreira’s method, one of the noteworthy stereospecific feature of the method was reported for the elimination with Boc-anhydride and 4- (N,N-dimethylamino) pyridine (DMAP) and TMG by Ferreira et al. (1999). Therefore, We treated N-Boc protected dipeptides with 3.3 equiv. of (Boc)2O in the presence of DMAP, gave O-tert-butyl carbonates of dipeptides, followed by direct reaction with N,N,N,N-tetra-methylguanidin (TMG) without isolation, afforded the corresponding dehydrodipeptides in good yields. This two-step method can be carried out as a one-pot procedure. However, In order to compare with Ferreira’s method, we tried to synthesize dehydrodipeptides by β-elimination of dipeptides containing β-hydroxyl group using Martin’s sulfurane. Unfortunately, the treatment of dipeptides 5 with Martin’s sulfurane did not led to the desired products. Therefore, Ferreira’s synthetic approach could be used efficiently stereoselective synthesis of dehydrodipeptides. In comparison with previous methods, this procedure is a one-step process, since separation of the intermediates is unnecessary and theatment of intermediates with TMG gave the corresponding dehydrodipeptides in good yield. This mildness of the mothod is compatible with the presence of a variety of functional groups.
Structure characterization
To further investigate the configuration of the dehydrodipeptides, 6c was subjected to the 2D NMR measurements. Since the NOE cross-peaks between the protons that are closer than 0.4 nm in space will be observed in NOESY spectrum and the relative intensities of these cross-peaks depend on the spaces between the corresponding protons. As can be seen from Fig. 1, the NOESY spectra of 6c showed clear NOE cross-peaks A of H1 of methyl group in double bonds and H2 of amino group in amide bonds, demonstrating that the substituents of methyl goup and amino group located on the same side of the double bond. As well as no correlation between H3 of double hand and H2 of amino group, would provide us further information about the orientation of the H proton and amino group in the double bonds. These indicated distinctly that the double bond in dehydrodipeptides was Z configuration. In addition, We obtained single crystals suitable for X-ray crystallography by slowly evaporating a ethyl acetate solution of 6c. Interestingly, X-ray crystallographic analysis of 6c reveals that the double bond C7–C8 in dehydrodipeptides was Z configuration (Fig. 2). Therefore, the dehydro-dipeptides was synthesized stereoselectively by β-elimination reaction using DMAP, (Boc)2O and tetramethylguanidine.
Fig. 1.
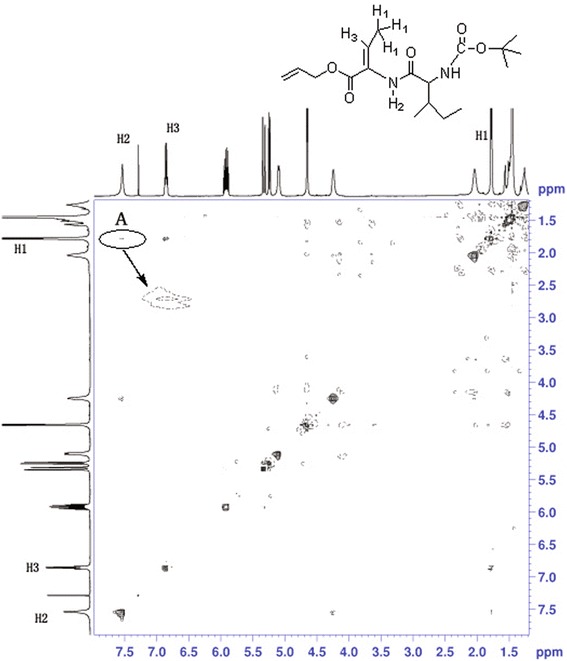
1H NOESY spectrum of 6c in CDCl3 at 293.2 K. Schematic representation of the polypyridyl bases and complexes 1 and 2
Fig. 2.
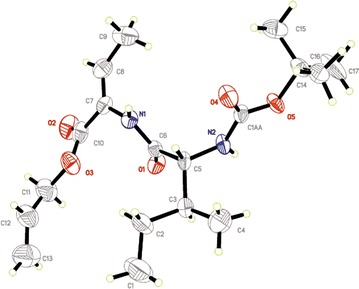
The X-ray crystal structure of 6c
Experimental
Materials and instrumentation
All of the org. solvents used in this study were dried over appropriate drying agents and distilled prior to use. All analytical grade chemicals were purchased commercially and used without further purification. Compound 2 (Goodall and Parsons 1995) was prepared according to the literature procedures.
1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE II500 instrument in CDCl3 solution, using tetramethylsilane as an internal reference. Elemental analyses were performed on a Perkin-Elmer 2400C instrument. The X-ray diffraction data were collected by using a Rigaku Mercury CCD AFC10 system with monochromated Mo Ka radiation.
