

Abstract
Powdery mildew disease caused by Blumeria graminis f. sp. tritici (Bgt) inflicts severe economic losses in wheat crops. A systematic understanding of the molecular mechanisms involved in wheat resistance to Bgt is essential for effectively controlling the disease. Here, using the diploid wheat Triticum urartu as a host, the genes regulated by immune (IM) and hypersensitive reaction (HR) resistance responses to Bgt were investigated through transcriptome sequencing. Four gene coexpression networks (GCNs) were developed using transcriptomic data generated for 20 T. urartu accessions showing IM, HR or susceptible responses. The powdery mildew resistance regulated (PMRR) genes whose expression was significantly correlated with Bgt resistance were identified, and they tended to be hubs and enriched in six major modules. A wide occurrence of negative regulation of PMRR genes was observed. Three new candidate immune receptor genes (TRIUR3_13045, TRIUR3_01037 and TRIUR3_06195) positively associated with Bgt resistance were discovered. Finally, the involvement of TRIUR3_01037 in Bgt resistance was tentatively verified through cosegregation analysis in a F2 population and functional expression assay in Bgt susceptible leaf cells. This research provides insights into the global network properties of PMRR genes. Potential molecular differences between IM and HR resistance responses to Bgt are discussed.
Powdery mildew fungi infect more than 10,000 plant species, and frequently decrease the grain yield and quality of agricultural crops1,2. Powdery mildew disease elicited by Blumeria graminis f. sp. tritici (Bgt) occurs worldwide in wheat crops, and can reduce grain yield by 5% to 45% depending on the severity of infestation3. Cultivation of resistant wheat varieties is the most economic measure for controlling the damage caused by Bgt4,5. In order to efficiently develop resistant varieties, it is necessary to have a detailed understanding of the genetic interactions and molecular mechanisms underlying Bgt resistance.
To date, more than 70 wheat genes and alleles conferring Bgt resistance have been identified and mapped at 49 chromosomal loci, most of which function in a race specific manner6. However, only four Bgt resistance genes (Pm3, Pm8, Pm21 and Pm38) have been molecularly characterized in detail7,8,9,10, and the signaling cascades and interacting proteins required for the functions of the four genes remain largely unknown. On a genome-wide scale, the gene networks and molecular interactions involved in wheat resistance to Bgt are also unclear at present. The slow progress in systematically dissecting the genes and functioning molecular interactions in Bgt resistance may partly be caused by the high genomic complexity of the polyploid wheat species, i.e., hexaploid common wheat (Triticum aestivum, AABBDD, 2n = 6x = 42) and tetraploid durum wheat (Triticum turgidum ssp. durum, AABB, 2n = 4x = 28), which have often been used in past genetic and molecular studies on wheat-Bgt interactions. According to a chromosome-based draft sequence published recently11, the hexaploid genome of common wheat is approximately 17 Gb in size and contains more than 120,000 genes.
Because of its well characterized genome and the availability of abundant functional genomic resources, the model plant Arabidopsis thaliana has been frequently used for studying the molecular mechanisms of plant resistance to fungal pathogens including the powdery mildews Golovinomyces cichoracearum, G. orontii and Erysiphe cruciferarum12. Through the studies on Arabidopsis and similar investigations in other plant species, e.g., rice (Oryza sativa), tomato (Solanum lycopersicum) and barley (Hordeum vulgare), it is now clear that plants generally employ a complex, two-tiered immune system to defend against pathogen attacks, namely microbe-associated molecular pattern (MAMP)-triggered immunity (MTI) and effector-triggered immunity (ETI)13,14,15,16,17,18,19. The former is a basal immune response initiated after sensing MAMPs by plant cell surface located pattern-recognition receptors (PRRs)13,14,15,16. The latter is activated after recognizing pathogen encoded avirulence factors (also called effector proteins) by nucleotide-binding domain, leucine-rich repeat (NLR) proteins, which are intracellularly located plant immune receptors17,18,19. ETI functions by enhancing MTI, and is frequently accompanied by restricted cell death and long distance defense signaling13,14,15,16,17,18,19. In higher plants, typical NLR proteins have been divided into two types17,18,19. The first type carries a N-terminal coiled-coil (CC) domain followed by nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains (like Pm3 protein). The second type also carries NBS and LRR domains but has a TOLL/interleukin 1 receptor (TIR) domain at the N-terminus. Moreover, plant species often contain multiple genes encoding different NLR proteins20. For example, the model grass species rice and purple false brome grass (Brachypodium distachyon) carry 458 and 212 NLR genes, respectively21.
Although previous studies have suggested that the mutation of a single NLR gene is sufficient to turn resistance to susceptibility for a given pathogenic race22,23, an increasing number of investigations have now revealed that the coordinated function between different NLR genes is required for successful resistance. For example, the paired NLR decoy receptor formed by RPS4 and RRS1 proteins is needed for resistance to several bacterial and fungal pathogens of Arabidopsis24,25. In this receptor complex, the decoy WRKY domain of RRS1 binds the pathogen effector and initiates resistance signaling together with RPS4. Concomitant to above studies, genome-wide expression profiling and transcriptome sequencing experiments have shown that the transcript levels of multiple NLR genes are significantly changed after attack by a given pathogen26,27,28. Apart from the NLR genes critical for resistance signaling, many other genes are also activated in the downstream defense events. In Arabidopsis, it has been estimated that approximately 14% of all annotated genes may be directly related to pathogen defense29. In barley, a recent study using transient-induced gene silencing identified 96 genes involved in the resistance to non-adapted or adapted powdery mildew fungi30. Clearly, a large number of host genes take part in resistance signaling and defense processes, and they may interact in a complex manner. Because of this situation, network analysis has emerged as a valuable approach for systematically uncovering and understanding the molecular complexities of plant immunity31.
So far, two main types of high throughput network analysis have been applied for systematically investigating the genes involved in plant resistance responses31. The first one is protein-protein interactome network analysis. Two representative studies of this type concern the construction and analysis of plant-pathogen immune networks (PPIN-1 and -2) in Arabidopsis using yeast two-hybrid experiment32,33. The network developed is composed of four categories of proteins, i.e., pathogen effectors, effector targets in host cells, known immune proteins (including NLR, receptor like kinase, and defense proteins), and immune interactors. Through analyzing the interactome, it was suggested that many pathogen effector targets are in fact important host proteins (i.e., cellular hubs) rather than NLR proteins. This supports the guard hypothesis of plant immunity, which proposes that most NLR proteins are indirectly linked with pathogen effectors, and that the signaling function of NLRs depends on sensing the host proteins modified by pathogen effectors32,33. The immune interactors are the host genes that show strong interactions with effector targets and known immune proteins. Thus, the proteins in PPIN-1 and -2 generally represent highly connected nodes in the entire plant protein network. The functions of effector targets and immune interactors are mostly unknown at present, but Gene Ontology (GO) annotation indicates that they participate in many molecular processes, such as regulation of transcription, metabolism, nuclear targeting and phytohormone signaling, and these proteins may form a range of modules during their function in plant immune response32,33.
The second approach is based on gene coexpression network (GCN) analysis. Robust GCNs can be developed with genome scale transcriptome data, which are then used to identify the coexpressed gene sets (i.e., modules) related to a specific resistance phenotype31. Subsequently, cofunctional gene clusters may be defined through functional enrichment analysis of the modules with GO. A cofunctional cluster is usually composed of a hub gene and multiple neighbors, some of which may represent potentially new regulators of pathogen resistance. Using GCN analysis and combined with experimental validation, three new genes regulating the MTI response controlled by rice PRR protein XA21 have been successfully identified34.
In contrast to above advances, little progress has been made on understanding the genetic networks involved in pathogen resistance in non-model crop species on a genome-wide scale, especially for common wheat and its related species. Therefore, the main objective of this study was to investigate the genes and major modules involved in resistance against Bgt through constructing and analyzing gene coexpression networks (GCNs) using genome scale transcriptomic data. To facilitate this study, we used the diploid wheat species Triticum urartu (AA, 2n = 2x = 14), rather than hexaploid common wheat or tetraploid durum wheat, as the host plant. T. urartu, a wild grass distributed in the Fertile Crescent region, is an ancestral species of polyploid wheat, which donated the A genome to durum wheat and common wheat35. T. urartu accessions differ in their response to Bgt infection, and a Bgt resistance locus has been identified in this species36. In 2013, the draft genome sequence of T. urartu (with an estimated genome coverage of 94.33%) was reported, with 34,879 protein-coding genes annotated37. By analyzing the draft genome sequence, the genome size of T. urartu (approximately 4.94 Gb) was shown to be substantially smaller than that of common wheat (~17 Gb), but the number of NLR genes in T. urartu is considerably higher than that of rice, B. distachyon, maize (Zea mays) and sorghum (Sorghum bicolor)37. Concomitantly, a draft genome sequence was published for Bgt38. These breakthroughs provide an opportunity for the systematic study of the molecular interactions between T. urartu and Bgt. Therefore, in the present work, a set of diverse T. urartu accessions were screened by Bgt inoculation, and high-quality transcriptomic data were obtained from 20 representative lines showing immune (IM), hypersensitive reaction (HR) or susceptible responses to Bgt inoculation. Four GCNs were subsequently developed, whose analysis allowed identification of the genes regulated by IM or HR. The network properties of these powdery mildew resistance regulated (PMRR) genes were examined. A wide occurrence of negative gene regulation and probable involvement of three new candidate NLR genes in powdery mildew resistance were shown. Lastly, GO analysis was employed to infer the main biological processes enriched for the coexpressed neighbors of the three NLR genes in IM and HR resistance, respectively.
