

Abstract
Retinal angiogenesis is tightly regulated to meet oxygenation and nutritional requirements. In diseases such as proliferative diabetic retinopathy and neovascular age-related macular degeneration, uncontrolled angiogenesis can lead to blindness. Our goal is to better understand the molecular processes controlling retinal angiogenesis and discover novel drugs that inhibit retinal neovascularization. Phenotype-based chemical screens were performed using the ChemBridge DiversetTM library and inhibition of hyaloid vessel angiogenesis in Tg(fli1:EGFP) zebrafish. 2-[(E)-2-(Quinolin-2-yl)vinyl]phenol, (quininib) robustly inhibits developmental angiogenesis at 4–10 μm in zebrafish and significantly inhibits angiogenic tubule formation in HMEC-1 cells, angiogenic sprouting in aortic ring explants, and retinal revascularization in oxygen-induced retinopathy mice. Quininib is well tolerated in zebrafish, human cell lines, and murine eyes. Profiling screens of 153 angiogenic and inflammatory targets revealed that quininib does not directly target VEGF receptors but antagonizes cysteinyl leukotriene receptors 1 and 2 (CysLT1–2) at micromolar IC50 values. In summary, quininib is a novel anti-angiogenic small-molecule CysLT receptor antagonist. Quininib inhibits angiogenesis in a range of cell and tissue systems, revealing novel physiological roles for CysLT signaling. Quininib has potential as a novel therapeutic agent to treat ocular neovascular pathologies and may complement current anti-VEGF biological agents.
Keywords: angiogenesis, drug discovery, eye, G protein-coupled receptor (GPCR), leukotriene, blindness, hyaloid vasculature development, ocular angiogenesis
Introduction
In the eye, developmental angiogenesis is a critical biological process enabling vision (1). Morphogenesis of the retinal vasculature is strictly controlled to balance high metabolic requirements while maintaining visual function. Uncontrolled pathological angiogenesis in the retina, choroid, and iris results in proliferative diabetic retinopathy, neovascular age-related macular degeneration, retinal vein occlusion, and retinopathy of prematurity, which are leading causes of blindness worldwide (2–5). Our understanding of the endogenous and exogenous factors regulating the overlapping but distinct phenotypes of developmental and pathological ocular angiogenesis is limited (6). Unbiased, phenotype-based chemical screens provide an opportunity to efficiently identify novel pharmacological inhibitors of angiogenesis in the eye (7, 8). These drugs and their molecular targets enhance our fundamental knowledge of the signaling networks regulating ocular angiogenesis and highlight alternative therapeutic interventions for angiogenesis-related disease.
An intricate balance of growth and inhibitory factors regulates angiogenesis (9). Imbalances can result in growth of abnormal, leaky vessels (6). Pathological angiogenesis is a hallmark of blinding ocular neovascular disease (3, 6), including neovascular age-related macular degeneration, which affects 2.6 million European Union and United States patients and whose prevalence is increasing because of an aging population (10, 11). Ocular neovascular disorders are regularly treated with biological agents (e.g. ranibizumab, bevacizumab, and aflibercept) targeting VEGF, a key proangiogenic mediator (12–14). Unfortunately, many patients do not respond clinically or become refractory to treatment (15). For example, ∼46% of diabetic macular edema patients require additional laser treatment, and limited improvement in visual acuity is achieved (16). This, coupled with an undesirable intraocular delivery route, large clinical burden and expensive biological therapies highlight the need for improved ocular neovascular therapeutic agents (10).
The retina is a highly metabolically active tissue needing significant nourishment and oxygen supply (17). The adult retina in most mammals is nourished by two vascular networks. The choroid vessels overlying the retinal pigmented epithelium (RPE) nourish the outer retina. The inner retinal vessels at the ganglion cell layer develop at late embryonic stages and complete their morphogenesis after birth (1, 18). During development, the inner mammalian retina is nourished by the hyaloid vasculature, a transient capillary network located between the lens and retina. Later, hyaloid vessels undergo programmed regression, and a retinal vasculature forms by angiogenesis (1, 18, 19). Defects in hyaloid vasculature regression, known as persistent fetal vasculature, result in pathological eye conditions (20). In zebrafish, intraocular vasculature development is initially similar to mammals. However, hyaloid vessels do not regress after embryonic development but progressively lose contact with the lens and, by 30 days after fertilization, adhere to the inner limiting membrane of the juvenile retina (21). In adult zebrafish, these vessels are found attached to the ganglion cell layer, exhibiting distinctive hallmarks of mammalian retinal vasculature (21, 22). Although the cellular morphogenesis of zebrafish hyaloid vasculature is well characterized, our understanding of the molecular regulators is limited to a small number of genetic and pharmacological studies (7, 8, 23).
Zebrafish are particularly amenable to phenotype-based drug discovery (24, 25). This “target-agnostic” approach focuses on a chosen phenotype and does not require prior selection of a molecular target. In this study, we identify unique drugs inhibiting developmental angiogenesis of the eye by performing an unbiased screen of ∼1800 small-molecule drugs in the zebrafish hyaloid vessel assay (7). The screen uncovered 2-[(E)-2-(quinolin-2-yl)vinyl]phenol (quininib)2 as a potent inhibitor of developmental angiogenesis in the zebrafish eye. Subsequently, quininib demonstrated significant anti-angiogenic activity in human endothelial cell, murine aortic ring, and murine oxygen-induced retinopathy models of angiogenesis. Target profiling identified quininib as a cysteinyl leukotriene 1 and 2 receptor (CysLT1–2) antagonist. The cysteinyl leukotrienes LTC4, LTD4, LTE4, and LTF4 are bioactive lipids synthesized from cell membrane arachidonic acid via a 5-hydroxyeicosatetraenoic acid intermediate and signal via G protein-coupled receptors (CysLT1, CysLT2, GPR17, and GPR99) (26–30). CysLT1 antagonists are commonly used to treat asthma and allergic rhinitis (31, 32). Previous studies have reported a role for CysLTs in inflammation, vascular permeability, immune responses, tissue repair, and regeneration (28, 33–36). CysLT2 and CysLT1 are expressed in the murine retina, and exogenous cysLTs are sufficient to induce retinal edema (37). Here quininib inhibits known cysteinyl leukotriene receptor signaling pathways, reducing ERK phosphorylation in response to leukotriene D4 agonism (38, 39). In summary, from unbiased chemical screens, we advance prior reports on cysLTs and demonstrate that a CysLT1–2 antagonist significantly attenuates angiogenesis in the eye.
Experimental Procedures
Animal Use
Procedures were approved by the University College Dublin Animal Research Ethics Committee and performed under a Department of Health license and adhered to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research. Wild-type C57BL/6J mice were obtained from Charles River Laboratories.
Phenotype-based Chemical Screens of Ocular Developmental Angiogenesis
ChemBridge DIVERSetTM library drug-like chemicals were screened (40). For all experiments, drugs were initially dissolved to 10 mm in DMSO and further diluted to the relevant concentration in double-distilled H2O. Transgenic Tg[fli1:EGFP] zebrafish embryos (41) were drug-treated as reported previously (7). A compound was designated a primary hit when more than three of five treated larvae exhibited >50% reduced primary hyaloid vessels. Compounds confirmed in replicate experiments were designated secondary hits. Chemical analogues were identified using the ChemBridge hit 2 lead search engine. The MEK-1/2 inhibitor TAK-733 and the broad-spectrum PKC inhibitor Gö 6983 were purchased from SelleckChem.
Zebrafish Optokinetic Response Assay
The optokinetic response assay was performed on larvae treated from 2–5 days post-fertilization (dpf) and 3–5 dpf (7, 42). The drug was removed prior to the optokinetic response assay, and the larvae were washed in embryo medium. The average number of saccades/minute was manually quantified (n = 30 zebrafish/data point).
Intravitreal Murine Maximum Tolerated Dose
C57BL/6J mice aged 3–6 months were anesthetized (ketamine, 67 mg/kg; medetomidine, 0.67 mg/kg), and 5-μl final concentrations of drug were injected intravitreally. Eyes were pierced below the pars planar using a 30-gauge needle, and the test drug was injected through this incision into the vitreous using a Nanofil syringe attached to a 33-gauge needle (World Precision Instruments). Post-injection, atipamezole (0.67 mg/kg) was administered. Mice were monitored and scored daily and culled 7 days after injection.
Histological Analysis of Zebrafish and Murine Eyes
Zebrafish larvae and mouse eyes were processed as reported previously (7). Mice were culled by carbon dioxide asphyxiation, and eyes were fixed in 2% paraformaldehyde/2.5% glutaraldehyde/0.1 M Sorenson's buffer. Prior to embedding, extraneous musculature was trimmed from the sclera and cornea, and the lens was removed, generating an eye cup that was bisected near the optic nerve. 500-nm sections were cut on a Leica EM UC6 microtome, stained with toluidine blue, and cover-slipped with DPX mounting medium. Sections from the central retina adjacent to the optic nerve were imaged and analyzed using NIS Elements BR on a Nikon E80i microscope.
Viability Assays in Human Cell Lines
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye reduction assays were performed according to the protocol of the manufacturer to determine the viability of dermally derived human microvascular endothelial cells (HMEC-1) or human retinal pigment epithelium cells (ARPE-19), which were maintained as described previously (43).