Synthesis
Boc-L-Thr-OAllyl (3)
21.9 g Boc-l-threonine-OH (0.1 mol) was dissolved in 70 mL DMF, 16.6 g (0.12 mol) K2CO3 was added and cooled to 0 °C in an ice bath. 14.4 g (0.12 mol) Allyl bromide was added dropwise with stirring by means of a separatory funnel. After the mixture was stirred for 1 h, the two phase solution was allowed to warm slowly to room temperature with vigorous stirring over 12 h. The solid residue was isolated by filtration. The solvent in the filtrate was removed in vacuo and the residue was taken up in 80 mL of a saturated NaCl solution, and the aqueous solution was extracted with ethyl acetate (30 mL × 5). The organics were combined and washed with 1 M KHSO4 solution, water, saturated KHCO3 and saturated NaCl, dried over MgSO4, the solvent removed under reduced pressure, evaporated in vacuo to give a viscous liquid, which was purified by means of silica-gel chromatography (Petroleum ether/Ethyl acetate = 5:1) to give Boc-L-Thr-OAllyl 3, the white solid with yield 91 %. 1H NMR (500 MHz, CDCl3) 1.26 (d, 3H), 1.45 (s, 9H), 2.89 (br, 1H), 4.26 (d, J = 8.5 Hz, 1H), 4.33 (m, 1H), 4.66 (m, 2H), 5.29 (dd, J = 1.5 Hz, J = 10.5 Hz, 1H), 5.37 (d, J = 1.5 Hz, J = 16 Hz 1H), 5.49 (d, J = 9.0, 1H), 5.91 (m, 1H). Anal. calc. for C12H21NO5 (259.14): C 55.58, H 8.16, N 5.40; found: C 55.63, H 8.19, N5.44.
Boc-N-AA-L-Thr-OAllyl (5)
1.29 g (5 mmol) Boc-L-Thr- OAllyl was taken up into a solution of trifluoroacetic acid (20 mL) in DCM (30 mL) and stirred for 3 h to remove the Boc group. After removing the solvent and TFA in vacuo, the crude product 4 was obtained (yield > 90 %), which was used to next step reaction without purification. The resulting compound was dissolved in 150 mL DCM and 15 mL DMF and 6 mmol BocNH-AA-OH and 2.25 g (6 mmol) HBTU were added, followed by gradual addition of 7.5 mL DIPEA, the mixture was stirred at room temperature overnight. The solution was concentrated in vacuo, then 100 mL water were added, and the aqueous solution was extracted with ethyl acetate (30 mL × 5). The organics were combined and washed with water, 5 % K2CO3, 2 % HCl and water, dried over MgSO4, the solvent removed under reduced pressure, evaporated in vacuo, the crude product was purified by means of silica-gel chromatography (Petroleum ether/Ethyl acetate = 5:1) to give white solids.
Boc-L-Leu-L-Thr-OAllyl (5a)
White solids, yield 95 %. 1H NMR (500 MHz, CDCl3): δ = 0.93 (d, J = 6.5, 3H), 0.95 (d, J = 6.0 Hz, 3H), 1.21 (d, J = 6.5 Hz, 3H), 1.43 (s, 9H), 1.49 (m, 1H), 1.66 (m, 2H), 3.41 (br, 1H), 4.17 (m, 1H), 4.37 (m, 2H), 4.65 (m, 2H), 5.17 (d, 1H), 5.24 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 17.0 Hz, 1H), 5.90 (m, 1 H), 7.05 (m, 1 H). Anal. calc. for C18H32N2O6 (372.23): C 58.05, H 8.66, N 7.52; found: C58.01, H 8.63, N7.54 (see Additional file 1: Figure S1).
Boc-L-Val-L-Thr-OAllyl (5b)
White solids, yield 97 %. 1HNMR (500 MHz, CDCl3) δ = 0.96 (d, J = 6.5, 3H), 0.98 (d, J = 7.0 Hz, 3H), 1.21 (d, J = 6.0 Hz, 3H), 1.43 (s, 9H), 2.07 (m, 1H), 3.46 (br, 1H), 3.95 (t, 1H), 4.38 (m, 2H), 4.64 (d, 1H), 4.66 (m, 2H), 5.24 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 17.0 Hz, 1H), 5.90 (m, 1 H), 6.99 (m, 1 H). Anal. calc. for C17H30N2O6 (358.21): C 56.97, H 8.44, N 7.82; found: C56.95, H 8.47, N7.88 (see Additional file 1: Figure S5).
Boc-L-ILeu-L-Thr-OAllyl (5c)
White solids, yield 93 %. 1HNMR (500 MHz, CDCl3) δ = 0.89 (d, J = 7.0, 3H), 0.95 (t, J = 7.0 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.23 (m, 2H), 1.43 (s, 9H), 1.49 (m, 1H), 2.05 (m, 1H), 2.14 (br, 1H), 4.19 (m, 1H), 4.38 (m, 2H), 4.62 (m, 1H), 4.67 (m, 1H), 4.94 (d, 1H), 5.26 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 17.0 Hz 1H), 5.90 (m, 1 H), 6.83 (m, 1H). Anal. calc. for C18H32N2O6 (372.23): C 58.05, H 8.66, N 7.52; found: C58.08, H 8.64, N7.57 (see Additional file 1: Figure S9).
Boc-L-Trp-L-Thr-OAllyl (5d)
White solids, yield 91 %. 1HNMR (500 MHz, CDCl3) δ = 0.99 (d, J = 6.5 Hz, 3H), 1.31 (s, 9H), 2.39 (br, 1H), 3.15 (m, 2H), 3.30 (br, 1H), 4.15 (m, 1H), 4.45 (m, 2H), 4.47 (m, 1H), 5.12 (d, J = 10.5 Hz, 1H), 5.19 (d, J = 17.0 Hz, 1H), 5.30 (d, J = 8.0 Hz, 1H), 5.74 (m, 1 H), 6.88 (d, J = 9.0 Hz, 1 H). 6.99 (m, 2H), 7.07 (m, 1H), 7.21 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 8.47 (s, 1H); Anal. calc. for C23H31N3O6 (445.22): C 62.01, H 7.01, N 9.43; found: C61.98, H 7.06, N9.44 (see Additional file 1: Figure S13).