Results
Assessment of the reaction phenotypes to powdery mildew and transcriptomic data
A total of 147 T. urartu accessions were inoculated with Bgt. Fourteen lines showed an immune (IM) type of resistance response. In these lines, Bgt spores germinated and produced primary germ tube (PGT) and an appressorium germ tube (AGT) at 4 h post inoculation (hpi), and the adjacent host cell exhibited little hydrogen peroxide (H2O2) accumulation and no cell death at 24 hpi (Fig. 1A). Fifty accessions displayed a hypersensitive reaction (HR) type of resistance response to Bgt. In these 50 accessions, Bgt spores germinated and produced PGT and AGT similar to those observed in the IM accessions at 4 hpi, but the infected cell accumulated H2O2 and underwent cell death at 24 hpi (Fig. 1B). In both IM and HR accessions, no Bgt hyphal growth in host intercellular space was observed at 48 hpi. In contrast, 83 T. urartu accessions exhibited a susceptible reaction following Bgt inoculation, with fungal haustoria and strong hyphal growth in the host intercellular space observed at 24 and 48 hpi, respectively (Fig. 1C,D). These observations indicated that, in T. urartu, the main molecular events determining IM or HR responses to Bgt occurred quite early, and many of them were accomplished by 24 hpi.
Figure 1. Reaction phenotypes of T. urartu accessions following Bgt inoculation.

(A) Immune (IM) reaction to powdery mildew. Bgt spore germinated, but neither H2O2 accumulation nor cell death were detected in the leaf area in contact with the spore at 24 hpi. (B) Hypersensitive reaction (HR) to powdery mildew. Bgt spore germinated and elicited a strong H2O2 accumulation (brown precipitates) and cell death at 24 hpi. (C) Susceptible reaction to powdery mildew. Bgt haustorium was observed in the infected host cells at 24 hpi. (D) Bgt hyphal growth in the intercellular space of susceptible T. urartu accessions at 48 hpi. H2O2 accumulation and cell death were detected by staining with 3,3′-diaminobenzidine and trypan blue, respectively. AGT, appressorium germ tube; H, haustorium; Hy, hyphae; PGT, primary germ tube; S, stomata; T, trichome. Bar indicates 25 μm.
Subsequently, RNA-sequencing (RNA-seq) was conducted for five IM, 11 HR, and four susceptible T. urartu accessions using RNA samples extracted from the leaves collected at 0 (harvested prior to Bgt inoculation), 4 and 24 hpi, respectively. The latter two sampling time points were selected for this study based on the data presented above. Most of these accessions were from different locations of Fertile Crescent region (Table S1), thus likely represented different genotypes. As an additional control, RNA-seq was also carried out with the RNAs prepared from Bgt hyphae and spore materials. After adaptor sequence trimming and low-quality read filtering, high-quality reads were obtained for each T. urartu sample (100 bp paired-end, 61.6 million on average) (Table S1). For all 60 pair-end T. urartu libraries, approximately 74.1% of the high-quality reads could be aligned to T. urartu reference genome sequence. Those reads were estimated to cover about 45 times of T. urartu transcriptome assuming a transcriptome size of 0.75 Gb (15% of the 4.94 Gb T. urartu genome). Of the aligned reads, approximately 88.3% were mapped to unique loci, with the remaining to multiple loci (Table S1). RNA-sequencing of Bgt materials yielded 57,218,644 high-quality fungal reads, and 81.6% of them could be mapped to the reference genome sequence of Bgt (isolate 96224)38, with the uniquely mapped reads being 78.2% of the total. After comparing T. urartu reads with those of Bgt, the 60 T. urartu transcriptomic datasets were each shown to contain a low percentage (<0.2%) of fungal reads (Table S1).
Construction of GCNs and identification of powdery mildew resistance regulated genes
After removing fungal reads, the 60 clean datasets were used to determine gene expression level by mapping to the 34,879 protein-coding genes of T. urartu reference genome. The fold change logarithm value for the inoculated (4 or 24 hpi)/uninoculated (0 hpi) was calculated for each expressed gene with the expression data from multiple T. urartu accessions, and used to construct scale-free GCNs by weighted gene correlation network analysis (WGCNA) as previously described39,40. WGCNA is a well-established method for constructing GCNs from mRNA expression data, and it considers not only the coexpression pattern between two genes, but also the overlap of neighboring genes. A weighted network retains more information and is more robust and accurate than an unweighted one in network analysis40,41. In total, four GCNs were developed, named as I4 and I24 (for IM) and H4 and H24 (for HR). The total nodes (genes) were 17,362 in I4 and H4, and 15,997 in I24 and H24, and the total edges varied substantially among the four GCNs, with the highest occurring in I4 and the lowest in H4 (Table 1). The two sets of GCNs provided the basis to investigate the genes regulating IM or HR resistance.
Table 1. Construction of GCNs and analysis of PMRR and hub-PMRR genes.
| GCNs | Total nodes | Total edges | PMRR genes |
Hub-PMRR genes |
||
|---|---|---|---|---|---|---|
| Nodes | Edges | Nodes | Edges | |||
| I4 | 17,362 | 84,781,451 | 3,864 | 43,149,830 | 174 | 2,363,001 |
| I24 | 15,997 | 55,685,354 | 424 | 4,347,422 | 21 | 232,928 |
| H4 | 17,362 | 16,910,530 | 145 | 202,415 | 0 | 0 |
| H24 | 15,997 | 49,927,774 | 4,089 | 30,469,907 | 151 | 1,668,098 |
As a first step, the number of PMRR genes was identified in each GCN. Here PMRR genes were defined as the ones whose mRNA expression levels were significantly correlated with the resistance either positively or negatively [false discovery rate (FDR) < 0.1]. The PMRR genes thus identified for I4, I24, H4 and H24 were 3,864 (P < 0.022), 424 (P < 0.003), 145 (P < 0.001) and 4,089 (P < 0.026), respectively (Fig. 2). The number of edges for the four sets of PMRR genes differed greatly, with the highest detected for the PMRR genes in I4 and the lowest for those in H4 (Table 1). The PMRR genes shared by I4 and I24 were 314, with the total number being 3,974 (Figure S1A). For H4 and H24, the shared PMRR genes were 125, and the total number of such genes was 4,109 (Figure S1A). The PMRR genes shared between IM and HR GCNs were 2,102, and the combined PMRR genes in the four GCNs were 5,982 (Figure S1A). The PMRR genes identified above showed great robustness through the assessment of subset samples (Figure S1B).
Figure 2. Identification of PMRR genes in the I4, I24, H4 and H24 GCNs.

The P-value distributions of the correlations between the expression levels of T. urartu genes and Bgt resistance responses in each GCN are shown. The PMRR genes were selected using a fixed FDR level (<0.1). The number of PMRR genes thus identified and the corresponding unadjusted P-value are displayed for each GCN.
Identification and analysis of network hubs and hub-PMRR genes
One key property for a node in a biological network is connectivity, which reflects how frequently a node interacts with other nodes. Based on node connectivity, genes can be classified into hub (with comparatively high level of connectivity) and non-hub types. Hub genes are very important nodes, because they often encode indispensable proteins39,40,41. Generally, hubs represent a small proportion of the genes in a GCN, but with relatively high information exchange with other nodes.
The connectivity distributions of all nodes were examined in each GCN (Figure S2), and 1% of nodes with the highest connectivity were defined as hub genes42,43,44,45. At this level, the numbers of hub genes in I4, I24, H4 and H24 were 174, 160, 174 and 160, respectively (Figure S2). From Fig. 3, it is clear that PMRR genes were significantly enriched in the hubs for I4 (P < 2.2 × 10−16), I24 (P < 3.1 × 10−9) and H24 (P < 2.2 × 10−16), but not H4 (P = 0.409). There was also significant enrichment of PMRR genes in the hub nodes for I4 (P < 2.2 × 10−16), I24 (P < 2.2 × 10−16) and H24 (P < 2.2 × 10−16) when defining the 5% of nodes with the highest connectivity as hubs (Figure S3).
Figure 3. Enrichment test of PMRR genes in the hubs in I4, I24, H4 and H24 GCNs.