In Vitro Tubule Formation in Human Microvascular Endothelial Cells
Microslide angiogenesis plates (IBIDI) were coated with Matrigel matrix (BD Biosciences), and tubule formation assays were performed according to the guidelines of the manufacturer. For all experiments, drugs were initially dissolved to 10 mm in DMSO and further diluted to the relevant concentration in MCDB 131 medium (Gibco). Total tubule length was quantified using Zeiss Axiovision image analysis software. Calcein AM stain (Invitrogen) 2 μg/ml was incubated with HMEC-1 cells following tubule formation for 30 min at 37 °C. Cells were imaged using brightfield and fluorescent microscopy.
Anti-angiogenic Activity in an ex Vivo Mouse Aortic Ring Model
The aortic ring angiogenesis assay was performed according to an established protocol (44). For all experiments, drugs were initially dissolved to 10 mm in DMSO and further diluted to the relevant concentration in medium. Aortic rings were drug-treated in 150 μl of DMEM supplemented with 10% FCS and incubated at 37 °C/5% CO2 for 6 days, when sprouts from the aortic ring perimeter were imaged using an Olympus CKX41 inverted phase-contrast microscope running ImageJ software and quantified as a percentage of vehicle control.
Mouse Ocular Angiogenesis Model
The mouse oxygen-induced retinopathy (OIR) model followed established protocols (45, 46). For all experiments, drugs were initially dissolved to 10 mm in DMSO and further diluted to the relevant concentration in Hanks' balanced salt solution. On postnatal day (P)12, each pup (>4 g) was administered oral acetaminophen (120 mg/5 ml) and anesthetized using ketamine (80 mg/kg) and medetomidine (0.67 mg/kg). Anesthetized pups were placed on heating pads and, using a Leica M651 ophthalmoscope, eyelids were opened by gently scoring with a beveled 30-gauge needle. A Dumont 7 forceps was used to gently proptose the eye, which was pierced just below the pars planar with a 33-gauge beveled needle attached to a Nanofil syringe (World Precision Instruments) angled behind the lens. 1 μl of drug or vehicle control was injected (rate of 1 μl/10 s), and the needle was left in situ for 60 s to allow pressure equilibration. Following intravitreal injection, atipamezole (0.67 mg/kg) was given. Uninjected controls were taken at P12 to confirm vascular regression. On P17, pups were culled by carbon dioxide asphyxiation, and the eyes were enucleated and fixed in 4% paraformaldehyde overnight at 4 °C.
Analysis of Murine Retinal Vascularization
Flat-mounted retinas were fluorescently stained with Griffonia simplicifolia isolectin (B4) and Alexa-streptavidin 568 as reported previously (47). Eyes were coverslipped using Aqua/Polymount and imaged using a Zeiss Axiovert 200 M fluorescent microscope running Andor IQ2 software and stitched using Andor montaging. Post-imaging, the total retinal area and total avascular area were measured using Fiji, and neovascular areas (measured from a 0.58 mm2 area for each quadrant; neovascular tufts were precisely selected using the magnetic lasso tool) were measured using Imaris software. The avascular or neovascular area was expressed as a percentage of the total or defined retinal area, respectively. For vascular density analysis, a representative quadrant was selected from a subset of OIR samples (n = 8–9), and images were taken in 4-μm slices from the vitreal surface 0 μm (superficial) to 84 μm (deep retinal vascular bed) according to Sidman et al. (48). Statistical analysis was performed using one-way ANOVA with Dunnett's comparison post hoc test for multiple comparisons.
Target Profiling
10 μm quininib was screened for activity in the SelectScreen® Kinase Profiling Service (Invitrogen), which profiled 22 kinases, and the Premier Screen of 140 protein kinase targets (Dundee). The profiled kinases are reported in Fig. 5A.
FIGURE 5.

Quininib is a cysteinyl leukotriene receptor 1 antagonist with an IC50 of 1.4 μm. Target profiling of 153 targets with 10 μm quininib (A) identified activity in the cysteinyl leukotriene pathway. Quininib blocked 0.1 nm LTD4-induced intracellular calcium release in CHO-K1 cells overexpressing CysLT1 with an average IC50 of 1.4 μm (three replicates). Plotted is the non-linear regression curve for a representative experiment (B, top panel). Quininib also acted as a very weak CysLT2 antagonist with an average IC50 of >38.5 μm (three replicates). Shown is the non-linear regression curve of a representative experiment (B, bottom panel). Upon further investigation, quininib appeared to act as an orthosteric CysLT1 antagonist, blocking 50% of LTD4-induced contraction in an ex vivo guinea pig lung strip assay (C) but exhibiting no agonist response (data not shown) and competing directly with the radiolabeled native agonist (LTD4). The genes encoding cysLT1R and cysLT2R were expressed in zebrafish at 3 and 5 dpf (D). Both CYSLT1R and CYSLT2R were expressed in a human dermally derived endothelial cell line (HMEC-1), a human retinal microvascular endothelial cell line (ACBRI-181), and a human retinal pigment epithelial cell line. E, Western blotting analysis showing the expression of cysteinyl leukotriene receptors 1 and 2 and α-tubulin in three human cell lines. Note that the antibody for CYSLT1R also binds to CYSLT2R protein. For CysLT1R, PDGFRα, and VEGFR1, target profiling was performed in triplicate to confirm results (10 μm quininib inhibited intracellular calcium release by 107% ± 8% (below baseline) in CHO-K1 cells overexpressing CysLT1, PDGFRα by 34% ± 13%, and VEGFR1 by 16% ± 18%. A hit was considered to inhibit >50% of the response).
Cysteinyl Leukotriene Receptor Assays
Drug antagonism was assessed in a guinea pig lung strip assay, where 3 nm LTD4 was used to induce lung strip contraction. For cell-based assays, basal agonism or antagonism of 0.1 nm LTD4-induced calcium mobilization was assessed in CHO-K1 cells overexpressing human CysLT1, and 30 nm LTC4-induced calcium mobilization was assessed in HEK 293 cells overexpressing human CysLT2. For ligand-binding assays, the percentage of bound [3H]LTD4 in human recombinant CHO-K1 cells overexpressing CysLT1 and human recombinant HEK 293 cells overexpressing CysLT2 was quantified by scintillation counting.
CysLT1–2 Expression Analyses
RNA was extracted from 3- and 5-dpf wild-type zebrafish eyes. The tissue was homogenized, and RNA was extracted using an RNeasy kit (Qiagen) and reverse-transcribed to cDNA using a Superscript III kit (Invitrogen). Primer sequences were as follows: zebrafish cysltr1 (forward, 5-GGCATCTTGCGCACTCTACT; reverse, 5-GCAAAGCGTGATGACCACAG), zebrafish cysltr2 (forward, 5-TGTTTGGAGCTCGCACATGA; reverse, 5-ATGATCACCAAAGCGCAAGC), and zebrafish actb (forward, 5-CGAGCAGGAGATGGGAACC; reverse, 5-CAACGGAAACGCTCATTGC).
Protein was extracted from human cell lines HMEC-1 (dermally derived endothelial cells), ACBRI-181 (primary retinal microvascular endothelial cells), and ARPE-19 (RPE cells). Cysteinyl leukotriene receptor expression was analyzed in immunoblots with primary antibodies (CysLT1R: Abcam, ab151484 (lot GR115651-4), 1:2000 with a goat anti-rabbit HRP-linked secondary antibody: Amersham Biosciences, NA934 (lot 9572648), 1:4000; CysLT2R: Santa Cruz Biotechnology, sc-27097 (lot F2514), 1:1000 with a donkey anti-goat IgG-HRP secondary antibody 1:2000 and α tubulin, Sigma, T9026, 1:10,000).
CysLT1–2 Pathway Analysis
HMEC-1 cells were seeded at 2.5 × 105/well. After 24 h, the medium was changed, and cells were serum-starved for 24 h. Cells were stimulated with LTD4 (10–1000 nm) for 5 min. For pathway inhibition studies, cells were pretreated with quininib or TAK-733 (10 μm) for 30 min, followed by 5-min stimulation with 500 nm LTD4. Control cells were stimulated with 0.1% DMSO and 0.25% ethanol. After treatments, cells were washed with ice-cold PBS, harvested, and lysed with lysis buffer as described previously (43). Protein concentration was determined using the BCA protein assay kit (Pierce). For immunoblotting, an equal amount of protein (20 μg) was prepared in 5× sample buffer and separated by 10% SDS/PAGE, transferred to PVDF membranes (Sigma), and probed with primary and anti-rabbit or anti-mouse HRP-labeled secondary antibody (anti-mouse, Amersham Biosciences, NA934, lot 9572648). The primary antibodies used were phospho-p44/42 MAPK (Erk1/2, Cell Signaling Technology, 4370S, lot 15, 1:2000) and p44/42 MAPK (Erk1/2, Cell Signaling Technology, 9102S, lot 26, 1:1000) and α tubulin (1:10,000). Chemiluminescent detection using Amersham Biosciences ECL Prime detection reagent (GE Healthcare, RPN2232) allowed visualization of protein. Quantitative measurement of the phosphorylated protein level was performed using ImageJ. Results are presented as relative densitometry ratios of phosphoprotein relative to the band intensity of total protein.