Boc-L- (p-Br)-Phe-L-Thr-OAllyl (5e)
White solids, yield 94 %. 1HNMR (500 MHz, CDCl3) δ = 1.17 (d, J = 6.0 Hz, 3H), 1.39 (s, 9H), 2.09 (br, 1H), 2.95 (m, 1H), 3.09 (m, 1H), 3.14 (br, 1H), 4.32 (m, 1H), 4.41 (m, 1H), 4.60 (m, 1H), 4.65 (m, 2H), 5.23 (m, 1H), 5.26 (d, J = 10.5 Hz, 1H), 5.33 (d, J = 17.0 Hz, 1H), 5.88 (m, 1 H), 6.97 (d, J = 9.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H). C21H29N2O6Br (484.12): C 51.97, H 6.02, N 5.77; found: C51.93, H 6.08, N5.81 (see Additional file 1: Figure S17).
Boc-L-Pro-L-Thr-OAllyl (5f)
White solids, yield 90 %. 1HNMR (500 MHz, CDCl3) δ = 1.20 (d, J = 6.5 Hz, 3H), 1.45 (s, 9H), 1.89 (m, 1H), 2.01 (m, 1H), 2.20 (m, 2H), 3.38 (m, 1H), 3.48 (m, 2H), 4.30 (m, 1H), 4.31 (m, 1H), 4.59 (m, 1H), 4.66 (m, 2H), 5.25 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 17.0 Hz, 1H), 5.90 (m, 1H), 7.24 (br, 1H), Anal. calc. for C17H28N2O6 (356.19): C 57.29, H 7.92, N 7.86; found: C 57.31, H 7.91, N7.89 (see Additional file 1: Figure S21).
Boc-L-His-L-Thr-OAllyl (5g)
White solids, yield 91 %. 1HNMR (500 MHz, CDCl3) δ = 1.14 (d, J = 6.0 Hz, 3H), 1.42 (s, 9H), 1.60 (s, 9H), 2.99 (dd, J = 6.0 Hz, J = 9.0 Hz, 1H), 3.06 (dd, J = 6.0 Hz, J = 9.0 Hz, 1H), 4.29 (m, 1H), 4.53 (m, 1H), 4.56 (m, 2H), 4.62 (m, 2H), 5.23 (d, J = 10.5 Hz, 1H), 5.31 (d, J = 17.0 Hz, 1H), 5.88 (m, 2H), 7.19 (s, 1H), 8.03 (s, 1H), Anal. calc. for C23H36N4O8 (496.25): C 55.63, H 7.31, N 11.28; found: C 55.60, H 7.35, N11.30 (see Additional file 1: Figure S25).
Boc-L-Phe-L-Thr-OAllyl (5h)
White solids, yield 96 %. 1HNMR (500 MHz, CDCl3) δ = 1.17 (d, J = 6.5 Hz, 3H), 1.40 (s, 9H), 2.51 (br, 1H), 3.06 (dd, J = 6.0 Hz, J = 9.0 Hz, 1H), 3.13 (dd, J = 6.0 Hz, J = 9.0 Hz, 1H) 4.29 (m, 1H), 4.39 (m, 1H), 4.59 (m, 1H), 4.63 (m, 1H), 4.64 (m, 1H), 5.05 (m, 1H), 5.25 (d, J = 10.5 Hz, 1H), 5.32 (d, J = 17.0 Hz, 1H), 5.88 (m, 1H), 6.73 (m, 1H), 7.21–7.30 (m, 5H). Anal. calc. for C21H30N2O6 (406.21): C 62.05, H 7.44, N 6.89; found: C 62.01, H 7.46, N6.93 (see Additional file 1: Figure S29).
Boc-L-Tyr-L-Thr-OAllyl (5i)
White solids, yield 93 %. 1HNMR (500 MHz, CDCl3) δ = 1.17 (d, J = 6.0 Hz, 3H), 1.41 (s, 9H), 2.98 (m, 2H), 4.33 (m, 2H), 4.57 (m, 1H), 4.623 (m, 2H), 5.14 (m, 1H), 5.25 (d, J = 10.5 Hz, 1H), 5.31 (d, J = 17.0 Hz, 1H), 5.88 (m, 1H), 6.07 (br, 1H), 6.72 (d, J = 8.5 Hz, 2H), 7.04 (m, J = 8.5 Hz, 2H), Anal. calc. for C21H30N2O7 (422.21): C 59.70, H 7.16, N 6.63; found: C 59.72, H 7.19, N6.68 (see Additional file 1: Figure S32).
Boc-L-β-Nap-L-Thr-OAllyl (5j)
White solids, yield 90 %. 1HNMR (500 MHz, CDCl3) δ = 1.12 (d, J = 6.5 Hz, 3H), 1.43 (s, 9H), 3.19 (dd, J1 = 6.0 Hz, J2 = 7.5 Hz, 1H), 3.27 (dd, J1 = 6.0 Hz, J2 = 7.5 Hz, 1H), 4.27 (br, 1H), 4.50 (m, 2H), 4.61 (d, J = 6.0 Hz, 2H), 4.98 (m, 3H), 5.13 (d, J = 8.0 Hz, 1H), 5.83 (m, 1 H), 6.85 (m, 1 H), 7.42 (m, 1H), 7.45 (m, 2H), 7.64 (m, 1H), 7.74 (m, 3H); Anal. calc. for C25H32N2O6 (456.23): C65.77, H 7.07, N6.14; found: C65.72, H 7.04, N6.17 (see Additional file 1: Figure S36).