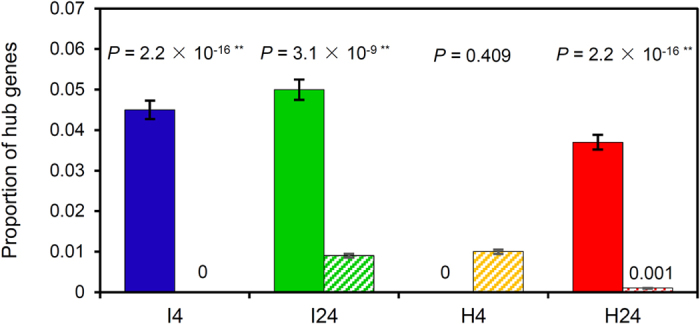
The test was conducted using the numbers of hub genes, PMRR genes and non-PMRR genes in each GCN. Solid bars represent the proportions of hub genes among PMRR genes; striped bars represent the proportions of hub genes among non-PMRR genes. Error bars indicate ±1 s.e.m. The P-values shown were calculated based on Fisher’s exact test.
The genes that were both hub and PMRR (designated hub-PMRR genes, hereafter) in I4, I24, H4 and H24 were 174, 21, 0 and 151, respectively (Figure S2, Table 1). The edges detected for the three sets of hub-PMRR genes differed substantially, with the highest number of edges occurring for the hub-PMRR genes of I4 and the lowest for the hub-PMRR genes of I24 (Table 1). I4 and I24 shared only one hub-PMRR gene, so the total number of hub-PMRR genes in the two GCNs were 194 (Figure S4). The hub-PMRR genes shared between IM (I4 and I24) and HR (H4) GCNs were 12, and the total number of hub-PMRR genes in the three GCNs were 333 (Figure S4).
The gene significance (GS) value (ranging from −1 to 1, see Methods) was investigated for each of the 333 hub-PMRR genes. Here, GS value reflected the correlation of a hub-PMRR gene expression profile with IM or HR resistance. The higher the GS value, the more significant the gene may be in Bgt resistance. In general, the GS values of the 333 hub-PMRR genes were higher than ±0.6 (FDR < 0.1) (Table S2). Notably, of the 333 genes, 238 (71.5%) displayed negative GS values, indicating that they were negatively correlated with Bgt resistance. Many of the 333 hub-PMRR genes shared identical annotations with the Arabidopsis genes interacting with G. orontii (Gor) effectors in PPIN-233 (Table S3). For example, the genes encoding RING/U-box superfamily proteins were among both Gor effector-interacting Arabidopsis genes and our hub-PMRR genes. The hub-PMRR genes specifying RING/U-box superfamily protein generally exhibited significant GS values whether they were found in I4 or H24 GCNs (Tables S2 and S3).
Investigation of major modules correlated with Bgt resistance
Another important property of a GCN is modularity, i.e., genes that are highly interconnected within the network are usually involved in the same biological module or pathway46,47. Therefore, the number of modules was examined for each GCN constructed here, and 49, 38, 28 and 53 modules were detected in I4, I24, H4 and H24, respectively (FDR < 0.1) (Figure S5). The number of modules containing PMRR genes in the four GCNs were 50, specifically, 12 in I4, five in I24, six in H4, and 27 in H24 (Table S4). The PMRR genes in the modules of I4, I24, H4 and H24 amounted to 3,827, 424, 139 and 3,956, respectively, accounting for 99%, 100%, 96% and 97%, respectively, of the total PMRR genes in the four GCNs (Table S4). The enrichment test showed that PMRR genes tended to belong to modules, especially for I4 (P < 1.43 × 10−8) and H24 (P = 6.35 × 10−5) GCNs (Fig. 4). Simulation analysis also revealed enrichment of PMRR genes in the modules for I4 (P = 0) and H24 (P = 0) (Figure S6).
Figure 4. Enrichment test of PMRR genes in the modules in I4, I24, H4 and H24 GCNs.
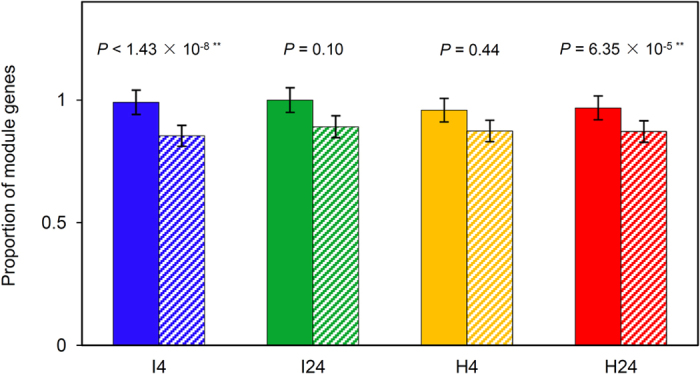
The test was executed using the numbers of module genes, PMRR genes and non-PMRR genes in each GCN. Solid bars represent the proportion of module genes among PMRR genes; striped bars represent the proportions of module genes among non-PMRR genes. Error bars indicate ± 1 s.e.m. P-values were calculated based on Fisher’s exact test.
The correlations between the 50 PMRR gene-containing modules and resistance responses (IM and HR) were assessed (see Methods). Based on a combined consideration of correlation coefficient and corresponding P-value (<0.05), the modules significantly correlated with Bgt resistance were three, five, four and six for I4, I24, H4 and H24, respectively, and among the 18 significantly correlated modules, both positive and negative correlations with Bgt resistance were found (Table S4). To focus on the major modules, the distribution of hub-PMRR genes was investigated in the 18 modules. The hub-PMRR genes were distributed in a highly biased manner among the modules in I4, I24 and H24 GCNs, with no hub-PMRR genes observed in any of the four modules of H4 (Table S4). The first three modules (MI41–MI43) of I4 were considered to be the major ones because together they contained all of the 174 hub-PMRR genes identified in this GCN. Similarly, MI241 (harboring 19 of the total 21 hub-PMRR genes of I24) was regarded as the major module in I24, and MH241 and MH242 (possessing 150 of the total 151 hub-PMRR genes of H24) as the major modules in H24 (Table S4).
Finding of disease resistance related genes in six major modules
To gain a further understanding of the six major modules, homologs of known resistance related genes in them were investigated. In total, 139 homologs were identified, which included 31 coding for putative NLR proteins, five for pathogenesis-related (PR) proteins, 10 for mitogen-activated protein (MAP) kinases, eight for WRKY transcription factors, seven for autophagy related proteins, 50 for GTP signaling related proteins, five for glutathione S-transferases, five for pectin metabolism related proteins, 14 for peroxidases, three for chitinases, and one for isochorismate synthase (Table S5). The 139 homologs were all PMRR genes, and exhibited significant GS values. Remarkably, a large proportion of these homologs (about 75.5%) exhibited negative GS values, which was especially apparent for the genes encoding NLR, MAP kinases, WRKY transcription factor, and autophagy proteins (Table S5).
To check the reliability of the negative gene regulations noticed above, the expression profiles of seven randomly chosen homologs before and after Bgt inoculation of the T. urartu accession PI428322 (showing HR resistance to Bgt, Table S1) were examined by quantitative RT-PCR (qRT-PCR). The seven homologs included four coding for NLR proteins, one for a MAP kinase, one for an autophagy related protein, and one for a protein involved GTP signaling (Table S5). From Figure S7, it is apparent that the expression level of the seven homologs was all decreased at 4 and 24 h after Bgt inoculation. These data suggested that the negative gene regulations computed for the homologs were reliable. Lastly, for each type of homologs, there generally existed both shared and unique gene members among the major modules. For example, among the 31 NLR genes, ten were found in both IM and HR modules, whereas another 14 and seven were specifically present in the relevant IM or HR modules (Table S5).
Analysis of three NLR genes positively correlated with Bgt resistance
Among the 31 NLR homologs, only three, TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195, exhibited positive GS values (Table S5), and were thus positively correlated with Bgt resistance. TRIUR3-13045 was present in both the I4 module MI41 and H24 module MH241, whereas TRIUR3-01037 and TRIUR3-06195 were found in only MH241 (Table S5). Considering the critical role of NLR proteins in plant disease resistance, and that only two NLR genes (Pm3 and Pm8) controlling Bgt resistance have so far been molecularly characterized in wheat, the involvement of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in Bgt resistance was examined in more detail. In T. urartu genome assembly, the scaffolds carrying the three genes were Scaffold68689 (41.479 kb), Scaffold25403 (332.744 kb) and Scaffold75600 (115.908 kb), respectively, and the deduced products of the three genes were all CC-NB-LRR proteins (Table S6). The common wheat orthologs of the three genes were Traes_6AS_5AEA068A7, Traes_7AL_0E46197BE and Traes_3AS_51B04A5B3, respectively, and the three orthologs resided on chromosomal arms 6AS, 7AL and 3AS, respectively (Table S6). At 4 hpi, the three genes were generally and more highly expressed in the IM and HR accessions than in the susceptible ones (Figure S8). At 24 hpi, TRIUR3-06195 remained more highly expressed in all resistant lines, but the expression levels of TRIUR3-13045 and TRIUR3-01037 decreased in some of the IM and HR accessions (Figure S8).