Results
Phenotype-based Screens in Zebrafish Uncover Novel Anti-angiogenic Compounds
In an unbiased anti-angiogenic drug discovery approach, 1760 randomized small-molecule compounds from the ChemBridge DiverSETTM library were screened in larval zebrafish (Fig. 1, A and B). Anti-angiogenic activity was quantified on the basis of the ability of 10 μm of each compound to significantly inhibit developmental angiogenesis of the primary hyaloid vessels (HVs) in the eye of TG[fli1:EGFP] larvae (21). Ten primary hits were identified on the basis of the criterion of >50% HV inhibition in >60% larvae. Of these, four secondary hits inhibited primary branch HV development in replicate experiments, a hit rate of 0.23% (Figs. 1, A–C, and 2). Dose-response experiments determined that quininib (CAS no. 1379458-56-6; Fig. 1D, inset) ranked with the highest efficacy and potency, inhibiting primary HV development by 85% with an IC50 of ∼4 μm (Fig. 1D). Quininib exerted a dose-dependent inhibition on the number of primary HVs (Fig. 1D), and fluorescent images of dissected lenses demonstrated quininib to robustly stunt growth of the entire basket-shaped hyaloid vasculature (Fig. 1E). Time-course analyses that looked at the effect of quininib on the initial formation of hyaloid vasculature at 2, 3, 4, and 5 dpf demonstrated that quininib initially completely abrogated primary vessel sprouting. Delayed primary vessels appeared at 4 dpf, but the branching pattern at 5 dpf was visibly reduced compared with vehicle controls at 3 dpf. Cell density appeared normal in the quininib-treated drug vessels (Fig. 1F).
FIGURE 1.

Hit quininib was identified in chemical screens of ocular developmental angiogenesis. 1760 compounds from the ChemBridge DiverSETTM library (A) were tested for the ability to attenuate developmental angiogenesis of zebrafish hyaloid vasculature (B). Ten primary hits were identified (>50% reduction in primary hyaloid vessels in three or more of five treated larvae), and four were selected for validation (C, the asterisk refers to the minimum effective dose). Quininib ranked highest on the basis of robust, reproducible anti-angiogenic activity at multiple doses (C). Quininib (D, inset) generates a dose-dependent, significant inhibition of the number (mean ± S.E.) of primary hyaloid vessels that develop (n = 10 zebrafish/dose). The number of primary hyaloid vessels is quantified and graphed as a percentage of control, which has an average of 3.4 primary vessels (D). Representative images of hyaloid vasculature formation on dissected zebrafish lenses (E) qualitatively demonstrate the anti-angiogenic activity of quininib on the entire hyaloid vasculature. Representative images from a time-course treatment carried out from 1–2, 3, 4, or 5 dpf (F) demonstrated that 5 μm quininib abolished and then delayed vessel sprouting. Statistics were performed using one-way ANOVA (with Dunnett's post hoc correction). ***, p < 0.001.
FIGURE 2.
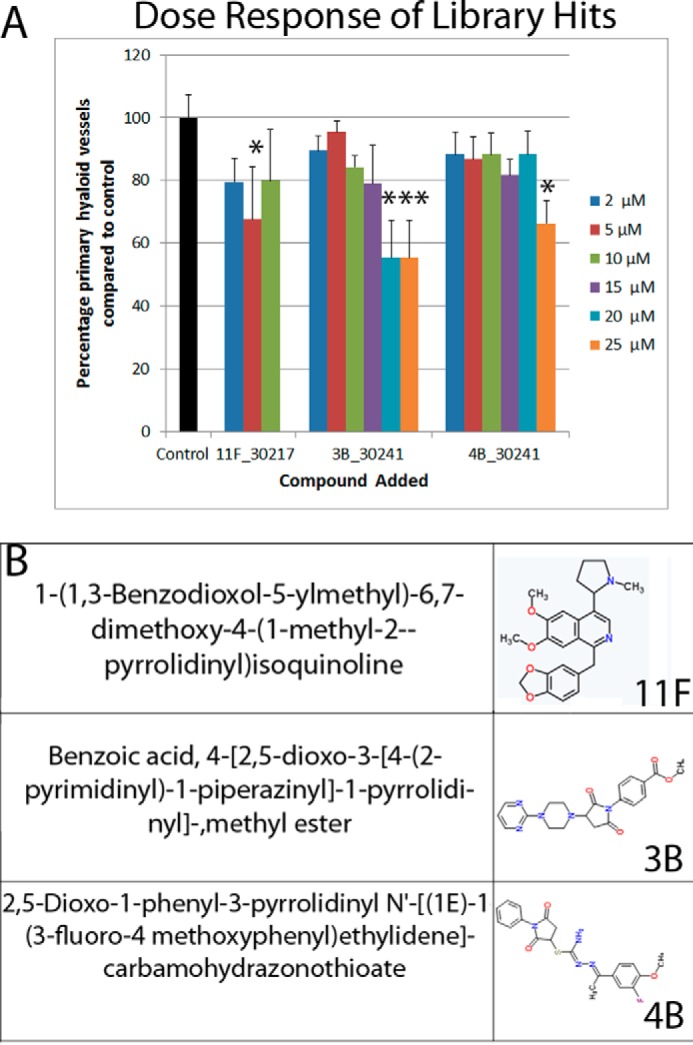
Dose-response activity of selected screen hits 11F, 3B, and 4B. Compounds 11F, 3B, and 4B showed efficacy at 10 μm on the basis of the initial screen criteria but not in validation experiments, wherein the average reduction in primary HV is graphed (A). 11F and 4B do not demonstrate a dose-dependent anti-angiogenic activity. 11F only demonstrated inhibition at 5 μm, whereas the minimal effective doses of 3B and 4B were 20 and 25 μm, respectively (n = 5–10 zebrafish/dose). These compounds were not progressed further. The number of primary hyaloid vessels is quantified and graphed as a percentage of control, which has an average of 3.4 primary vessels. Data shown are mean ± S.E. Statistics were performed using one-way ANOVA (with Dunnett's post hoc correction). *, p < 0.05; ***, p < 0.001. The chemical names and two-dimensional structures of 11F, 3B, and 4B are given in B.
A Structural Analogue of Quininib Significantly Inhibits HV Development
To corroborate the specific pharmacological response produced by quininib, commercial analogues were assayed for anti-angiogenic activity. 4-Bromo-2-(2-quinolin-2-yl-vinyl)-phenol (QB-590, CAS no. 337352-14-4) had inclusion of a 5-bromo substituent. (4-[(E)-2-Vinyl]phenol (QB-663, CAS no. 430459-22-6) changed the hydroxy group to the 4′ from the 2′ position and inclusion of an ester linkage at C-8. QB-771 changed the hydroxy group to the 4′ from the 2′ position and included a 6′-bromo substituent and an ester linkage at C-3′. 4-bromo-2-(2-quinolin-2-yl-vinyl)-phenol, 4-[(E)-2-vinyl]phenol, and QB-771 did not exhibit biological activity in the HV assay (Fig. 3). However, the quininib Z-isomer and the other most structurally related analogue, 4-[(Z)-2-(4-quinolinyl)vinyl]phenol (QB-799, CAS no. 1379458-54-4), which contains a Z-isomer configuration and relocation of the phenol ring hydroxy group to the 4′ position, phenocopied quininib and reduced primary zebrafish HV development by ∼34% (Fig. 3).
FIGURE 3.