Boc-D-Leu-L-Thr-OAllyl (5k)
White solids, yield 92 %. 1H NMR (500 MHz, CDCl3): δ = 0.95 (d, J = 6.0 Hz, 3H), 0.96 (d, J = 4.5 Hz, 3H), 1.23 (d, J = 6.5 Hz, 3H), 1.45 (s, 9H), 1.49 (m, 1H), 1.69 (m, 3H), 4.18 (br, 1H), 4.36 (br, 1H), 4.59 (m, 1H), 4.66 (m, 2H), 4.90 (m, 1H), 5.25 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 17 Hz 1H), 5.90 (m, 1 H), 6.92 (m, 1 H). Anal. calc. for C18H32N2O6 (372.23): C 58.05, H 8.66, N 7.52; found: C58.03, H 8.69, N7.56 (see Additional file 1: Figure S40).
Boc-D-β-Nap-L-Thr-OAllyl (5l)
White solids, yield 93 %. 1HNMR (500 MHz, CDCl3) δ = 1.00 (d, J = 6.0 Hz, 3H), 1.38 (s, 9H), 3.24 (dd, J1 = 6.0 Hz, J2 = 7.0 Hz, 1H), 3.29 (dd, J1 = 6.0 Hz, J2 = 7.0 Hz, 1H), 4.22 (br, 1H), 4.54 (m, 2H), 4.62 (d, J = 5.5 Hz, 2H), 5.03 (m, 1H), 5.24 (d, J = 10.5 Hz, 1H), 5.15 (d, J = 17.0 Hz 1H), 5.87 (m, 1 H), 6.69 (m, 1 H), 7.37 (m, 1H), 7.45 (m, 2H), 7.67 (m, 1H), 7.79 (m, 3H); Anal. calc. for C25H32N2O6 (456.23): C65.77, H 7.07, N6.14; found: C65.75, H 7.09, N6.20 (see Additional file 1: Figure S43).
Synthesis of Boc-AA-Z-∆Abu-OAllyl
Boc-N-AA-L- Thr-OAllyl 5 (2.0 mmol) was dissolved in 2 mL dry acetonitrile, DMAP (0.2 mmol) was added to above solution, followed by di-tert-butyl dicarbonate (3.3 equiv.) under rapid stirring at room temperature 2 h. Then TMG (0.4 mL) was added dropwise to above solution, the reaction was stirred overnight. Evaporation at reduced pressure gave a residue that was partitioned between diethyl ether (100 mL) and KHSO4 (50 mL, 1 mol dm−3). The organic phase was thoroughly washed with KHSO4 (1 mol dm−3), NaHCO3 (1 mol dm−3) and saturated brine (3×50 mL, each), and dried with MgSO4. Removal of the solvent afforded 8c as an oil. The crude product was purified by means of silica-gel chromatography (Petroleum ether/Ethyl acetate = 8:1) to give the product.
Boc-L-Leu-Z-ΔAbu-OAllyl (6a)
Yellow viscous liquid, yield 90 %. 1H NMR (500 MHz, CDCl3): δ = 0.96 (m, 6H), 1.45 (s, 9H), 1.54 (m, 1H), 1.76 (d, J = 7.0 Hz, 3H), 1.77 (m, 2H), 4.27 (br, 1H), 4.65 (d, J = 4.5 Hz, 2H), 5.05 (br, 1H), 5.24 (d, J = 5.5 Hz, 1H), 5.33 (d, J = 17.0 Hz, 1H), 5.92 (m, 1 H), 6.83 (m, 1 H), 7.66 (br, 1H). 13C NMR (500 MHz, CDCl3): δ 14.20 (1C), 14.54 (1C), 22.92 (1C), 24.73 (1C), 28.28 (3C), 40.99 (1C), 53.24 (1C), 60.42 (1C), 65.89 (1C), 118.50 (1C), 125.97 (1C), 131.86 (1C), 134.47 (1C), 155.82 (1C), 163.96 (1C), 171.02 (1C), 173.84 (1C); EI-MS m/z 377.3 (M+ + Na) (see Additional file 1: Figures S2–S4).
Boc-L-Val-Z-ΔAbu-OAllyl (6b)
White solids, m.p. 88–89 °C, yield 92 %. 1H NMR (500 MHz, CDCl3): δ = 0.98 (t, J = 7.0 Hz, 3H), 1.03 (d, J = 7.0 Hz, 3H), 1.44 (s, 9H), 1.77 (d, J = 7.0 Hz, 3H), 2.20 (br, 1H), 4.12 (br, 1H), 4.64 (d, J = 5.5 Hz, 2H), 5.22 (d, J = 10.5 Hz, 1H), 5.24 (br, 1H), 5.32 (d, J = 17.0 Hz, 1H), 5.91 (m, 1H), 6.837 (m, 1H), 7.730 (br, 1H). 13C NMR (500 MHz, CDCl3): δ 14.47 (1C), 17.84 (1C), 19.29 (1C), 28.27 (3C), 28.40 (1C), 30.88 (1C), 59.98 (1C), 65.87 (1C), 118.51 (1C), 126.15 (1C), 131.84 (1C), 134.67 (1C), 155.99 (1C), 163.95 (1C), 170.45 (1C), EI-MS m/z 363.2 (M+ + Na) (see Additional file 1: Figures S6–S8).