To investigate the participation of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in Bgt resistance, two association analysis experiments were conducted using single nucleotide polymorphisms (SNPs). The first experiment was accomplished using 97 T. urartu accessions showing IM (14) or susceptible (83) responses to powdery mildew for testing the involvement of TRIUR3-13045 in the IM resistance. A total of 15 SNPs were observed in the Scaffold68689 carrying TRIUR3-13045, of which ten of them were located in the first and second exons of TRIUR3-1304 (Fig. 5). The ten genic SNPs were all significantly associated with IM resistance to Bgt, with SNP-28252 showing the lowest P-value (5.33 × 10−11) (Fig. 5). Moreover, this SNP site exhibited substantial linkage disequilibrium (LD) with the other associated SNPs based on pairwise r2 levels (Fig. 5). The second experiment was executed with 133 T. urartu accessions showing HR (50) or susceptible (83) responses to Bgt for testing the involvement of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in the HR resistance. The genic SNPs significantly associated with HR resistance were identified for all three genes, specifically, seven for TRIUR3-13045, 10 for TRIUR3-01037 and two for TRIUR3-06195 (Fig. 6). The significantly associated SNPs generally exhibited medium to high levels of LD (Fig. 6). Of all three NLR genes, TRIUR3-01037 exhibited the highest association signal (Fig. 6).
Figure 5. Association test of the involvement of TRIUR3_13045 in IM resistance to Bgt.

The test was carried out with 97 T. urartu accessions exhibiting IM (14) or susceptible (83) responses to Bgt challenge, and 15 SNPs detected in TRIUR3_13045 genomic region. (A) Association plot for TRIUR3_13045. The 15 SNPs resided in a 40 kb genomic region. The significance threshold (P) was set as 0.01/total SNPs (-Log10P = 3.18, dashed line). The 10 SNPs in TRIUR3_13045 open reading frame (ORF) (boxed) were all significantly associated with IM resistance to Bgt. (B) Linkage disequilibrium (LD) plot of the 15 SNPs used in association test. The exon (rectangle) and intron (line between rectangles) structure of TRIUR3_13045 is shown on the top, with the 15 SNPs displayed below. The 10 SNPs, colored in blue and located in TRIUR3_13045 ORF, were associated with IM resistance. The LD between the SNPs is measured using r2, which varies from 0 (white) to 1 (black).
Figure 6. Association test of the involvement of TRIUR3_13045, TRIUR3-01037 and TRIUR3-06195 in HR resistance to Bgt.

The test was carried out with 133 T. urartu accessions exhibiting HR or susceptible responses to Bgt, and various numbers of SNPs detected in TRIUR3_13045, TRIUR3-01037 or TRIUR3-06195 genomic regions. The significance threshold (P) was set as 0.01/total SNPs (−Log10P = 4.13, dashed line). To facilitate presentation, the exon (rectangle) and intron (line between rectangles) patterns of the three genes are provided. (A,C,E) Association plots for TRIUR3_13045, TRIUR3-01037 and TRIUR3-06195, respectively. The SNPs located in the ORF of the three genes are boxed, with the ones above the threshold (dashed line) being significantly associated with HR resistance. (B,D,F) LD plots for the SNPs in TRIUR3_13045, TRIUR3-01037 and TRIUR3-06195 genomic regions, respectively. The SNPs, marked in blue and located in the respective genomic ORFs, were significantly associated with HR resistance. The LD between the SNPs is measured using r2 ranging from 0 to 1 (as indicated by the inset).
To further examine the involvement of TRIUR3-01037 in powdery mildew resistance, a F2 population developed from the crossing of two T. urartu accessions, PI428198 and PI428322, which showed susceptibility and HR resistance to Bgt, respectively (Table S1), were genotyped at two selected SNP sites. These two sites differed between the resistance associated allele (RAA) and susceptibility associated allele (SAA) of TRIUR3-01037; site 1 involved a C (in RAA) to T (in SAA) change whereas site 2 was a G (in RAA) to A (in SAA) change (Fig. 7A). The SNP in site 1 caused the substitution of a serine residue (in RAA) by phenoalanine (in SAA), while that in site 2 led to the replacement of an alanine serine residue (in RAA) by threonine (in SAA) (Fig. 7A). A total of 62 F2 progenies were inoculated with Bgt, and the ratio of resistant and susceptible individuals were identified to be 47: 15 (Table S7). These data indicated that, in this F2 population, resistance was dominant whereas susceptibility was recessive. The genotypic data of the 62 individuals were obtained by examining SNPs at sites 1 and 2 (Fig. 7A), followed by comparison with Bgt reaction phenotype data. The results showed a positive match between Bgt reaction phenotype and TRIUR3-01037 SNP genotype for the 62 individuals (Table S7). This analysis indicated that the RAA of TRIUR3-01037 cosegregated with powdery mildew resistance. Subsequently, the effects of ectopic expression of RAA and SAA on Bgt growth in susceptible leaf cells were assessed using a transient functional expression assay (detailed in Methods). Compared with the control, expression of RAA, but not SAA, significantly decreased the growth of Bgt haustorium (as judged from haustorium index, Fig. 7B). The results of this functional expression assay, plus the cosegregation analysis data described above, supported a functional involvement of the RAA of TRIUR3-01037 in powdery mildew resistance.
Figure 7. Comparative analysis of resistance associated allele (RAA) and susceptibility associated allele (SAA) of the NLR gene TRIUR3_01037.

(A) A diagram illustrating the two SNP sites (Sites 1 and 2) in the coding region of RAA and SAA. These two SNPs were used to genotype F2 individuals in the cosegregation analysis. The first SNP caused a serine to phenoalanine substitution whereas the second one rendered an alanine to threonine replacement. (B) The effects of ectopically expressing RAA or SAA on Bgt haustorium index in single-cell functional expression assay. The cells were transiently transformed by pUbi-GUS alone (as control), pUbi-RAA + pUbi-GUS (for expressing RAA) or pUbi-SAA + pUbi-GUS (for expressing SAA), followed by Bgt inoculation. About 200 infected cells were examined for haustorium growth in each treatment. Haustorium index (mean ± SD) was calculated as the percentage of examined cells with haustorium presence. The means marked by different letters are statistically different (ANOVA, P < 0.05).
Finally, the coexpressed neighbors of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in the relevant modules were identified, and the main biological processes enriched by the neighbors were investigated through GO analysis (see Methods). In the IM module MI41, the coexpressed partners of TRIUR3-13045 were 549, 76 of which were hub-PMRR genes (Figure S9A). The biological process (BP), molecular function (MF) and cell component (CC) terms enriched by the 549 coexpressed genes were mainly related to protein translation, although the CC terms ‘plastid’ and ‘thylakoid’ were also significantly enriched (Table S8). In the HR module MH241, the total number of coexpressed partners of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 were 397, 56 of which were hub-PMRR genes (Figure S9B). The BP, MF and CC terms enriched by the 397 coexpressed genes mainly concerned photosynthesis, though the CC term ‘Golgi apparatus’ was also significantly enriched (Table S8).
Discussion
The signaling and defense processes of plant disease resistance are highly complex and involve multiple genes. Several recent studies have elegantly demonstrated that network analysis is very effective for uncovering the genes and their interactions functioning in plant disease resistance32,33,34. Here GCN analysis was conducted in order to reveal the genes and major modules involved in the two types of wheat resistance responses (IM and HR) to powdery mildew. The use of transcriptomic data from multiple T. urartu accessions showing IM, HR or susceptible phenotypes permitted the construction of robust GCNs, thus facilitated the identification of PMRR and hub-PMRR gene sets and the major modules that were correlated with Bgt resistance. This provided a reliable basis for further investigations into the key mechanisms and genes that are likely important in the IM and HR responses to Bgt.
One of the basic questions in studying the systems biology of plant resistance to pathogen attack concerns the number of host genes regulated by disease resistance. In Arabidopsis, an earlier estimation suggests that about 3,000 genes (approximately 14% of all annotated Arabidopsis genes) may be directly involved in pathogen defense29. In line with this estimation, 2,043 Arabidopsis proteins were predicted to function in the interaction with the bacterial pathogen Pseudomonas syringae using domain and interolog-based computation approaches48. In citrus, 3,507 genes have been suggested to act in the defense response to the bacterial pathogen Candidatus Liberibacter asiaticus through GCN analysis49. In this study, the above question was approached by estimating the number of PMRR genes in four GCNs, two (I4 and I24) for IM and two (H4 and H24) for HR. For both types of responses, the GCNs developed harbored gene expression information at two different time points after Bgt inoculation (4 and 24 hpi), thus facilitating a more objective assessment of the genes regulated by IM or HR. Based on the total number of PMRR genes in I4 and I24 and that in H4 and H24 (Figure S1A), we suggest that in T. urartu the genes regulated by IM and HR resistance to Bgt may exceed 3,900 and 4,100, respectively. Because the total number of protein-coding genes annotated for T. urartu stands at 34,879 currently, it is likely that about 11%∼12% of T. urartu protein-coding genes may take part in its IM or HR resistance to Bgt. However, these values are conservative estimates because this GCN analysis could not detect the genes that did not show significant expression changes in the samples analyzed but still function in Bgt resistance. In addition, miRNAs and long noncoding RNAs were not considered in this GCN analysis, both of which have been shown to modulate plant pathogen resistance50,51. Nevertheless, our estimates represent a genome-wide assessment of the number of protein-coding genes likely taking part in IM or HR responses to Bgt in T. urartu. Considering the high synteny of genome organization among polyploid wheat and its diploid ancestral species and the likely conservation of the genetic mechanism of powdery mildew resistance among these species11,52, it is possible that the number of PMRR genes may exceed 8,000 in tetraploid wheat and 12,000 in hexaploid common wheat.