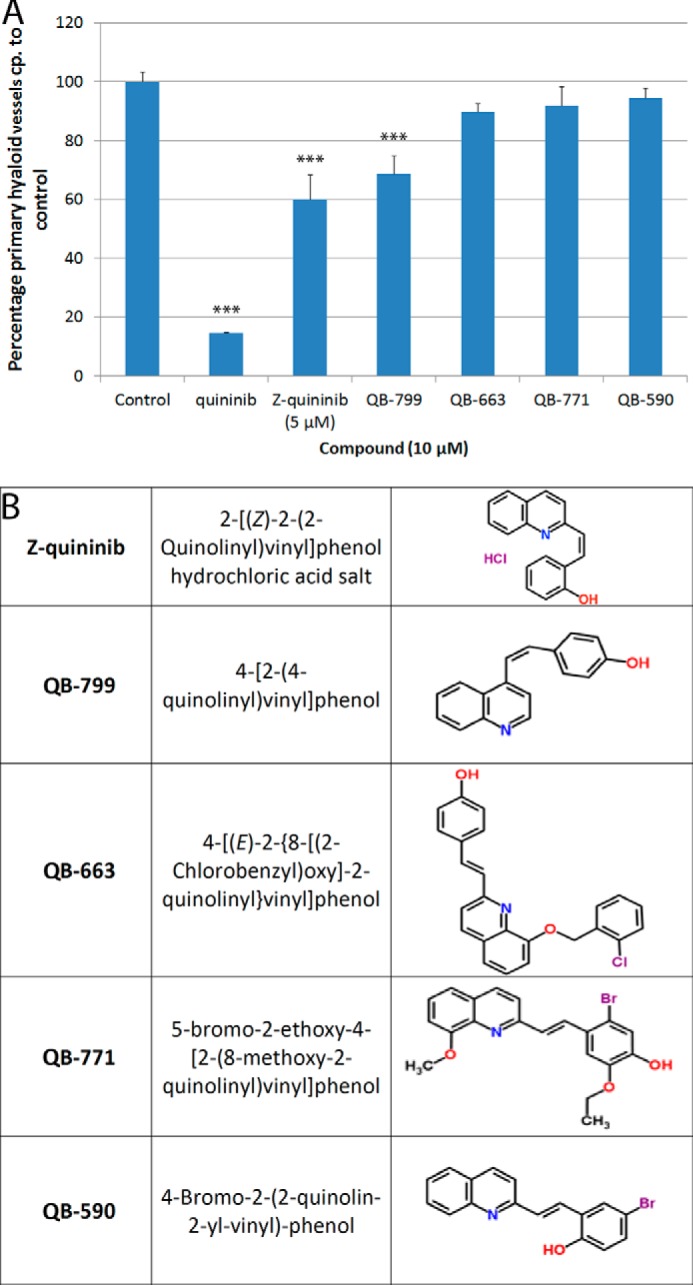
Quininib analogs can inhibit ocular developmental angiogenesis in zebrafish. Commercially available quininib analogues were tested in the zebrafish hyaloid vasculature assay. Two Z conformation analogues, Z-quininib and 4-[(Z)-2-(4-quinolinyl)vinyl]phenol, at 5 and 10 μm respectively, exhibited significant inhibition of ocular developmental angiogenesis, whereas three analogues were inactive (A). The number of primary hyaloid vessels is quantified and graphed as a percentage of control, which has an average of 3.4 primary vessels. The chemical names and two-dimensional structures of five tested analogues are shown in B. Data are given as mean ± S.E. Statistics were performed using one-way ANOVA (with Dunnett's post hoc test). ***, p <0.001 (n = 10 zebrafish/compound).
Safety Pharmacology
Quininib is well tolerated at concentrations that elicit anti-angiogenic responses (Fig. 4). 2- 5-dpf zebrafish treated with 10 μm quininib developed normally, except for some pericardial edema, hypopigmentation, and reduced eye size (Fig. 4A). In addition, treatment of zebrafish larvae with 10 μm quininib from 2–5 dpf did not affect the existing intersegmental vasculature but did affect formation of the subintestinal vessels (which form after 2.5 dpf), suggesting that quininib preferentially inhibits newly forming vessels (Fig. 4, B and C). Quininib-treated zebrafish eyes have equivalent ocular histology to vehicle controls, with comparable central retinal morphology, diameter, lamination, RPE pigmentation, and absence of pyknotic nuclei (Fig. 4E). In contrast, zebrafish visual behavior was significantly or moderately reduced with 10 or 5 μm quininib, respectively (Fig. 4D). In human cell lines, <25 μm quininib was well tolerated for 96 h in retinal pigment epithelium cells (ARPE-19) and 24 h in human microvascular endothelial cells (HMEC-1) (Fig. 4F). In contrast, 5–20 μm quininib reduced HMEC-1 viability by 20–30% at 96 h, consistent with an anti-angiogenic profile (Fig. 4F). In mice, intravitreally administered quininib had a maximum tolerated dose of 200 μm on the basis of ocular morphology. (Fig. 4G). Gross ocular morphology was unaffected. No cataracts, inflammation, or infections occurred up to 7 day post-delivery. Retinal histology was equivocal to controls with respect to retinal cell types, cell numbers, lamination, RPE pigmentation, and absence of pyknotic nuclei (Fig. 4G).
FIGURE 4.

Quininib is safe and well tolerated in zebrafish, human cell lines, and mouse retina. A, dorsal and lateral images of zebrafish treated with 10 μm quininib from 2–5 dpf showing normal gross morphology with the exception of some pericardial edema, a marginally smaller eye diameter, and a slight reduction in pigmentation. Quininib treatment from 2–5 dpf showed no disruption of established vasculature, as exemplified by intact intersegmental vessels, which formed before 1 dpf (B), but did inhibit formation of the subintestinal vessels (SIV) formed from 2–5 dpf (C). 5 and 10 μm quininib reduced visual behavior (n = 3 replicates, 10 zebrafish/replicate) recorded from the optokinetic response (D). Light microscopy showed normal retinal morphology, including the presence of expected cell types, proper lamination, and lack of pyknotic nuclei in 2- 5-dpf quininib-treated larvae (E). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assays showed that quininib is well tolerated at 20 μm for 24 h in a human endothelial cell line (n = 3) and at 25 μm for 96 h in a human retinal pigment epithelium cell line (n = 1, 4 replicates) (F). The quininib intravitreal maximum tolerated dose was determined in mouse eyes (G and H). Light microscopy (×40) of the central retina (near the optic nerve) showed a normal retinal morphology in 200 μm quininib-injected eyes: the presence of all cell types, similar thickness of layers, normal lamination, and lack of pyknotic nuclei (G). Mice were scored on a daily basis (for 7 days after injection) for nine general welfare traits. A score of 3 was normal. All scores were normal, with the exception of one mouse at 100 μm that had a corneal bleed visible 1 day after injection (H) (n = 3 mice/dose). ON, optic nerve; OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Target Profiling and CysLT1 Antagonism
To elucidate the molecular mechanism of action of quininib, literature searches and target profile assays were undertaken. Previously, quininib was reported as a weak cysteinyl leukotriene 1 receptor (CysLT1) antagonist (inhibited binding of [3H]LTD4 to guinea pig lung membrane, IC50, >50 μm) and an endothelin-converting enzyme 2 inhibitor (IC50, 6.42 μm) (49–51). Here target profiling of 153 putative targets revealed that only CysLT1 was inhibited more than the 50% threshold criterion by 10 μm quininib (Fig. 5A). Of note, quininib did not significantly inhibit human endothelin-converting enzyme (ECE-1) or any of the human vascular endothelial growth factor receptors (VEGFR1–3) that are clinically targeted in neovascular disease (Fig. 5, A–C).
Quininib Significantly Attenuates Angiogenesis in Mammalian Models
To determine whether quininib has an evolutionarily conserved anti-angiogenic bioactivity, it was tested in human and mouse models. In HMEC-1 cells, 3.16 μm quininib significantly inhibited (∼44.2%) in vitro endothelial cell tubule formation (Fig. 6, A and B). In the ex vivo mouse aortic ring assay of angiogenesis, 10 μm quininib administered for 6 days significantly inhibited (35.9% ± 13%) sprout formation compared with vehicle controls (Fig. 6, C and D). To assess whether quininib inhibited ocular angiogenesis in vivo, the murine OIR model was utilized. Quininib was injected once intravitreally at the P12 transition from hyperoxia to normoxia (relative hypoxia), and analyses at P17 demonstrated inhibition of retinal revascularization (normal intraretinal vessel regrowth similar to developmental angiogenesis; Fig. 6, E and F) and, at higher quininib concentrations, increased retinal neovascularization (preretinal pathological neovascularization; Fig. 6, G–I). Compared with vehicle-injected controls, there was a 1.5-fold increase in avascular area in quininib-treated eyes (25%) versus vehicle-injected controls (16%) (Fig. 6, E and F). 0.5 μm quininib had no effect on neovascularization, whereas 3 μm quininib resulted in an increased neovascular area compared with vehicle controls (Fig. 6G). Relative vascular density analysis showed that, at P17, the retinal blood vessels were mainly found in the superficial layer and that vascular density was increased in 3 μm quininib-injected mice, reflecting the increase in neovascularization (Fig. 6, H and I).
FIGURE 6.

Quininib is anti-angiogenic in mammalian models of angiogenesis. Investigation of the anti-angiogenic activity of quininib and montelukast, a CysLT1 antagonist, in mammalian models of angiogenesis showed that quininib inhibited the formation of tubules in vitro, of sprouts ex vivo, and of blood vessels in vivo. A and B, in a quininib-treated human microvascular endothelial cell line (HMEC-1), 3.16 μm quininib inhibited the formation of tubules by 44% ± 9% (p < 0.01). A, the number of tubules qualitatively in a brightfield image (top panel) and stained with Calcein AM (bottom panel, green, a vital dye to show cell viability). B, quantification of total tubule length (n = 3 independent replicates; *, p < 0.05; **, p < 0.01). C and D, in a quininib-treated ex vivo mouse aortic ring model, 10 μm quininib reduced sprout formation (35.9 ± 13%) comparable to 10 m montelukast (43.9 ± 15.4%), whereas vehicle control did not reduce sprout formation (0%) (n = 3 independent replicates with 6 aortic rings/replicate). E and F, in a drug-treated mouse model of ocular angiogenesis, the oxygen-induced retinopathy model, both 0.5, and 3 μm quininib, injected on P12, prevented the revascularization of the central retina at P17, as evidenced by a larger avascular area (spaces enclosed within the lines) in 0.5 μm (25.5%, p <0.0001) and 3 μm (23.9%, p < 0.001) quininib-treated eyes compared with vehicle control (16% avascular area). 0.5 μm montelukast-treated eyes did not show a statistically significant increase in avascular area compared with the control. Shown are qualitative representative isolectin-stained flat-mount retina images of vehicle control, 0.5 μm quininib, and 0.5 μm montelukast (E) and a scatter dot plot with lines denoting mean ± S.E. (F). Statistics were performed using one-way ANOVA (with Dunnett's post hoc test; **, p < 0.01; ***, p < 0.001), and n > 18 mice across three or more independent OIR experiments (vehicle control, quininib 0.5 μm and 3 μm) and four or five mice in one OIR experiment (0.5 μm montelukast and 0.05 μm quininib). A subset (n = 8–9) of the quininib- and vehicle-treated OIR samples was further analyzed for percentage neovascular area (G) and vascular density (H and I). G, scatter dot plot showing the amount of retinal neovascularization as a percentage of total retinal area measured from a 0.58 mm2 area for each quadrant. Drug treatment with 3 μm quininib resulted in an increase in the area of neovascularization (25.8%) compared with vehicle (12.8%). Statistics were performed using one-way ANOVA (with Dunnett's post hoc test). **, p < 0.01. H, a subset (n = 8–9) of the quininib- and vehicle-treated OIR samples was further analyzed for vascular density (H and I). A single quadrant was selected per sample, and the relative vascular staining (isolectin) was quantified as a percentage of the entire quadrant every 4 μm from 0–84 μm (representative data from a single quadrant are graphed in H). The maximum staining in all groups was seen in the superficial retina ∼20 μm from the vitreous. Vascular density was highest in the 3 μm quininib-treated sample and lowest in 0.5 μm quininib-treated animals when compared with vehicle. Representative quantitative data (H) and representative qualitative data from a z-slice 24 μm from the vitreous (I) is shown. *, p < 0.05; **, p < 0.01.