Boc-L-ILeu-Z-ΔAbu-OAllyl (6c)
White solids, m.p. 107–109 °C, yield 94 %. 1H NMR (500 MHz, CDCl3): δ = 0.93 (t, J = 7.0 Hz, 3H), 0.96 (d, J = 7.5 Hz, 3H), 1.26 (m, 2H), 1.45 (s, 9H), 1.46 (m, 1H), 1.77 (d, J = 7.0 Hz, 3H), 2.05 (br, 1H), 4.22 (br, 1H), 4.66 (d, J = 5.5 Hz, 2H), 5.019 (br, 1H), 5.24 (d, J = 10.5 Hz, 1H), 5.33 (d, J = 17.0 Hz, 1H), 5.92 (m, 1H), 6.86 (m, 1H), 7.41 (br, 1H). 13C NMR (500 MHz, CDCl3): δ11.71 (1C), 14.30 (1C), 14.72 (1C), 26.37 (1C), 28.28 (3C), 28.28 (1C), 37.07 (1C), 58.43 (1C), 65.96 (1C), 118.62 (1C), 125.82 (1C), 131.82 (1C), 134.69 (1C), 155.92 (1C), 164.00 (1C), 170.39 (1C). EI-MS m/z 377.4 (M+ + Na) (see Additional file 1: Figures S10–S12).
Boc-L-Trp-Z-ΔAbu-OAllyl (6d)
White solids, m.p. 126–127 °C, yield 88 %. 1H NMR (500 MHz, CDCl3): δ = 1.42 (s, 9H), 1.67 (t, J = 7.0 Hz, 3H), 3.30 (m, 2H), 4.61 (m, 3H), 5.13 (m, 1H), 5.23 (d, J = 10.5 Hz, 1H), 5.31 (d, J = 17.0 Hz, 1H), 5.90 (m, 1H), 6.80 (m, 1H), 7.14 (m, 2H), 7.21 (m, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H); 8.10 (br, 1H). 13C NMR (500 MHz, CDCl3): δ14.54 (1C), 28.22 (3C), 29.68 (1C), 55.27 (1C), 61.61 (1C), 65.81 (1C), 111.14 (1C), 118.41 (1C), 118.83 (1C), 119.78 (1C), 122.27 (1C), 123.32 (1C), 125.73 (1C), 127.49 (1C), 128.5 (1C), 131.83 (1C), 134.45 (1C), 136.17 (1C), 163.77 (1C), 170.09 (1C), 170.09 (1C). EI-MS m/z 450.3 (M+ + Na) (see Additional file 1: Figures S14–S16).
Boc-L- (p-Br) -Phe-Z-ΔAbu-OAllyl (6e)
Yellow viscous liquid, yield 88 %. 1H NMR (500 MHz, CDCl3): δ = 1.11 (d, J = 7.0 Hz, 3H), 1.59 (s, 9H), 3.27 (dd, J = 2.5 Hz, J = 12.0 Hz, 1H), 3.56 (dd, J = 2.5 Hz, J = 12.0 Hz, 1H), 4.64 (m, 2H), 4.817 (m, 1H), 5.23 (d, J = 10.5 Hz, 1H), 5.28 (d, J = 17.0 Hz, 1H), 5.87 (m, 1H), 7.04 (d, J = 8.0 Hz, 2H), 7.19 (m, 1H), 7.40 (d, J = 8.0 Hz, 2H). 13C NMR (500 MHz, CDCl3): δ13.35 (1C), 27.85 (1C), 28.09 (3C), 34.15 (1C), 60.45 (1C), 66.29 (1C), 118.71 (1C), 121.89 (1C), 131.47 (1C), 131.63 (2C), 131.99 (2C), 132.49 (1C), 144.39 (1C), 148.67 (1C), 149.82 (1C), 161.39 (1C), 168.68 (1C). EI-MS m/z 491.2 (M+ + H + Na) (see Additional file 1: Figures S18–S20).
Boc-L-Pro-Z-ΔAbu-OAllyl (6f)
Yellow viscous liquid, yield 90 %. 1H NMR (500 MHz, CDCl3): δ = 1.48 (s, 9H), 1.78 (d, J = 6.5 Hz 3H), 1.94 (m, 3H), 2.20 (m, 1H), 3.42 (m, 2H), 4.38 (m, 1H), 4.65 (m, 2H), 5.24 (d, J = 10.5 Hz 1H), 5.33 (d, J = 17.0 Hz 1H), 5.92 (m, 1H), 6.82 (m, 1H). 13C NMR (500 MHz, CDCl3): δ 14.75 (1C), 24.61 (1C), 28.35 (3C), 31.36 (1C), 47.16 (1C), 60.04 (1C), 61.49 (1C), 65.79 (1C), 118.69 (1C), 126.63 (1C), 131.97 (1C), 133.57 (1C), 155.89 (1C), 163.94 (1C), 171.17 (1C). EI-MS m/z 361.3 (M+ + Na) (see Additional file 1: Figures S22–S24).
Boc-L-His-Z-ΔAbu-OAllyl (6g)
Yellow viscous liquid, yield 88 %. 1H NMR (500 MHz, CDCl3): δ = 1.58 (s, 18H), 1.73 (d, J = 7.5 Hz, 3H), 3.34 (dd, J = 6.0 Hz, J = 8.5 Hz, 1H), 3.56 (dd, J = 6.0 Hz, J = 8.5 Hz, 1H), 4.66 (m, 2H), 4.75 (m, 1H), 5.23 (d, J = 10.5 Hz 1H), 5.311 (d, J = 17.0 Hz 1H), 5.90 (m, 1H), 7.14 (s, 1H), 7.33 (m, 1H), 7.90 (s, 1H). 13CNMR (500 MHz, CDCl3): δ12.82 (1C), 27.00 (3C), 26.83 (3C), 26.69 (1C), 28.67 (1C), 58.18 (1C), 65.19 (1C), 114.39 (1C), 117.55 (1C), 121.45 (1C), 130.55 (1C), 135.49 (1C), 136.04 (1C), 143.58 (1C), 147.46 (1C), 149.45 (1C), 160.83 (1C), 168.29 (1C). EI-MS m/z 479.3 (M+ + H) (see Additional file 1: Figures S26–S28).