Previous network studies involving Arabidopsis and several bacterial and fungal pathogens suggest that the host genes involved in pathogen resistance are more likely to be hubs that are enriched in modules32,33,34. Here, the enrichment and simulation investigations revealed that the PMRR genes in only I4 and H24, but not in I24 and H4, were significantly enriched in GCN hubs and modules. Interestingly, this finding corresponds closely to the presence of more numerous PMRR and hub-PMRR genes in I4 and H24. Together, these data suggest that the PMRR genes and their interactions in I4 and H24 are probably more fundamental to the IM and HR responses to Bgt, respectively, when compared to those in I24 and H4. Furthermore, the 333 hub-PMRR genes identified in I4, I24 and H24 generally exhibited high GS values (Table S2), indicating that they may be strongly involved in IM or HR resistance. Supporting this proposition, the deduced products of many hub-PMRR genes resembled the proteins that have been reported to be important in the resistance response of Arabidopsis to the powdery mildew pathogen G. orontii33 (Table S3). According to the findings made in previous plant-pathogen interaction network studies32,33,34, the hub-PMRR genes identified in this study may include key players in the immunity of T. urartu to Bgt, such as the targets of Bgt effectors. Therefore, these hub-PMRR genes may assist in the identification of Bgt effector targets guarded by NLR proteins in future research. In this context, it is interesting to note that multiple hub-PMRR genes were present among the coexpressed neighbors of the three NLR genes likely involved in Bgt resistance in this study (Figure S9, see also below).
The six major modules identified in this study may reflect the main molecular interactions and processes central to IM or HR responses to Bgt, because 1) they generally contained a large number of PMRR and hub-PMRR genes, and 2) they all showed high level (R > ±0.8) and highly significant (P < 0.001) module-trait correlation values (Table S4). Further support for the six major modules comes from the presence of multiple types and members of resistance related gene homologs in them (Table S5). The deduced proteins of these homologs are either known to be active in resistance signaling (e.g., NLR proteins, MAP kinases, and WRKY transcription factors) or have been shown to act in defense processes (e.g., PR proteins, glutathione S-transferases, and peroxidases). Remarkably, 50 genes encoding various components of GTP signaling were present in the six modules, and all of them showed significant GS values (Table S5). This indicates that GTP signaling may play a vital role in IM and HR resistance to Bgt. Previous investigations in barley have identified a small GTPase (HvRacB) and its interacting proteins (HvMAGAP1 and HvELMOD_C) as important modulators of the host response to powdery mildew53,54,55,56. Recent studies in rice have established that the OsRac1 GTPase and its signaling partners, SPIN6 (a RhoGAP protein) and OsRacGEF1 (a Rho guanine nucleotide exchange factor), positively regulate immunity to both fungal and bacterial pathogens57,58. Therefore, the role of GTP signaling in Bgt resistance is well worth investigating. The 50 genes encoding GTP signaling related proteins, together with the major modules to which they belong, should aid such investigations.
In this research, negative correlations with Bgt resistance were repeatedly observed among hub-PMRR genes, major modules and resistance related gene homologs. The majority of the hub-PMRR genes (71.5%) and resistance related gene homologs (75.5%) exhibited significant negative GS values, and four out of the six major modules were negatively correlated with Bgt resistance. These data indicate wide occurrence of negative regulation of a large proportion of the PMRR genes in IM and HR responses to Bgt. Hence, it is highly possible that negative gene regulation is a key mechanism involved in Bgt resistance in T. urartu. Previous molecular genetic and network studies have also revealed the occurrence of negative gene regulation in plant-microbe interactions59,60,61,62,63. It has been suggested that down-regulation of gene expression facilitates effective orchestration of the key cellular processes normally under negative control (such as autophagy and cell death), fine-control of the magnitude and specificity of defense output, and proper adjustment of host physiology during and after pathogen attack59,60,61,62,63. Together, these actions lead to a dynamic balance between defense and growth, with efficient resource allocation and utilization. The importance of negative gene regulation in plant resistance to powdery mildews was well demonstrated by the repressive function of WRKY transcription factors in controlling the immune response mediated by barley MLA immune receptors59,60. However, judging from the large number of PMRR genes showing negative GS values in IM and HR responses, substantial efforts will be needed to fully understand the involvement of negative gene regulation in T. urartu resistance to Bgt.
Based on the GCN modeling and association analysis data gathered in this study, we propose that TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195, all showing positive GS values (Table S5), represent newly identified candidate NLR genes involved in T. urartu resistance to Bgt. Specifically, TRIUR3-13045 may represent a potent determinant of IM resistance, whereas the three of them may all be important for HR resistance. The involvement of all three candidate NLR genes in HR resistance is consistent with the emerging concept on the control of plant disease resistance through molecular interactions between different NLR proteins24,25. The GCN modeling and association analysis in this study used diverse T. urartu accessions showing HR response to Bgt (11 in GCN modeling, and 50 in association analysis). This is conducive for revealing the function of multiple NLR genes in resistance response when compared to a typical map-based cloning approach that normally uses a limited number of genotypes. On the other hand, only TRIUR3-13045 was implicated in IM resistance to Bgt. This may be due to the fact that relatively fewer T. urartu accessions exhibiting IM response were used in the GCN modeling (5) and association analysis (14), and a possibility that the molecular and functional diversities of NLR genes in these IM lines may be limited.
In addition to its positive association with HR response to Bgt (Fig. 6), functional involvement of TRIUR3-01037 in powdery mildew resistance was further supported by cosegregation analysis in a F2 population and the expression assay of its RAA and SAA (Table S7, Fig. 7). The finding that the RAA of TRIUR3-01037, but not its SAA, could significantly decrease Bgt haustorium growth in a susceptible background (Fig. 7B) provides a positive indication of the action of this gene in the initiation and execution of Bgt resistance. However, more experiments, such as development and examination of loss-of-function mutants and transgenic wheat plants stably expressing the different alleles of TRIUR3-01037, are needed to finally validate the function of TRIUR3-01037 in powdery mildew resistance.
Apart from TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195, 28 NLR genes exhibited negative GS values in the major modules (Table S5). The expression of multiple NLR genes has frequently been observed in the plants grown under normal conditions or challenged by artificial pathogen inoculation26,27,28. These NLR genes may be involved in monitoring potential pathogenic microbes in the surrounding environments, or have alternative functions in other physiological processes32,64. When encountering a strong pathogen challenge (such as artificial inoculation of Bgt spores), the expression of specific NLR gene(s) is up-regulated, with concomitant down-regulation of other NLR genes. Under this scenario, down-regulation of the 28 NLR genes may aid in the proper function of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in Bgt resistance. Further research is required to verify this hypothesis.
Based on comparisons of the 14 accessions showing IM and 50 accessions exhibiting HR, this study indicates that IM resistance arrests Bgt growth in the absence of host cell death (Fig. 1). This type of resistance response has also been observed and studied in other pathosystems. For example, pathogen resistance controlled by barley Mla1 and Rdg2a proteins, both of which are NLRs, occur without HR65,66. The potato NLR protein Rx confers strong resistance to potato virus x in the absence of HR67. Similarly, natural alleles of two Arabidopsis NLRs, RPS4 and RPS6, inhibit pathogen growth without involving HR68. More importantly, our GCN modeling suggests that IM and HR involve many differences in the expression of PMRR genes. For IM, PMRR gene expression and their interactions may mainly occur soon after infection (i.e., before 4 hpi). This is supported by the finding of substantially more PMRR genes and hub-PMRR genes in I4 rather than I24 (Table 1). On the other hand, for HR, the expression and genetic interactions of PMRR genes may largely occur at a comparatively later stage (probably later than 4 hpi, but before 24 hpi) because considerably more PMRR genes and hub-PMRR genes were detected in H24 rather than H4 (Table 1). Lastly, as discussed above, the NLR genes involved in IM and HR responses may be different, with one (TRIUR3-13045) implicated in IM and three (TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195) in HR.
The differential involvement of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in IM and HR may provide a valuable basis for further investigation of the different molecular interactions and processes in the two types of resistance responses. It is important to point out that the network characteristics of the coexpressed neighbors of TRIUR3-13045 in IM differed considerably from those of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in HR (Table S8, Figure S9). The main biological process enriched for the coexpressed neighbors of TRIUR3-13045 in IM (protein translation) was clearly different from that for the coexpressed neighbors of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in HR (photosynthesis) (Table S8). According to the guard hypothesis and previous studies32,33, the coexpressed neighbors of a NLR protein may contain the pathogen effector targets and the protein(s) required for the normal function of effector targets. The NLR protein monitors the binding of pathogen effector to its target, thereby controlling resistance signaling and downstream defense events. Thus, in IM, the target of Bgt effector may be certain component of host translation machinery that is guarded by TRIUR3-13045. On the other hand, in HR, the target of Bgt effector may be certain component of host photosynthesis process guarded by one or more of the three NLR genes (TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195).