CysLT1 Antagonism
CysLT1 was corroborated as a relevant target of quininib by competitive antagonist binding and activity assays and target expression profiling (Fig. 5, B–E). In guinea pig lung strip contraction assays, 30 μm quininib alone did not act as an agonist (data not shown). However, 30 μm quininib antagonized LTD4-induced contraction by 51% (Fig. 5C). In radio-ligand binding assays, 10 μm quininib inhibited binding of [3H]LTD4 to CysLT1 and CysLT2 by 65% and 15%, respectively (Fig. 5C). Cell-based assays of CysLT1 or CysLT2 activity confirmed that quininib preferentially antagonized the LTD4-induced reporter activity of human CysLT1 (IC50, 1.4 μm; Emax, 137%) compared with CysLT2 (IC50, 38.5 μm; Emax, 45%) (Fig. 5B). Previous studies have confirmed retinal expression of CysLT1 in rodent models of retinal neovascularization (37), and we demonstrated here ocular expression of the zebrafish cysltr1 and cysltr2 genes during hyaloid vessel development (Fig. 5D) and expression of cysteinyl leukotriene receptors 1 and 2 in human endothelial cells (HMEC-1), human retinal microvascular endothelial cells (ACBRI-181), and human RPE cells (ARPE-19) (Fig. 5E).
Other established CysLT1 antagonists are clinically used for the treatment of upper and lower respiratory tract disorders (31, 32). A commonly prescribed CysLT1 antagonist is montelukast (15)-3-[2-(2-hydroxy-2-propanyl) phenyl]propyl{sulfanyl)methyl]cyclopropyl}acetic acid. Montelukast is less effective than quininib at attenuating hyaloid vasculature development in zebrafish eyes. 20 μm montelukast is required to produce a significant reduction (15% fewer hyaloid vessels) versus 4 μm quininib (∼50% fewer hyaloid vessels) (Fig. 1D, data not shown). In contrast, 30 μm montelukast was more effective than 30 μm quininib at antagonizing LTD4-induced guinea pig lung strip contraction (89% versus 51%), and 10 μm montelukast or quininib reduced mouse aortic ring angiogenesis by an equivalent ∼40% (Fig. 6, C and D). Interestingly, 0.5 μm quininib significantly attenuated revascularization in the OIR model, whereas 0.5 μm montelukast had a negligible effect (Fig. 6E). In summary, CysLT1 antagonists can reduce angiogenesis, but, in the eye, quininib is significantly more effective than montelukast at attenuating ocular angiogenesis or revascularization in zebrafish and mice.
Downstream Inhibitors in the Cysteinyl Leukotriene Pathway Phenocopy Quininib
Downstream inhibitors of the cysteinyl leukotriene pathway (Fig. 7A) were tested to see whether they inhibited developmental angiogenesis in zebrafish. Drug treatment with the broad-spectrum PKC inhibitor Gö 6983 (10 μm) or the MEK 1/2 inhibitor TAK-733 (10 μm) inhibited the formation of primary hyaloid vessels in zebrafish (Fig. 7, B–E) by 43% and 38%, respectively, phenocopying quininib.
FIGURE 7.

Quininib inhibits cysteinyl leukotriene receptor signaling pathways. Investigation of signaling pathways reported to be downstream of cysteinyl leukotriene receptors (A) showed that known pharmacological inhibitors of this signaling pathway phenocopy quininib and that quininib can inhibit leukotriene D4 up-regulation of phospho-ERK. A, schematic (modified from Savari et al. (69)) showing the signaling pathway downstream of CysLTR (shown in cell membrane, cytoplasmic, and nuclear localization). B—E, drug treatment of transgenic Tg[fli1:EGFP] zebrafish from 2–5 dpf with 10 μm pan-PKC inhibitor Gö 6983 (C) or 10 μm MEK 1/2 inhibitor TAK-733 (D) inhibited the development of primary hyaloid vessels (green) compared with vehicle control (0.1% DMSO, B) in a significant manner (E, n = 20–30 larvae/compound, ***, p < 0.001). F, treatment of endothelial HMEC-1 cells with 10, 100, and 1000 nm LTD4 for 5 min up-regulated downstream ERK phosphorylation. G, pretreatment of HMEC-1 cells for 30 min with vehicle (V) control (0.1% DMSO), quininib, or the MEK 1/2 inhibitor TAK-733 followed by a 5-min stimulation with 500 nm LTD4 or vehicle (0.25% ethanol) showed that quininib and TAK-733 inhibited LTD4 induced phospho-ERK up-regulation. Shown is a representative Western blotting analysis. H, densitometry analysis of phospho-ERK expression normalized to ERK expression showing that quininib inhibited LTD4-induced phospho-ERK up-regulation (a decrease of 46% in phospho-ERK expression in vehicle versus quininib LTD4-stimulated HMEC-1 cells, n = 4 repetitions). One-way ANOVA showed that quininib inhibited LTD4-induced phospho-ERK up-regulation in a statistically significant manner. *, p < 0.05).
Quininib Affects CysLT1–2 Signaling
HMEC-1 endothelial cells exhibit an up-regulation of cysteinyl leukotriene signaling pathways, as evidenced by an increase in phospho-ERK 44/42 expression, when stimulated with 10, 100, or 1000 nm LTD4 for 5 min (Fig. 7F). Leukotriene D4-induced phospho-ERK 44/42 up-regulation is inhibited in the presence of quininib and the downstream MEK 1/2 inhibitor TAK-733 (Fig. 7G). Western blotting analyses show that quininib inhibits LTD4-induced phospho-ERK 44/42 up-regulation by 29% compared with vehicle control (0.1% DMSO and 0.25% ethanol) (Fig. 7H). This is in comparison with the MEK 1/2 inhibitor TAK-733, which completely abolishes phospho-ERK expression with or without the presence of LTD4.
Discussion
From phenotype-based screens of a randomized small molecule library, quininib was uncovered as a novel, robust inhibitor of angiogenesis in vivo. Quininib is an orthosteric cysteinyl leukotriene receptor 1 antagonist and a weak cysteinyl leukotriene receptor 2 antagonist. Overall, this research enhanced our understanding of the molecular pathways controlling ocular angiogenesis and identified drugs and molecular targets with potential to be developed into therapeutic agents for angiogenesis-related diseases.
Two studies previously identified inhibitors of zebrafish hyaloid vasculature development using phenotype-based chemical screens. Alvarez et al. (7) identified LY 294,002, a PI3K inhibitor, in a screen of 11 known angiogenic modulators. In a screen of ∼2000 bioactive drugs from the MicroSource Spectrum collection, Kitambi et al. (8) identified pyrogallin, an ATP-competitive inhibitor of JAK3, plus albendazole and mebendazole, both anti-helminthic medications that inhibit microtubule assembly. Thus, both studies screened existing bioactive compounds. Our study differs in that a randomized library (ChemBridge DiverSETTM) of ∼1800 molecules with predicted drug-like physiochemical properties was screened with the intent of identifying novel regulators of developmental angiogenesis in the eye.
Analysis in zebrafish, human, and rodent models demonstrates that quininib is an effective and well tolerated inhibitor of angiogenesis. Efficacy in the zebrafish model upon administration into the larval medium confirms desirable in vivo pharmacokinetic properties that support bioavailability beyond effective threshold concentrations in ocular tissue. Quininib inhibits the formation of newly forming but not existing vessels and so fits the properties of a vascular targeting agent and not a vascular disrupting agent (52). The anti-angiogenic effect of quininib is a specific pharmacological response and cannot be attributed to toxic effects or developmental delay in the models investigated. Quininib-treated larvae, however, present with a defective optokinetic response. In agreement, previous genetic studies report that zebrafish lacking ocular vasculature (cloche and silent heart mutants or VEGF-A morphants) exhibit reduced differentiation of retinal neurons and impaired synaptic processes (53, 54). The reduced visual behavior of quininib-treated zebrafish larvae is not an overt concern for further drug development because it likely reflects an indirect developmental defect that would not be encountered in adult eyes.