Boc-L-Phe-Z-ΔAbu-OAllyl (6h)
White solids, m.p. 108–109 °C, yield 91 %. 1H NMR (500 MHz, CDCl3): δ = 1.41 (s, 9H), 1.70 (d, J = 7.0 Hz, 3H), 3.06 (dd, J = 6.5 Hz, J = 7.5 Hz, 1H), 3.09 (dd, J = 6.5 Hz, J = 7.5 Hz, 1H), 4.49 (m, 1H), 4.64 (m, 2H), 5.00 (m, 1H), 5.246 (d, J = 11.5 Hz 1H), 5.312 (d, J = 17.0 Hz, 1H), 5.90 (m, 1H), 6.83 (q, J = 7.0 Hz, 1H), 7.25 (m, 5H). 13C NMR (500 MHz, CDCl3): δ = 14.55 (1C), 28.24 (3C), 38.63 (1C), 52.31 (1C), 55.90 (1C), 70.26 (1C), 125.75 (1C), 126.99 (1C), 128.69 (3C), 129.40 (3C), 134.53 (1C), 136.45 (1C), 155.55 (1C), 164.63 (1C), 169.72 (1C). EI-MS m/z 411.3 (M+ + Na) (see Additional file 1: Figures S30, S31)
Boc-L-Tyr-Z-ΔAbu-OAllyl (6i)
White solids, m.p. 87–89 °C, yield 92 %. 1H NMR (500 MHz, CDCl3): δ = 1.41 (s, 9H), 1.69 (d, J = 7.0 Hz, 3H), 3.03 (dd, J1 = 6.0 Hz, J2 = 8.0 Hz, 1H), 3.17 (dd, J1 = 6.0 Hz, J2 = 8.0 Hz, 1H), 4.48 (m, 1H), 4.63 (d, J = 6.0 Hz, 2H), 5.07 (m, 1H), 5.23 (d, J = 10.5 Hz, 1H), 5.32 (d, J = 17.0 Hz, 1H), 5.90 (m, 1H), 6.82 (m, 1H), 7.10 (d, J = 8.0 Hz, 2H), 7.5 (d, J = 8.0 Hz, 2H). 7.48 (br, 1H), 13C NMR (500 MHz, CDCl3): δ14.59 (1C), 27.70 (3C), 28.25 (1C), 37.14 (1C), 55.76 (1C), 65.93 (1C), 118.55 (1C), 121.48 (2C), 125.77 (1C), 130.37 (2C), 131.85 (1C), 134.74 (1C), 150.09 (1C), 151.84 (1C), 155.53 (1C), 163.81 (1C), 169.60 (1C). EI-MS m/z 405.2 (M+ + 1) (see Additional file 1: Figures S33–S35).
Boc-L-β-Nap-Z-Δabu–OAllyl (6j)
White solids, m.p. 108–109 °C, yield 93 %. 1H NMR (500 MHz, CDCl3): δ = 0.64 (d, J = 7.0 Hz, 3H), 1.67 (s, 9H), 3.50 (dd J = 2.5 Hz, J = 11.5 Hz, 1H), 3.77 (d, J = 5.0 Hz, J = 9.0 Hz, 1H), 4.63 (m, 2H), 4.91 (m, 1H), 5.22 (d, J = 11.5 Hz, 1H), 5.30 (d, J = 17.0 Hz, 1H), 5.87 (m, 1H), 7.06 (q, J = 7.5 Hz, 1H), 7.20 (m, 1H), 7.44 (m, 2H), 7.70 (s, 1H), 7.74 ( (m, 3H).13C NMR (500 MHz, CDCl3): δ12.97 (1C), 27.73 (1C), 28.15 (3C), 34.87 (1C), 60.92 (1C), 66.22 (1C), 118.65 (1C), 122.11 (1C), 126.11 (1C), 126.38 (1C), 127.56 (1C), 128.55 (1C), 129.16 (1C), 130.98 (1C), 131.50 (1C), 132.67 (1C), 133.40 (1C), 144.45 (1C), 148.77 (1C), 149.88 (1C), 161.45 (1C), 168.99 (1C), 171.17 (1C). EI-MS m/z 461.3 (M+ + Na) (see Additional file 1: Figures S37–S39).
Boc-D-Leu-Z-ΔAbu-OAllyl (6k)
Yellow viscous liquid, yield 89 %. 1H NMR (500 MHz, CDCl3): δ = 0.96 (t, J = 6.5 Hz, 6H), 1.46 (s, 9H), 1.56 (m, 2H), 1.66 (m, 2H), 1.75 (d, J = 7.0 Hz, 3H), 4.22 (br, 1H), 4.65 (d, J = 6.0 Hz, 2H), 4.90 (br, 1H), 5.26 (d, J = 10.5 Hz, 1H), 5.35 (d J = 17.0 Hz, 1H), 5.92 (m, 1H), 6.85 (m, 1H), 7.48 (br, 1H). 13C NMR (500 MHz, CDCl3): δ14.21 (1C), 14.53 (1C), 22.92 (1C), 24.73 (1C), 28.29 (3C), 41.00 (1C), 53.24 (1C), 60.44 (1C), 65.89 (1C), 118.50 (1C), 125.97 (1C), 131.83 (1C), 134.47 (1C), 155.84 (1C), 163.99 (1C), 171.02 (1C), 173.88 (1C); EI-MS m/z 355.2 (M+ + H) (see Additional file 1: Figures S41, S42).