Previous studies have shown that protein translation elongation factors of higher plants are often targeted by viral effectors, and that recessive mutations of translation elongation factors can confer strong resistance to viral pathogens69. Plant photosynthesis machinery, especially its oxygen evolving complex (OEC), has been shown to be required for developing HR70, and a bacterial pathogen effector, HopN1, targets the PsbQ protein of tomato OEC to suppress host defense71. At present, little has been reported on the interaction of powdery mildew effectors with host translation or photosynthesis components. The finding that photosynthesis was enriched for the coexpressed neighbors of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in HR, but not those of TRIUR3-13045 in IM, is consistent with the observed H2O2 accumulation and presence of cell death in HR, but not IM, response (Fig. 1). Therefore, further studies of the coexpressed neighbors of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 may reveal the roles of translation and photosynthetic processes in wheat immunity to Bgt.
In summary, this study has shed new light on T. urartu genes regulated by two types of resistance responses to Bgt on a genome-wide scale. The GCNs developed and the PMRR gene sets and major modules identified are useful for finding the wide occurrence of negative gene regulation, and the involvement of three new NLR genes, in Bgt resistance. The information generated has increased the understanding on IM and HR responses to Bgt infection, and may continue to aid further research into the molecular differences between the two types of resistance responses. This study has benefited from the lower ploidy level of T. urartu and the availability of a reference genome sequence for this species. These advantages, together with the fact that more and more information is becoming available for Bgt effector proteins38, lead us to suggest that T. urartu represents an efficient model for further and deeper systems biology studies of wheat-Bgt interactions.
Methods
Plant materials, Bgt inoculation and microscopic examination
A total of 147 T. urartu accessions collected from Armenia, Iran, Iraq, Lebanon, Syria and Turkey were subjected to two independent Bgt inoculation tests with the isolate E09 as previously reported36. In each test and for each accession, approximately 25 seeds were planted in compost in a growth chamber set at 25 °C (with a photoperiod of 18 h light/6 h dark and a relative humidity of 80%). At the two-leaf stage, 10–15 well developed seedlings were selected in each accession and inoculated with Bgt. The growth temperature was adjusted to 20 °C following the inoculation. The reactions to Bgt inoculation were examined visually, and classified into three types, IM, HR or susceptible reaction, based on the presence (or absence) of hypersensitive necrotic spots and powdery mildew colonies on the leaf surface. Highly similar results were obtained between the two separate inoculation tests. Subsequently, Bgt spore germination and host cell change were examined in more detail in 20 representative accessions (Table S1) showing IM, HR or susceptible responses at 4, 24, 48 and 96 hpi. Detection of H2O2 accumulation and cell death by staining with 3,3′-diaminobenzidine or trypan blue were accomplished as detailed in previous studies72,73. The stained leaf materials were examined under a compound microscope (Leica DMRE, Wetzlar, Germany), with the images collected using a digital camera (EXi Aqua, QImaging, BC, Canada).
RNA sequencing and preparation of high-quality transcriptomic reads
The 20 T. urartu accessions used in transcriptome sequencing exhibited IM (5), HR (11) or susceptible (4) reactions to Bgt infection (Table S1). Leaf samples were collected from the 20 accessions prior to Bgt inoculation and at 4 and 24 hpi of Bgt. For each accession and at each time point, leaf samples from five uniform seedlings were combined for total RNA extraction using Illumina TruSeq RNA Sample Prep Kit (Illumina, Inc., San Diego, USA). A total of 60 paired-end libraries were constructed with Illumina Paired-End Sample Prep Kit, followed by sequencing on Illumina HiSeq 2000 platform. Adaptor sequence trimming and removal of low-quality reads were performed with the ngsShoRT algorithms74. A paired-end library was also constructed using the total RNA extracted from Bgt hyphae and spore materials collected from a susceptible T. urartu accession (PI428198, Table S1) at 168 hpi. The library was sequenced and high-quality reads were obtained as described above. Genome mapping of high-quality reads was executed using the software TopHat275. The references used for genome mapping were either T. urartu draft genome assembly (http://gigadb.org/dataset/100050, for mapping the reads obtained from 60 T. urartu libraries) or the whole genome assembly of Bgt isolate 96224 (www.ncbi.nlm.nih.gov/Traces/wgs/?val=ANze&display=contigs&search=ANze01000000, for mapping the reads from Bgt hyphae and spore library).
While designing the transcriptome sequencing scheme described above, it was considered that the inclusion of multiple T. urartu accessions showing IM, HR or susceptible phenotypes could facilitate realistic estimations of gene expression levels. Therefore, no biological replicates were collected and sequenced. Previous studies have also used a similar strategy in examining gene expression levels through transcriptome sequencing76,77,78.
Calculation of gene expression level and network construction
The expression level of T. urartu genes, measured as fragments per kilobase of transcript per million fragments sequenced, was determined using the reads with positive genome mapping by DEGseq79. The reference used was the coding sequence of 34,879 T. urartu genes (downloaded from http://gigadb.org/dataset/100050). To construct robust GCNs in this study, mRNA expression data of the 34,879 protein-coding genes in the 60 libraries were filtered in two ways. First, the genes whose expression was not detected in one of the 60 libraries were discarded. Second, the genes with aligned reads lower than 50 were also discarded to reduce background noise. After these filtering steps, 17,362 and 15,997 expressed genes were retained for constructing the GCNs at 4 and 24 hpi, respectively. The WGCNA (weighted gene correlation network analysis) R package was then used to build GCNs, investigate main network properties (hubs and modules), calculate the significance values of genes, and examine the correlations between modules and Bgt resistance phenotypes (IM or HR) as described in previous publications39,40.
Identification of powdery mildew resistance regulated (PMRR) genes
The PMRR genes in the four GCNs (I4, I24, H4 and H24) were identified following the strategy reported previously39,40. The genes, whose FDR (false discovery rate)-corrected P-value was smaller than 0.1, were regarded as PMRR genes. The robustness of the PMRR genes thus identified was assessed using 100 random samples (each involving 75% of the PMRR genes in the relevant GCN). The hub-PMRR genes were located in two steps. First, the hub genes in each GCN were identified based on gene connectivity (i.e., sum of the weights across all the edges of a node) and the top 1% of the genes with the highest connectivity in the network were considered as hubs39,40. Second, for each GCN, the set of hub genes was compared to that of PMRR genes, leading to the identification of hub-PMRR genes. In each GCN, the enrichment of PMRR genes in hub nodes and in modules was examined by Fisher’s exact test45. Simulation tests for hub and module enrichment of PMRR genes were accomplished as described previously45.
Search of homologs of known resistance related genes
The PMRR genes, which were detected in the 6 major modules (Table S4), were selected out from the 34,897 protein-coding gene set of T. urartu37, and their annotations were carefully checked. They were then subjected to a text search using 11 plant disease resistance associated keywords, including “NLR protein”, “pathogenesis-related (PR) protein”, “mitogen-activated protein (MAP) kinase”, “WRKY transcription factor”, “autophagy related protein”, “GTP signaling”, “glutathione S-transferase (GST)”, “isochorismate synthase”, “chitinase”, “pectin metabolism”, and “peroxidase”. Positive hits were identified based on the presence of key word in the annotation, and a GS (gene significance) value was computed for each hit as described above.
Investigations of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195
The scaffolds (i.e., Scaffold68689, Scaffold25403 and Scaffold75600) carrying the three NLR genes (TRIUR3-13045, TRIUR3-01037 or TRIUR3-06195) were found in T. urartu draft genome assembly (http://gigadb.org/dataset/100050). Because the scaffolds in T. urartu draft genome sequence were not completely ordered along chromosomes, the orthologs of the three NLR genes were identified in the draft genome assembly of common wheat (https://urgi.versailles.inra.fr/download/iwgsc/) by a blastN search, with positive hits judged by identity (>90%) and coverage (>90%) values. Hierarchical clustering of the expression data of the three NLR genes at 4 and 24 hpi of Bgt was generated using the software Cluster 3.080.
To evaluate the involvement of TRIUR3-13045, TRIUR3-01037 and TRIUR3-06195 in Bgt resistance, two sets of SNP marker based case/control type of association test were performed using PLINK81. To facilitate the finding of SNPs, RNA-seq was carried out for 127 more T. urartu accessions with total RNA samples extracted from Bgt inoculated leaf materials as described above. This led to the availability of transcriptomic data for all 147 T. urartu accessions that had been evaluated for Bgt response in this research. In the first test, the participation of TRIUR3-13045 in IM response was examined. The 14 T. urartu accessions showing IM response to Bgt were cases whereas the 83 susceptible accessions were controls. In the second test, the involvement of all three NLR genes in HR response was examined. The 50 T. urartu accessions displaying HR after Bgt infection were cases whereas the 83 susceptible accessions were controls. SNPs for the three scaffolds carrying TRIUR3-13045, TRIUR3-01037 or TRIUR3-06195 were called by jointly using TopHat2 (for mapping transcriptomic reads) and SAMtools (for nucleotide variant detection)82. The SNPs found were 15 for Scaffold68689, 92 for Scaffold25403, and 28 for Scaffold75600. The significance threshold of the association tests was set as 0.01/total SNPs. The total SNPs used for calculating significance threshold in the first and second association tests were 15 and 135, respectively. The LD plots of SNPs were drawn using the LDheatmap package83.