In mammalian models, quininib is well tolerated in mice and is effective at inhibiting angiogenesis in the mouse oxygen-induced retinopathy model of ocular angiogenesis via an intravitreal delivery route. Two phases of blood vessel growth occur following the relative hypoxia that occurs when these mice are returned to normoxia on P12. These phases are a revascularization (normal intraretinal vessel regrowth), which is similar to a recapitulation of developmental angiogenesis, and a preretinal pathological neovascularization (55). Both 0.5 and 3 μm quininib are effective at blocking retinal revascularization in the OIR model, with equivalent responses observed with 0.5 and 3 μm quininib, suggesting attainment of a maximum response plateau. Curiously, neovascularization in drug-treated eyes shows that the lower quininib dose slightly reduces neovascularization, whereas the higher dose increases the relative neovascular area. This may be due to the drug targeting one specific CysLT receptor at lower concentrations and more than one CysLT receptor at higher concentrations. In agreement, Barajas-Espinosa et al. (37) reported a similar finding when Cyslt2r knockout mice were exposed to the OIR experimental paradigm. P17 flat-mount retinas, the same time point analyzed in our study, exhibited both reduced revascularization and increased neovascularization. They postulated that the increased neovascularization is due to the compensatory up-regulation of CysLT1 in the knockout.
Adult Cysltr2 knockout mice show no retinal vascular phenotype compared with wild types (37). In agreement, preliminary analysis of isolectin-stained retinal flat mounts from P8 Cysltr1 or Cysltr2 knockout mice suggest that neither receptor alone is required for retinal developmental angiogenesis (data not shown). This supports the theory that cysteinyl leukotriene receptors exhibit functional redundancy.
Quininib acts as an orthosteric antagonist of the G protein-coupled receptors cysteinyl leukotriene receptor 1 and 2. Experimentally, we demonstrated quininib to significantly compete with the endogenous ligand LTD4 for binding to CysLT1 preferentially over CysLT2. Additionally, quininib significantly attenuated reporter activity from CysLT1 more than CysLT2. It may be that a higher concentration of quininib is required to elicit a response through CysLT2. These findings are in agreement with a medicinal chemistry report in 1992 by Zamboni et al. (51) who generated a series of CysLT1 antagonists by structural modification of 3(-2-quinolinyl-(E)-ethenyl)pyridine. A compound equivalent to the free-base version of quininib generates an IC50 of >50 μm for competition with LTD4 binding to guinea pig lung strips (51). The significantly lower IC50 of 1.4 μm for quininib reported here likely reflects that the human CysLT1- or CysLT2-overexpressing cells more specifically report on these receptors compared with the more heterogeneous guinea pig lung strips. Alternatively, the quininib hydrochloric salt used here may result in greater activity than the amine form. Finally, the chemical structure of quininib (molecular weight, 283.75 g/mol) extensively overlaps with a portion of the much larger, clinically approved CysLT1 antagonist montelukast (Merck, MW 586.18g/mol) (32). Recently montelukast has been reported to reduce neuroinflammation in an aged rat model, acting via inhibition of a separate cysteinyl leukotriene receptor, GPR17 (56), and a fourth cysteinyl leukotriene receptor, GPR99, has been proposed (26). In genetic loss-of function models, cysteinyl leukotriene receptors demonstrate cognate compensatory up-regulation (37, 39, 57). Thus, there are exciting future opportunities to decipher the cysLT receptors exhibiting co-regulatory expression to understand the underlying control mechanisms and to determine the combinations of cysLT receptors that regulate physiological phenotypes.
Cysteinyl leukotrienes have been reported previously to up-regulate ERK phosphorylation (38, 39). Here we show that drug treatment with quininib inhibits LTD4-induced phospho-ERK up-regulation, which is completely blocked by the more downstream MEK 1/2 inhibitor TAK-733. TAK-733 and the broad-spectrum PKC inhibitor Gö 6983 phenocopy the effects of quininib on developing hyaloid vessels in zebrafish.
On the basis of a virtual drug screen, the quininib E-isomer was reported in 2008 to inhibit endothelin-converting enzyme 2 (ECE-2) (49). This target is unlikely to mediate the anti-angiogenic activity of quininib because alternative endothelin receptor antagonists do not phenocopy quininib (data not shown). In addition, recently Gupta et al. (50) reported that the quininib Z-isomer more potently inhibits ECE-2 than the E-isomer, which does not correlate with our finding of greater anti-angiogenic activity of the E-isomer (50). Also of note is that quininib does not directly inhibit the activity of any VEGF receptor. VEGF, particularly in the eye, is a key target to treat ocular neovascularization using humanized antibodies (e.g. Avastin®, Lucentis®) or soluble decoy receptors (Eylea®), and many small-molecule VEGF receptor inhibitors are available (e.g. the tyrosine kinase inhibitors AL 39324, PTK787, and Tg 100801; reviewed in Ref. 58). Thus, the distinct cysteinyl leukotriene pathway-mediated anti-angiogenic mechanism of action of quininib offers potential for additive effects with anti-VEGFs or alternative therapeutic targets for patients non-responsive to current anti-VEGFs (15).
Although this is the first report demonstrating a significant anti-angiogenic effect of CysLT1 antagonists on ocular angiogenesis, accumulating evidence confirms a key role for cysteinyl leukotrienes in angiogenesis. For example, the cysteinyl leukotrienes LTD4 and LTC4 stimulate angiogenesis in endothelial cells (59, 60). Of particular relevance, the CysLT1 antagonist montelukast, but not the CysLT2 antagonist BayCysLT2, blocks the LTD4-induced migratory phenotype of the human endothelial cell line EA.hy926 (61). Similarly, montelukast, but not BayCysLT2, inhibits basal microvessel outgrowth from rat thoracic aortic rings, and both drugs inhibit an LTD4-induced model of aortic ring angiogenesis (62). In vivo, Savari et al. (63) recently reported CysLT1 receptor antagonists to reduce tumor angiogenesis in a mouse xenograft model of colorectal cancer, and this was concomitant with reduced tumor VEGF levels. Interestingly, the recognized anti-asthmatic effect of montelukast is modulated by VEGF polymorphisms, and montelukast has been postulated to attenuate airway inflammation by reducing VEGF expression (64, 65). Focusing on the eye, Barajas-Espinosa et al. (37), in defining a role for CysLT2 in retinal permeability and neovascularization, also demonstrated expression of CysLT1 in the retina, which is significantly increased upon knockout of Cysltr2. Here we demonstrate that the zebrafish cysltr1 and cysltr2 genes are expressed in the eye during hyaloid vasculature development and that the CysLT1 antagonist quininib can attenuate ocular angiogenesis. Intriguingly, quininib is significantly more potent than montelukast at inhibiting angiogenesis in the eye. Pharmacokinetic reasons potentially explain these differences. Quininib is significantly smaller than montelukast, which may facilitate greater absorption into the zebrafish eye and greater penetration across the mouse retinal layers, resulting in enhanced bioavailability and efficacy. Additionally, montelukast is known to undergo light-induced isomerization, which produces structurally related but inactive products (66).
In summary, we demonstrate a novel role for cysteinyl leukotriene receptor antagonists in attenuating ocular angiogenesis. Cysteinyl leukotrienes are proinflammatory agents that increase vascular permeability (26, 29) through receptor-mediated activation of phospholipase C, phosphoinositide-3-kinase, and/or extracellular signal-related kinases (29, 67, 68). This increased permeability can increase extravasation of proangiogenic factors that can remodel the extracellular environment and promote growth of new vessels. CysLTs provide a novel pathway from which to enhance our understanding of developmental angiogenesis and a novel therapeutic target in angiogenesis-driven disease.
Author Contributions
A. L. R. conducted validation, visual behavior, analogue, and time-course experiments in zebrafish; safety and efficacy experiments in mice; and target profiling analyses, analyzed and interpreted the results, and wrote the paper with Y. A. and B. N. K. Y. A. and N. W. conducted the initial drug discovery and safety experiments in zebrafish and analyzed the results. T. S. performed zebrafish behavioral assays, expression analyses, and downstream cysLT signaling experiments and analyzed the results. C. B. performed HMEC-1 cell viability studies and the in vitro tubule formation assay. C. K. performed the target profiling analysis and helped with mouse OIR experiments. A. J. S. performed screening of downstream cysteinyl leukotriene inhibitors in zebrafish. C. M. and V. H. Y. W. performed a subset of the mouse OIR experiments, analyzed the results, and assisted with the interpretation of results. O. G. performed the mouse aortic ring experiments. S. M. performed the ARPE-19 cell viability studies. J. O. and A. S. provided animal models. G. G. performed analysis of OIR mouse retinas. A. W. S. provided animal models and assisted with analysis and interpretation of the results. B. N. K. conceived the idea for the project, assisted with analysis and interpretation of the results, and wrote the paper with A. L. R. and Y. A.