Boc-D-β-Nap-Z-Δabu–Oallyl (6l)
White solids, m.p. 109–110 °C, yield 92 %. 1H NMR (500 MHz, CDCl3): δ = 0.65 (d, J = 7.5 Hz, 3H), 1.61 (s, 9H), 3.49 (dd, J = 2.5 Hz, J = 11.5 Hz, 1H), 3.770 (d, J = 5.0 Hz, J = 9.0 Hz, 1H), 4.62 (m, 2H), 4.90 (m, 1H), 5.22 (d, J = 10.5 Hz, 1H), 5.26 (d, J = 17.0 Hz, 1H), 5.86 (m, 1H), 7.05 (q, J = 7.5 Hz, 1H), 7.21 (m, 1H), 7.44 (m, 2H), 7.71 (s, 1H), 7.74 (m, 3H). 13C NMR (500 MHz, CDCl3): δ12.97 (1C), 21.07 (1H), 28.14 (3C), 34.87 (1C), 60.92 (1C), 66.21 (1C), 118.63 (1C), 122.10 (1C), 126.11 (1C), 126.38 (1C), 127.63 (1C), 128.54 (1C), 129.15 (1C), 130.96 (1C), 131.50 (1C), 132.68 (1C), 133.40 (1C), 144.43 (1C), 148.77 (1C), 149.87 (1C), 161.44 (1C), 168.98 (1C), 171.18 (1C). EI-MS m/z 461.2 (M+ + Na) (see Additional file 1: Figures S44, S45).
Conclusions
α,β-Dehydroamino acids play an important role in the design and synthesis of biological peptide and the study of its structure-activity relationship, while The synthesis of peptides containing dehydroamino acids were a challenge. A variety of dehydroamino acid derivatives were synthesized. The results showed that this methods could be carried out as a one-pot procedure, and had high stereoselectivity. 2D NMR NOESY and X-ray crystal diffraction determined Z configuration of double bond.
Supplementary information
All additional information pertaining to characterization of the complexes using 1H NMR, 13C NMR and EI-MS spectra are given in the supporting information available at XXX.
Authors’ contributions
XT, LL, JH, XZ, SL were involved in the study design and manuscript preparation, data collection, data analysis and revisions. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful for the financial assistance received from the National Natural Science Foundation of China (NSFC) (Grant numbers 21272052, 21472034), the National Basic Research Program of China (Grant numbers 2011CB512007 and 2012CB723501), the Hebei Province Natural Science Foundation (Grant numbers B2014208138, 12966737D), Hebei province Science and Technology Support Program (Grant numbers 14272604D), and the Foundation of the Education Department of Hebei Province (Grant numbers ZH2012025, ZD2014017).
Competing interests
None declared under financial, general, and institutional competing interests. I wish to disclose a competing interest (s) such as those defined above or others that may be perceived to influence the results and discussion reported in this paper.
Additional file
10.1186/s40064-016-2005-z 1H NMR, 13C NMR and MS spectrums of some intermediates and all dehydrodipeptides.
Contributor Information
Xia Tian, Email: kdtxxyzxyz@126.com.
Linna Li, Email: 1101903925@qq.com.
Jianrong Han, Email: han_jianrong@126.com.
Xiaoli Zhen, Email: zhenxlleilei@126.com.
Shouxin Liu, Email: chsxLiu@hotmail.com.
References
- Bierbaum G. Chemotherapie J. 1999;6:204. [Google Scholar]
- Bonauer C, Walenzyk T, König B. α, β-dehydroamino acids. (2006) Synthesis 1:1
- Botes DP, Tuinman AA, Wessels PL, Viljoen CC, Kruger H, Williams DH, Santikarn S, Smith RJ, Hammond SJ. The structure of cyanoginosin-LA, a cyclic heptapeptide toxin from the cyanobacterium Microcystis aeruginosa. J Chem Soc Perkin Trans. 1984;1:2311. doi: 10.1039/p19840002311. [DOI] [Google Scholar]
- Chang LC, Bhat KPL, Pisha E, Kennelly EJ, Fong HHS, Pezzuto JM, Kinghorn AD. Activity-Guided isolation of steroidal alkaloid antiestrogen-binding site inhibitors from Pachysandra procumbens. J Nat Prod. 1998;10:1257. doi: 10.1021/np980162x. [DOI] [PubMed] [Google Scholar]
- Chen DJ, Guo L, Liu JY, Kirtane S, Cannon JF, Li GG. Functionalization of α, β-unsaturated esters and ketones: a facile and highly stereoselective one-pot approach to N-protected α, β-dehydroamino acid derivatives. Org Lett. 2005;7:921. doi: 10.1021/ol050002u. [DOI] [PubMed] [Google Scholar]
- Duhamel L, Duhamel P, Combrisson S. Configuration D’enamines B substituees etude par effet overhauser intramoleculaire. Tetrahedron Lett. 1972;34:3603. doi: 10.1016/S0040-4039(01)94110-3. [DOI] [Google Scholar]