Quantitative RT-PCR assay
The coding sequence of the seven genes, which were randomly chosen to verify the negative regulations computed for resistance related gene homologs by qRT-PCR (Table S5), was downloaded from http://gigadb.org/dataset/100050. Specific nucleotide primers (Table S9) were designed for each of the seven genes using the software Primer3 (Primer3.ut.ee). Fourty seedlings of PI428322 (with HR resistance to Bgt, Table S1) were inoculated with Bgt isolate E09 as described above. Three samples were collected at 0, 4 and 24 hpi, respectively, with each sampling involving 6 different seedlings. The extraction of total RNAs, preparation of cDNAs, and execution of qRT-PCR were conducted following a previous study84. A common wheat actin gene (GenBank accession AB181991) was used as the internal reference for qRT-PCR. The assay was conducted twice using independent biological replicates.
Cosegregation analysis of TRIUR3_01037 in F2 population
The Bgt reaction phenotype of the 62 F2 individuals (Table S7) was identified by inoculation with Bgt isolate E09 as described above. For genotyping the 62 individuals using the SNPs at sites 1 and 2 (Fig. 7a), amplicons containing the SNP at site 1 or 2 were obtained by genomic PCR, followed by DNA sequencing. The amplicon sequences were then analyzed to determine the genotypes of different F2 plants. The primers used for obtaining the amplicons are listed in Table S9.
Single-cell functional expression assay
The single-cell transient functional expression assay was conducted essentially as described59. The cDNA coding region of TRIUR3_01037 was amplified from PI428198 and PI428322, respectively, with the primers listed in Table S9. The resultant cDNA sequences, designated as RAA and SAA, respectively, were confirmed to be error-free by DNA sequencing. They were then cloned into a plasmid vector driven by the maize polyubiquitin promoter, resulting in the expression constructs pUbi-RAA and pUbi-SAA, respectively. A β-glucuronidase (GUS) reporter gene construct (pUbi-GUS) was also used in this assay, whose expression facilitated the visualization of transformed cells59.
The RAA and SAA constructs were each mixed with pUbi-GUS at a 1:1 molar ratio for coating the DNA microcarrier. As control for the assay, only pUbi-GUS was used for coating the DNA microcarrier. The plasmid DNA coated on the microcarrier was delivered into the leaf epidermal cells of the T. urartu accession PI428252, which was susceptible to Bgt isolate E09, through particle bombardment. The leaf segments were inoculated with E09 conidial spores at 4 h post bombardment. At 48 hpi of Bgt, the leaf segments were stained in a β-glucuronidase staining solution for 8 h at 37 °C, followed by destaining for 72 h at 25 °C. Before mounting for microscopic examination, the leaf segments were stained with Coomassie blue to visualize Bgt structures. For assessing the effects of each construct (pUbi-RAA, pUbi-SAA or pUbi-GUS), approximately 200 cells (in 6 to 8 leaf segments) with GUS signals and Bgt mycelia were examined for haustorium growth, with haustorium index calculated as the percentage of examined cells with haustorium presence59.
Gene ontology analysis of the coexpressed neighbors of NLR genes
The coexpressed interactors of TRIUR3-13045 in IM and all three NLR genes in HR were extracted from relevant GCNs (I4 or H24) and visualized using Cytoscape85. To aid GO enrichment analysis of the extracted neighbors, GO term annotation of T. urartu genes was improved using Blast2GO86 and InterProScan87. This led to the assignment of GO terms to 25,437 genes, accounting for 73% of the 34,879 genes annotated by T. urartu draft genome sequence. The enrichment analysis was subsequently conducted using agriGO (http://bioinfo.cau.edu.cn/agriGO/)88. Only the terms with a significance level lower than 0.05 and a minimum of 5 annotated genes in the input list were selected for further analysis.
Additional Information
Accession codes: The transcriptomic data generated for 20 T. urartu accessions at 0, 4 and 24 hpi of Bgt have been submitted to the BioProject database of National Center for Biotechnology Information (accession number PRJNA289598).
How to cite this article: Zhang, J. et al. Coexpression network analysis of the genes regulated by two types of resistance responses to powdery mildew in wheat. Sci. Rep. 6, 23805; doi: 10.1038/srep23805 (2016).
Supplementary Material
Acknowledgments
This study was supported by the Ministry of Science and Technology of China (grants 2012AA10A308 and 2013CB127703) and the State Key Laboratory of Plant Cell and Chromosome Engineering (grant PCCE-TD-2012-02). We thank Drs Dingzhong Tang, Qianhua Shen and Congcong Jiang for constructive comments on this research and critical reading of the manuscript. Dr. Xinyun Han is thanked for guidance in single-cell functional expression assay.
Footnotes
Author Contributions J.Z., L.D. and D.W. perceived and designed the research; J.Z., L.D. and H.Z. produced and analyzed the data; Y.L., H.L., X.L. and H.Q. managed the samples and the data; J.Z., L.D. and D.W. wrote the paper; D.W. and L.D. revised the manuscript. All authors read and approved the final manuscript.
References
- Glawe D. A. The powdery mildews: a review of the world’s most familiar (yet poorly known) plant pathogens. Annu Rev Phytopathol 46, 27–51 (2008). [DOI] [PubMed] [Google Scholar]
- Braun U. & Cook R. T. Taxonomic manual of the Erysiphales (powdery mildews). (CBS-KNAW Fungal Biodiversity Center, 2012). [Google Scholar]
- Marshall D. in Wheat science and trade (ed. Carver B. F.) 155–169 (Wiley-Blackwell, 2009). [Google Scholar]
- Bennett F. G. A. Resistance to powdery mildew in wheat: A review of its use in agriculture and breeding programmes. Plant Pathol 33, 279–300 (1984). [Google Scholar]
- Huang X. Q. & Röder M. S. Molecular mapping of powdery mildew resistance genes in wheat: a review. Euphytica 137, 203–223 (2004). [Google Scholar]
- Hao Y. et al. Molecular characterization of a new powdery mildew resistance gene Pm54 in soft red winter wheat. Theor Appl Genet 128, 465–476 (2015). [DOI] [PubMed] [Google Scholar]
- Yahiaoui N., Srichumpa P., Dudler R. & Keller B. Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J 37, 528–538 (2004). [DOI] [PubMed] [Google Scholar]
- Hurni S. et al. Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J 76, 957–969 (2013). [DOI] [PubMed] [Google Scholar]
- Cao A. et al. Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci USA 108, 7727–7732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krattinger S. G. et al. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323, 1360–1363 (2009). [DOI] [PubMed] [Google Scholar]
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 6194 (2014). [DOI] [PubMed] [Google Scholar]
- Micali C., Göllner K., Humphry M., Consonni C. & Panstruga R. In The Arabidopsis Book 6 e0115 (The American Society of Plant Biologists, 2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm S. T., Coaker G., Day B. & Staskawicz B. J. Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803–814 (2006). [DOI] [PubMed] [Google Scholar]
- Jones J. D. & Dangl J. L. The plant immune system. Nature 444, 323–29 (2006). [DOI] [PubMed] [Google Scholar]
- Zipfel C. Early molecular events in PAMP-triggered immunity. Curr Opin Plant Biol 12, 414–420 (2009). [DOI] [PubMed] [Google Scholar]
- Dodds P. N. & Rathjen J. P. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11, 539–548 (2010). [DOI] [PubMed] [Google Scholar]
- Elmore J. M., Lin Z. J. & Coaker G. Plant NB-LRR signaling: Upstreams and downstreams. Curr Opin Plant Biol 14, 365–371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonardi V. & Dangl J. L. How complex are intracellular immune receptor signaling complexes? Front Plant Sci 3, 237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. A. & Yeom S. I. Plant NB-LRR proteins: Tightly regulated sensors in a complex manner. Brief Funct Genomics 14, 233–242 (2015). [DOI] [PubMed] [Google Scholar]
- Jacob F., Vernaldi S. & Maekawa T. Evolution and conservation of plant NLR functions. Front Immunol 4, 297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol Genet Genomics 283, 427–438 (2010). [DOI] [PubMed] [Google Scholar]
- Van der Biezen E. A. & Jones J. D. Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem Sci 23, 454–456 (1998). [DOI] [PubMed] [Google Scholar]
- Dodds P. N. et al. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc Natl Acad Sci USA 103, 8888–8893 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux C. et al. A receptor pair with an integrated decoy converts pathogen disabling of transcription factors to immunity. Cell 161, 1074–1088 (2015). [DOI] [PubMed] [Google Scholar]
- Sarris P. F. et al. A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell 161, 1089–1100 (2015). [DOI] [PubMed] [Google Scholar]
- Arya P., Kumar G., Acharya V. & Singh A. K. Genome-wide identification and expression analysis of NBS-encoding genes in Malus x domestica and expansion of NBS genes family in Rosaceae. PLoS One 9, e107987 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. et al. Genome-wide comparative analysis of NBS-encoding genes between Brassica species and Arabidopsis thaliana. BMC Genomics 15, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodamilans B. et al. Transcriptomic analysis of Prunus domestica undergoing hypersensitive response to plum pox virus infection. PLoS One 9, e100477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan M. et al. Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391, 485–488 (1998). [DOI] [PubMed] [Google Scholar]
- Douchkov D. et al. Discovery of genes affecting resistance of barley to adapted and non-adapted powdery mildew fungi. Genome Biol 15, 518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windram O., Penfold C. A. & Denby K. J. Network modeling to understand plant immunity. Annu Rev Phytopathol 52, 93–111 (2014). [DOI] [PubMed] [Google Scholar]