Acknowledgments
We thank the Conway Institute Imaging Core Technology, especially Dimitri Scholz, for assistance with montaging and Tiina O'Neill and Kasia Welzel for assistance with embedding and sectioning of samples; Pat Guiry for help with interpreting chemical structures; Ken O'Halloran for access to hyperoxia equipment; and Adrian Murphy, Jacintha O'Sullivan, Nils Ohnesorge, and Oliver Blacque for discussions and comments on the manuscript.
This work was supported by funding from an Enterprise Ireland proof of concept award, an Enterprise Ireland Commercialisation Fund award, a Health Research Board Ireland Health Research Award, and a Science Foundation Ireland Technology innovation development award. B. N. K. and Y. A. are authors on granted patent WO2012095836 A1. A. R., C. K., and B. N. K. are authors on patent filing WO2014012889 A1.
- quininib
- 2-[(E)-2-(quinolin-2-yl)vinyl]phenol
- CysLT
- cysteinyl leukotriene
- DMSO
- dimethyl sulfoxide
- OIR
- oxygen-induced retinopathy
- P12
- postnatal day 12
- ANOVA
- analysis of variance
- HV
- hyaloid vessel
- dpf
- day(s) post-fertilization.
References
- 1. Fruttiger M. (2007) Development of the retinal vasculature. Angiogenesis 10, 77–88 [DOI] [PubMed] [Google Scholar]
- 2. Cheung N., Mitchell P., and Wong T. Y. (2010) Diabetic retinopathy. Lancet 376, 124–136 [DOI] [PubMed] [Google Scholar]
- 3. Jager R. D., Mieler W. F., and Miller J. W. (2008) Age-related macular degeneration. N. Engl. J. Med. 358, 2606–2617 [DOI] [PubMed] [Google Scholar]
- 4. Hellström A., Smith L. E., and Dammann O. (2013) Retinopathy of prematurity. Lancet 382, 1445–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rehak M., and Wiedemann P. (2010) Retinal vein thrombosis: pathogenesis and management. J. Thromb. Haemost. 8, 1886–1894 [DOI] [PubMed] [Google Scholar]
- 6. Gariano R. F., and Gardner T. W. (2005) Retinal angiogenesis in development and disease. Nature 438, 960–966 [DOI] [PubMed] [Google Scholar]
- 7. Alvarez Y., Astudillo O., Jensen L., Reynolds A. L., Waghorne N., Brazil D. P., Cao Y., O'Connor J. J., and Kennedy B. (2009) Selective inhibition of retinal angiogenesis by targeting PI3 kinase. PLoS ONE 4, e7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitambi S. S., McCulloch K. J., Peterson R. T., and Malicki J. J. (2009) Small molecule screen for compounds that affect vascular development in the zebrafish retina. Mech. Dev. 126, 464–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Folkman J. (1997) in Regulation of Angiogenesis (Goldberg I., and Rosen E., eds.) pp. 1–8, Birkhäuser, Basel, Switzerland [Google Scholar]
- 10. Frost & Sullivan (June 7, 2011) Analysis of the US Retinal Therapeutics Market: Improvements in Administration and Efficacy Drive Growth, Frost & Sullivan Report NC77–52
- 11. Frost & Sullivan (July 3, 2010) European Ophthalmic Pharmaceuticals Market, Frost & Sullivan Report M4AC-52
- 12. CATT Research Group, Martin D. F., Maguire M. G., Ying G. S., Grunwald J. E., Fine S. L., and Jaffe G. J. (2011) Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 364, 1897–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heier J. S., Brown D. M., Chong V., Korobelnik J.-F., Kaiser P. K., Nguyen Q. D., Kirchhof B., Ho A., Ogura Y., Yancopoulos G. D., Stahl N., Vitti R., Berliner A. J., Soo Y., Anderesi M., Groetzbach G., Sommerauer B., Sandbrink R., Simader C., Schmidt-Erfurth U., and VIEW 1 and VIEW 2 Study Groups. (2012) Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 119, 2537–2548 [DOI] [PubMed] [Google Scholar]
- 14. Rosenfeld P. J., Brown D. M., Heier J. S., Boyer D. S., Kaiser P. K., Chung C. Y., Kim R. Y., and MARINA Study Group (2006) Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 355, 1419–1431 [DOI] [PubMed] [Google Scholar]
- 15. Rofagha S., Bhisitkul R. B., Boyer D. S., Sadda S. R., Zhang K., and SEVEN-UP Study Group (2013) Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 120, 2292–2299 [DOI] [PubMed] [Google Scholar]
- 16. Diabetic Retinopathy Clinical Research Network, Elman M. J., Qin H., Aiello L. P., Beck R. W., Bressler N. M., Ferris F. L. 3rd, Glassman A. R., Maturi R. K., and Melia M. (2012) Intravitreal ranibizumab for diabetic macular edema with prompt vs. deferred laser treatment: 3-year randomized trial results. Ophthalmology 119, 2312–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wangsa-Wirawan N. D., and Linsenmeier R. A. (2003) Retinal oxygen: fundamental and clinical aspects. Arch. Ophthalmol. 121, 547–557 [DOI] [PubMed] [Google Scholar]
- 18. Saint-Geniez M., and D'Amore P. (2004) Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 48, 1045–1058 [DOI] [PubMed] [Google Scholar]
- 19. Fruttiger M. (2002) Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest. Ophthalmol. Vis. Sci. 43, 522–527 [PubMed] [Google Scholar]
- 20. Shastry B. S. (2009) Persistent hyperplastic primary vitreous: congenital malformation of the eye. Clin. Exp. Ophthalmol. 37, 884–890 [DOI] [PubMed] [Google Scholar]
- 21. Alvarez Y., Cederlund M. L., Cottell D. C., Bill B. R., Ekker S. C., Torres-Vazquez J., Weinstein B. M., Hyde D. R., Vihtelic T. S., and Kennedy B. N. (2007) Genetic determinants of hyaloid and retinal vasculature in zebrafish. BMC Dev. Biol. 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hartsock A., Lee C., Arnold V., and Gross J. M. (2014) In vivo analysis of hyaloid vasculature morphogenesis in zebrafish: a role for the lens in maturation and maintenance of the hyaloid. Dev. Biol. 394, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalén M., Wallgard E., Asker N., Nasevicius A., Athley E., Billgren E., Larson J. D., Wadman S. A., Norseng E., Clark K. J., He L., Karlsson-Lindahl L., Häger A.-K., Weber H., Augustin H., Samuelsson T., Kemmet C. K., Utesch C. M., Essner J. J., Hackett P. B., and Hellström M. (2009) Combination of reverse and chemical genetic screens reveals angiogenesis inhibitors and targets. Chem. Biol. 16, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peterson R. T., Link B. A., Dowling J. E., and Schreiber S. L. (2000) Small molecule developmental screens reveal the logic and timing of vertebrate development. Proc. Natl. Acad. Sci. U.S.A. 97, 12965–12969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rennekamp A. J., and Peterson R. T. (2015) 15 years of zebrafish chemical screening. Curr. Opin. Chem. Biol. 24, 58–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanaoka Y., Maekawa A., and Austen K. F. (2013) Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J. Biol. Chem. 288, 10967–10972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh R. K., Gupta S., Dastidar S., and Ray A. (2010) Cysteinyl leukotrienes and their receptors: molecular and functional characteristics. Pharmacology 85, 336–349 [DOI] [PubMed] [Google Scholar]
- 28. Kanaoka Y., and Boyce J. A. (2004) Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J. Immunol. 173, 1503–1510 [DOI] [PubMed] [Google Scholar]
- 29. Bäck M., Powell W. S., Dahlén S. E., Drazen J. M., Evans J. F., Serhan C. N., Shimizu T., Yokomizo T., and Rovati G. E. (2014) Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br. J. Pharmacol. 171, 3551–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ciana P., Fumagalli M., Trincavelli M. L., Verderio C., Rosa P., Lecca D., Ferrario S., Parravicini C., Capra V., Gelosa P., Guerrini U., Belcredito S., Cimino M., Sironi L., Tremoli E., Rovati G. E., Martini C., and Abbracchio M. P. (2006) The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 25, 4615–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meltzer E. O., Malmstrom K., Lu S., Prenner B. M., Wei L. X., Weinstein S. F., Wolfe J. D., and Reiss T. F. (2000) Concomitant montelukast and loratadine as treatment for seasonal allergic rhinitis: a randomized, placebo-controlled clinical trial. J. Allergy Clin. Immunol. 105, 917–922 [DOI] [PubMed] [Google Scholar]
- 32. Reiss T. F., Altman L. C., Chervinsky P., Bewtra A., Stricker W. E., Noonan G. P., Kundu S., and Zhang J. (1996) Effects of montelukast (MK-0476), a new potent cysteinyl leukotriene (LTD4) receptor antagonist, in patients with chronic asthma. J. Allergy Clin. Immunol. 98, 528–534 [DOI] [PubMed] [Google Scholar]
- 33. Kyritsis N., Kizil C., Zocher S., Kroehne V., Kaslin J., Freudenreich D., Iltzsche A., and Brand M. (2012) Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338, 1353–1356 [DOI] [PubMed] [Google Scholar]
- 34. Qian X.-D., Wei E.-Q., Zhang L., Sheng W.-W., Wang M.-L., Zhang W.-P., and Chen Z. (2006) Pranlukast, a cysteinyl leukotriene receptor 1 antagonist, protects mice against brain cold injury. Eur. J. Pharmacol. 549, 35–40 [DOI] [PubMed] [Google Scholar]