- Ferreira PMT, Maia HLS, Pereira G. Reactivity of dehydroamino acids and dehydrodipeptides towards N-bromosuccinimide: synthesis of β-bromo-and β,β-dibromodehydroamino acid derivatives and of substituted 4-imidazolidinones. Eur J Org Chem. 2007;35:5934. doi: 10.1002/ejoc.200700669. [DOI] [Google Scholar]
- Ferreira PMT, Maia HLS, Monteiro LS. Efficient synthesis of dehydroamino acid derivatives. Tetrahedron Lett. 1998;39:9575. doi: 10.1016/S0040-4039(98)02164-9. [DOI] [Google Scholar]
- Ferreira PMT, Maia HLS, Monteiro LS, Sacramento J. High yielding synthesis of dehydroamino acid and dehydropeptide derivatives. J Chem Soc Perkin Trans. 1999;1:3697. doi: 10.1039/a904730a. [DOI] [Google Scholar]
- Goodall K, Parsons AF. A new and efficient preparation of α, β-dehydroamino acids. Tetrahedron Lett. 1995;18:3259. doi: 10.1016/0040-4039(95)00459-P. [DOI] [Google Scholar]
- Gross E, Meienhofer J (1983) The peptides: analysis, synthesis, biology, vol 4. Elsevier Inc, p 285
- Harburn JJ, Lofius GC, Marples BA. Synthesis of novel steroidal inhibitors of HIV-I protease. Tetrahedron. 1998;39:11907. doi: 10.1016/S0040-4020(98)83048-2. [DOI] [Google Scholar]
- Kohno J, Nishio M, Kawano K, Nakanishi N, Suzuki S, Chida T, Komatsubara S. TMC-1 A, B, C and D, new antibiotics of the manumycin group produced by Streptomyces sp. Taxonomy, production, isolation, physico-chemical properties, structure elucidation and biological properties. J Antibiot. 1996;12:1212. doi: 10.7164/antibiotics.49.1212. [DOI] [PubMed] [Google Scholar]
- Kolasa T. An effective synthesis of α, β-dehydro-α-amino acid derivatives from N-acyl-N-hydroxy-α-amino acid esters. Synthesis. 1983;7:539. doi: 10.1055/s-1983-30412. [DOI] [Google Scholar]
- Li KW, Wu J, Xing WN, Simon JA. Total synthesis of the antitumor depsipeptide FR-901,228. J Am Chem Soc. 1996;30:7237. doi: 10.1021/ja9613724. [DOI] [Google Scholar]
- Maekawa K, Kanno Y, Kubo K, Igarashi T, Sakurai T. A novel ring-opening reaction of (Z)-2-methyl-4-arylmethylene-5(4H)-oxazolone derivatives with acylhydrazines. Heterocycles. 2004;63:1273. doi: 10.3987/COM-04-10061. [DOI] [Google Scholar]
- Miller MJ. Isourea-mediated preparation of dehydro amino acids. J Org Chem. 1980;15:313l. [Google Scholar]
- Nagano T, Kinoshita H. A new and convenient method for the synthesis of dehydroamino acids starting from ethyl N-Boc- and N-Z-α-tosylglycinates and various nitro compounds. Bull Chem Soc Jpn. 2000;73:1605. doi: 10.1246/bcsj.73.1605. [DOI] [Google Scholar]
- Poisel H, Schmidt U. Synthesis of α,β-dehydro amino acid esters and N-tert-butoxycarbonyl α,β-dehydro amino acids. Angew Chem Int Ed. 1976;5:294. doi: 10.1002/anie.197602942. [DOI] [PubMed] [Google Scholar]
- Rappoport Z. The chemistry of enamines, Part 1. New York: Wiley; 1994. p. 612. [Google Scholar]
- Schmidt U, Griesser H, Leitenberger V, Lieberknecht A, Mangold R, Meyer R, Riedl B. Diastereoselective formation of (Z)-didehydroamino acid esters. Synthesis. 1992;5:487–490. doi: 10.1055/s-1992-26143. [DOI] [Google Scholar]
- Shimohigashi Y, Nitz TJ, Stammer CH. E/Z-Configurational assignment of dehydropeptides: differential noe enhancement between the vinyl and amide protons of an α,β-dehydro amino acid derivative. Tetrahedron Lett. 1982;32:3235. doi: 10.1016/S0040-4039(00)87578-4. [DOI] [Google Scholar]
- Somekh L, Shanzer A. Stereospecific synthesis of α,β-dehydroamino acids from β-hydroxy α-amino acid derivatives. J Org Chem. 1983;6:907. doi: 10.1021/jo00154a040. [DOI] [Google Scholar]
- Stohimeyer MM, Tanaka H, Wandless TJ. A stereospecific elimination to form dehydroamino acids: synthesis of the phomopsin tripeptide side chain. J Am Chem Soc. 1999;121:6100. doi: 10.1021/ja991037q. [DOI] [Google Scholar]
- Trost BM, Dake GRJ. Nucleophilic α-addition to alkynoates. A synthesis of dehydroamino acids. J Am Chem Soc. 1997;119:7595. doi: 10.1021/ja971238z. [DOI] [Google Scholar]
- Valentekovich RJ, Schreiber SL. Enantiospecific total synthesis of the protein phosphatase inhibitor motuporin. J Am Chem Soc. 1995;117:9069. doi: 10.1021/ja00140a025. [DOI] [Google Scholar]
- Yokokawa F, Shioiri T. Novel stereospecific dehydration of β-hydroxy-α-amino acids using Martin’s sulfurane. Tetrahedron Lett. 2002;48:8679. doi: 10.1016/S0040-4039(02)02179-2. [DOI] [Google Scholar]