- Mukhtar M. S. et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weßling R. et al. Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 16, 364–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. et al. Genetic dissection of the biotic stress response using a genome-scale gene network for rice. Proc Natl Acad Sci USA 108, 18548–18553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamini F., Ozkan H., Brandolini A., Schäfer-Pregl R. & Martin W. Genetics and geography of wild cereal domestication in the Near East. Nat Rev Genet 3, 429–441 (2002). [DOI] [PubMed] [Google Scholar]
- Qiu Y. C., Zhou R. H., Kong X. Y., Zhang S. S. & Jia J. Z. Microsatellite mapping of a Triticum urartu Tum. derived powdery mildew resistance gene transferred to common wheat (Triticum aestivum L.). Theor Appl Genet 111, 1524–1531 (2005). [DOI] [PubMed] [Google Scholar]
- Ling H. Q. et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 87–90 (2013). [DOI] [PubMed] [Google Scholar]
- Wicker T. et al. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat Genet 45, 1092–1096 (2013). [DOI] [PubMed] [Google Scholar]
- Langfelder P. & Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P. & Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw 46, 1–17 (2012). [PMC free article] [PubMed] [Google Scholar]
- Liang H. & Li W. H. Gene essentiality, gene duplicability and protein connectivity in human and mouse. Trends Genet 23, 375–378 (2007). [DOI] [PubMed] [Google Scholar]
- Sun J. C. & Zhao Z. M. A comparative study of cancer proteins in the human protein-protein interaction network. BMC Genomics 11, S5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Kim P. M., Sprecher E., Trifonov V. & Gerstein M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput Biol 3, e59 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. Y., Greenbaum D., Lu H. X., Zhu X. W. & Gerstein M. Genomic analysis of essentiality within protein networks. Trends Genet 20, 227–231 (2004). [DOI] [PubMed] [Google Scholar]
- Yang Y. et al. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun 5, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. D. et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93 (2004). [DOI] [PubMed] [Google Scholar]
- Stuart J. M., Segal E., Koller D. & Kim S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003). [DOI] [PubMed] [Google Scholar]
- Sahu S. S., Weirick T. & Kaundal R. Predicting genome-scale Arabidopsis-Pseudomonas syringae interactome using domain and interolog-based approaches. BMC Bioinformatics 15, S13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z. L. & Zhao Y. Transcriptome comparison and gene coexpression network analysis provide a systems view of citrus response to ‘Candidatus Liberibacter asiaticus’ infection. BMC Genomics 14, 27 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. The miR9863 family regulates distinct Mla alleles in barley to attenuate NLR receptor-triggered disease resistance and cell-death signaling. PLoS Genet 10, e1004755 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q. H., Stephen S., Taylor J., Helliwell C. A. & Wang M. B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol 201, 574–584 (2014). [DOI] [PubMed] [Google Scholar]
- Gill B. S., Friebe B. R. & White F. F. Alien introgressions represent a rich source of genes for crop improvement. Proc Natl Acad Sci USA 108, 7657–7658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss H., Dechert C., Kogel K. H. & Hückelhoven R. A small GTP-binding host protein is required for entry of powdery mildew fungus into epidermal cells of barley. Plant Physiol 128, 1447–1454 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathuri I. P., Zellerhoff N., Schaffrath U., Hensel G. & Kumlehn J. Constitutively activated barley ROPs modulate epidermal cell size, defense reactions and interactions with fungal leaf pathogens. Plant Cell Rep 27, 1877–1887 (2008). [DOI] [PubMed] [Google Scholar]
- Hoefle C. et al. A barley ROP GTPase ACTIVATING PROTEIN associates with microtubules and regulates entry of the barley powdery mildew fungus into leaf epidermal cells. Plant Cell 23, 2422–2439 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefle C. & Hückelhoven R. A barley Engulfment and Motility domain containing protein modulates Rho GTPase activating protein HvMAGAP1 function in the barley powdery mildew interaction. Plant Mol Biol 84, 469–478 (2014). [DOI] [PubMed] [Google Scholar]
- Kawano Y., Kaneko-Kawano T. & Shimamoto K. Rho family GTPase-dependent immunity in plants and animals. Front Plant Sci 5, 522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. The RhoGAP SPIN6 associates with SPL11 and OsRac1 and negatively regulates programmed cell death and innate immunity in rice. PLoS Pathog 11, e1004629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q. H. et al. Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. Science 315, 1098–1103 (2007). [DOI] [PubMed] [Google Scholar]
- Chang C. et al. Barley MLA immune receptors directly interfere with antagonistically acting transcription factors to initiate disease resistance signaling. Plant Cell 25, 1158–1173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei dit Frey N. et al. Functional analysis of Arabidopsis immune-related MAPKs uncovers a role for MPK3 as negative regulator of inducible defences. Genome Biol 15, R87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segonzac C. et al. Negative control of BAK1 by protein phosphatase 2A during plant innate immunity. EMBO J 33, 2069–2079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M. et al. Network modeling reveals prevalent negative regulatory relationships between signaling sectors in Arabidopsis immune signaling. PLoS Pathog 6, e1001011 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonardi V. et al. Expanded functions for a family of plant intracellular immune receptors beyond specific recognition of pathogen effectors. Proc Natl Acad Sci USA 108, 16463–16468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. et al. Cell-autonomous expression of barley Mla1 confers race-specific resistance to the powdery mildew fungus via a Rar1-independent signaling pathway. Plant Cell 3, 337–350 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli D. et al. The CC-NB-LRR-type Rdg2a resistance gene confers immunity to the seed-borne barley leaf stripe pathogen in the absence of hypersensitive cell death. PLoS One 5, e12599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahmane A., Kanyuka K. & Baulcombe D. C. The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11, 781–791 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann W. Natural variation in the Arabidopsis response to the avirulence gene hopPsyA uncouples the hypersensitive response from disease resistance. Mol Plant-Microbe Interact 18, 1054–1060 (2005). [DOI] [PubMed] [Google Scholar]
- Wang A. & Krishnaswamy S. Eukaryotic translation initiation factor 4E-mediated recessive resistance to plant viruses and its utility in crop improvement. Mol Plant Pathol 13, 795–803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangasjärvi S., Tikkanen M., Durian G. & Aro E. M. Photosynthetic light reactions-an adjustable hub in basic production and plant immunity signaling. Plant Physiol Biochem 81, 128–134 (2014). [DOI] [PubMed] [Google Scholar]
- Rodríguez-Herva J. J. et al. A bacterial cysteine protease effector protein interferes with photosynthesis to suppress plant innate immune responses. Cell Microbiol 14, 669–681 (2012). [DOI] [PubMed] [Google Scholar]
- Thordal-Christensen H., Zhang Z., Wei Y. & Collinge D. B. Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during barley-powdery mildew interaction. Plant J 11, 1187–1194 (1997). [Google Scholar]
- Wäspi U., Schweizer P. & Dudler R. Syringolin reprograms wheat to undergo hypersensitive cell death in a compatible interaction with powdery mildew. Plant Cell 13, 153–161 (2001). [PMC free article] [PubMed] [Google Scholar]
- Chen C., Khaleel S. S., Huang H. & Wu C. H. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med 9, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrin R. et al. Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol 10, R55 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta B. et al. A network-based, integrative study to identify core biological pathways that drive breast cancer clinical subtypes. Br J Cancer 106, 1107–1116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogelman L. J. et al. Identification of co-expression gene networks, regulatory genes and pathways for obesity based on adipose tissue RNA Sequencing in a porcine model. BMC Med Genomics 7, 57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Feng Z., Wang X., Wang X. & Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010). [DOI] [PubMed] [Google Scholar]
- de Hoon M. J., Imoto S., Nolan J. & Miyano S. Open source clustering software. Bioinformatics 20, 1453–1454 (2004). [DOI] [PubMed] [Google Scholar]
- Purcell S. et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Blay S., McNeney B. & Graham J. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J Stat Softw 16, 1–9 (2006). [Google Scholar]
- Wang G. F. et al. Molecular analysis of common wheat genes encoding three types of cytosolic heat shock protein 90 (Hsp90): Functional involvement of cytosolic Hsp90s in the control of wheat seedling growth and disease resistance. New Phytol 191, 418–431 (2011). [DOI] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A. et al. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- Zdobnov E. M. & Apweiler R. InterProScan – and integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2005). [DOI] [PubMed] [Google Scholar]
- Du Z., Zhou X., Ling Y., Zhang Z. & Su Z. AgriGO: A GO analysis toolkit for the agricultural community. Nucl Acids Res 38, W64–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.