- 35. Wang X. Y., Tang S. S., Hu M., Long Y., Li Y. Q., Liao M. X., Ji H., and Hong H. (2013) Leukotriene D4 induces amyloid-β generation via CysLT1R-mediated NF-κB pathways in primary neurons. Neurochem. Int. 62, 340–347 [DOI] [PubMed] [Google Scholar]
- 36. Kanaoka Y., and Boyce J. A. (2014) Cysteinyl leukotrienes and their receptors: emerging concepts. Allergy Asthma Immunol. Res. 6, 288–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barajas-Espinosa A., Ni N. C., Yan D., Zarini S., Murphy R. C., and Funk C. D. (2012) The cysteinyl leukotriene 2 receptor mediates retinal edema and pathological neovascularization in a murine model of oxygen-induced retinopathy. FASEB J. 26, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 38. Duah E., Adapala R. K., Al-Azzam N., Kondeti V., Gombedza F., Thodeti C. K., and Paruchuri S. (2013) Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT(2) and CysLT(1) receptors. Sci. Rep. 3, 3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang Y., Borrelli L. A., Kanaoka Y., Bacskai B. J., and Boyce J. A. (2007) CysLT(2) receptors interact with CysLT(1) receptors and down-modulate cysteinyl leukotriene-dependent mitogenic responses of mast cells. Blood 110, 3263–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lipinski C., Lombardo F., Dominy B., and Feeney P. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–26 [DOI] [PubMed] [Google Scholar]
- 41. Lawson N. D., and Weinstein B. M. (2002) In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307–318 [DOI] [PubMed] [Google Scholar]
- 42. Brockerhoff S. (2006) Measuring the optokinetic response of zebrafish larvae. Nat. Protoc. 1, 2448–2451 [DOI] [PubMed] [Google Scholar]
- 43. Sasore T., and Kennedy B. (2014) Deciphering combinations of PI3K/AKT/mTOR pathway drugs augmenting anti-angiogenic efficacy in vivo. PLoS ONE 9, e105280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baker M., Robinson S. D., Lechertier T., Barber P. R., Tavora B., D'Amico G., Jones D. T., Vojnovic B., and Hodivala-Dilke K. (2012) Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 7, 89–104 [DOI] [PubMed] [Google Scholar]
- 45. Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., and Smith L. E. (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D'Amato R., Sullivan R., and D'Amore P. A. (1994) Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- 47. Gardiner T. A., Gibson D. S., de Gooyer T. E., de la Cruz V. F., McDonald D. M., and Stitt A. W. (2005) Inhibition of tumor necrosis factor-α improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am. J. Pathol. 166, 637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sidman R. L., Li J., Lawrence M., Hu W., Musso G. F., Giordano R. J., Cardó-Vila M., Pasqualini R., and Arap W. (2015) The peptidomimetic Vasotide targets two retinal VEGF receptors and reduces pathological angiogenesis in murine and nonhuman primate models of retinal disease. Sci. Transl. Med. 7, 309ra165–309ra165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gagnidze K., Sachchidanand, Rozenfeld R., Mezei M., Zhou M.-M., and Devi L. A. (2008) Homology modeling and site-directed mutagenesis to identify selective inhibitors of endothelin-converting enzyme-2. J. Med. Chem. 51, 3378–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gupta A., Gomes I., Wardman J., and Devi L. A. (2014) Opioid receptor function is regulated by post-endocytic peptide processing. J. Biol. Chem. 289, 19613–19626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zamboni R., Belley M., Champion E., Charette L., DeHaven R., Frenette R., Gauthier J. Y., Jones T. R., Leger S., and Masson P. (1992) Development of a novel series of styrylquinoline compounds as high-affinity leukotriene D4 receptor antagonists: synthetic and structure-activity studies leading to the discovery of (+−)-3-[[[3-[2-(7-chloro-2-quinolinyl)-(E)-ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propionic acid. J. Med. Chem. 35, 3832–3844 [DOI] [PubMed] [Google Scholar]
- 52. Lorusso P. M., Boerner S. A., and Hunsberger S. (2011) Clinical development of vascular disrupting agents: what lessons can we learn from ASA404? J. Clin. Oncol. 29, 2952–2955 [DOI] [PubMed] [Google Scholar]
- 53. Dhakal S., Stevens C., Weiss O., Inbal A., and Stenkamp D. (2013) Role of the early ocular vasculature in regulation of retinal neurogenesis. Invest. Ophthalmol. Vis. Sci. 54, Abstr. 5145 [Google Scholar]
- 54. Dhakal S., Stevens C. B., Sebbagh M., Weiss O., Frey R. A., Adamson S., Shelden E. A., Inbal A., and Stenkamp D. L. (2015) Abnormal retinal development in Cloche mutant zebrafish. Dev. Dyn. 244, 1439–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., and Smith L. E. (2010) The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marschallinger J., Schaffner I., Klein B., Gelfert R., Rivera F. J., Illes S., Grassner L., Janssen M., Rotheneichner P., Schmuckermair C., Coras R., Boccazzi M., Chishty M., Lagler F. B., Renic M., Bauer H.-C., Singewald N., Blumcke I., Bogdahn U., Couillard-Despres S., Lie D. C., Abbracchio M. P., and Aigner L. (2015) Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 6, 8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maekawa A., Kanaoka Y., Xing W., and Austen K. F. (2008) Functional recognition of a distinct receptor preferential for leukotriene E(4) in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 16695–16700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reynolds A. L., Kent D., and Kennedy B. N. (2014) Ocular neovascularisation: current and emerging therapies. Adv. Exp. Med. Biol. 801, 797–804 [DOI] [PubMed] [Google Scholar]
- 59. Tsopanoglou N. E., Pipili-Synetos E., and Maragoudakis M. E. (1994) Leukotrienes C4 and D4 promote angiogenesis via a receptor-mediated interaction. Eur. J. Pharmacol. 258, 151–154 [DOI] [PubMed] [Google Scholar]
- 60. Kanayasu T., Nakao-Hayashi J., Asuwa N., Morita I., Ishii T., Ito H., and Murota S. (1989) Leukotriene C4 stimulates angiogenesis in bovine carotid artery endothelial cells in vitro. Biochem. Biophys. Res. Commun. 159, 572–578 [DOI] [PubMed] [Google Scholar]
- 61. Yuan Y.-M., Fang S.-H., Qian X.-D., Liu L.-Y., Xu L.-H., Shi W.-Z., Zhang L.-H., Lu Y.-B., Zhang W.-P., and Wei E.-Q. (2009) Leukotriene D4 stimulates the migration but not proliferation of endothelial cells mediated by the cysteinyl leukotriene CysLT1 receptor via the extracellular signal-regulated kinase pathway. J. Pharmacol. Sci. 109, 285–292 [DOI] [PubMed] [Google Scholar]
- 62. Xu L., Zhang L., Liu L., Fang S., Lu Y., Wei E., and Zhang W. (2010) Involvement of cysteinyl leukotriene receptors in angiogenesis in rat thoracic aortic rings. Pharmazie 65, 750–754 [PubMed] [Google Scholar]
- 63. Savari S., Liu M., Zhang Y., Sime W., and Sjölander A. (2013) CysLT1R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS ONE 8, e73466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Balantic M., Rijavec M., Skerbinjek Kavalar M., Suskovic S., Silar M., Kosnik M., and Korosec P. (2012) Asthma treatment outcome in children is associated with vascular endothelial growth factor A (VEGFA) polymorphisms. Mol. Diagn. Ther. 16, 173–180 [DOI] [PubMed] [Google Scholar]
- 65. Lee K. S., Kim S. R., Park H. S., Jin G. Y., and Lee Y. C. (2004) Cysteinyl leukotriene receptor antagonist regulates vascular permeability by reducing vascular endothelial growth factor expression. J. Allergy Clin. Immunol. 114, 1093–1099 [DOI] [PubMed] [Google Scholar]
- 66. Smith G. A., Rawls C. M., and Kunka R. L. (2004) An automated method for the determination of montelukast in human plasma using dual-column HPLC analysis and peak height summation of the parent compound and its photodegradation product. Pharm. Res. 21, 1539–1544 [DOI] [PubMed] [Google Scholar]
- 67. Kim M. H., Lee Y. J., Kim M. O., Kim J. S., and Han H. J. (2010) Effect of leukotriene D4 on mouse embryonic stem cell migration and proliferation: involvement of PI3K/Akt as well as GSK-3β/β-catenin signaling pathways. J. Cell Biochem. 111, 686–698 [DOI] [PubMed] [Google Scholar]
- 68. Salim T., Sand-Dejmek J., and Sjölander A. (2014) The inflammatory mediator leukotriene D4 induces subcellular β-catenin translocation and migration of colon cancer cells. Exp. Cell Res. 321, 255–266 [DOI] [PubMed] [Google Scholar]
- 69. Savari S., Vinnakota K., Zhang Y., and Sjölander A. (2014) Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J. Gastroenterol. 20, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]