

Abstract
Cholesterol can be a major carbon source for Mycobacterium tuberculosis during infection, both at an early stage in the macrophage phagosome and later within the necrotic granuloma. KstR is a highly conserved TetR family transcriptional repressor that regulates a large set of genes responsible for cholesterol catabolism. Many genes in this regulon, including kstR, are either induced during infection or are essential for survival of M. tuberculosis in vivo. In this study, we identified two ligands for KstR, both of which are CoA thioester cholesterol metabolites with four intact steroid rings. A metabolite in which one of the rings was cleaved was not a ligand. We confirmed the ligand-protein interactions using intrinsic tryptophan fluorescence and showed that ligand binding strongly inhibited KstR-DNA binding using surface plasmon resonance (IC50 for ligand = 25 nm). Crystal structures of the ligand-free form of KstR show variability in the position of the DNA-binding domain. In contrast, structures of KstR·ligand complexes are highly similar to each other and demonstrate a position of the DNA-binding domain that is unfavorable for DNA binding. Comparison of ligand-bound and ligand-free structures identifies residues involved in ligand specificity and reveals a distinctive mechanism by which the ligand-induced conformational change mediates DNA release.
Keywords: bacterial metabolism, cholesterol metabolism, Mycobacterium tuberculosis, protein structure, transcription repressor, CoA thioester
Introduction
Tuberculosis remains a global threat to human health due to the emergence of extremely drug-resistant forms of Mycobacterium tuberculosis and co-infection with HIV (1). Elucidation of metabolic networks essential to the pathogenesis of M. tuberculosis is an important part of the quest to identify new drug target candidates that can be exploited to tackle extremely drug-resistant tuberculosis. In particular, M. tuberculosis possesses an unusual ability to metabolize lipids, and its genome is heavily orientated toward this task with the involvement of a remarkable number of genes, ∼250, constituting around 6% of its genome (2).
Cholesterol, the dominant lipid accumulated in M. tuberculosis-induced foamy macrophages, is a key growth substrate for M. tuberculosis in the intraphagosomal environment and is also important to the bacterium in necrotizing granulomas in late stage infection (3–5). M. tuberculosis is capable of utilizing cholesterol as a sole carbon source for both energy synthesis and assimilation into membrane lipids (3, 6). Many genes in the cholesterol degradation pathway are up-regulated or are essential for M. tuberculosis infection in various models of disease (3, 4, 7–11). Disruption of genes encoding cholesterol catabolic enzymes, either through genetic manipulation or chemically, inhibits growth of the bacterium in both macrophage and animal models, and in vitro experiments suggest that this growth inhibition may be due to the toxicity of accumulated intermediates (4, 9, 12).
The cholesterol degradation pathway is regulated by two TFRs,7 KstR and KstR2 (13, 14). These regulate two distinct regulons and negatively autoregulate their own expression. KstR regulates the expression of the genes involved in the transmembrane transport of cholesterol, β-oxidation of the cholesterol aliphatic side chain, and opening and removal of steroidal rings A and B, whereas KstR2 regulates subsequent steps that presumably degrade the C and D rings of the steroid (15). As is common in TFRs, KstR and KstR2 show highest sequence similarity in their N-terminal DNA-binding domains (DBDs) and are less similar in their C-terminal ligand-binding domains (LBDs). KstR and KstR2 act independently of each other, indicating that they are triggered by different ligands (14). Intriguingly, KstR itself is essential for survival in mouse models (16). Although the reason for the essentiality of KstR is not fully understood, it may be because cholesterol catabolism is essential for virulence (12).
TetR family members are known for their ability to bind diverse ligands: one of the best characterized examples is QacR, the regulator of a multidrug export pump, which responds to more than 16 different ligands (17, 18). The unusually large regulon of KstR (74 genes in M. tuberculosis) and the presence of genes within it that are involved in growth on palmitate suggest that the repressor may also control the metabolism of lipids other than just cholesterol (13) and is therefore likely to be induced by more than one compound. In the first investigation of possible cognate ligands for KstR in Mycobacterium smegmatis, García-Fernández et al. (19) determined that cholesterol and cholest-4-en-3-one, the entry compound and initial metabolite of the pathway (Fig. 1), respectively, were unable to induce the release of KstR from its operator. In contrast, M. smegmatis KstR primarily responded to 3-oxocholest-4-en-26-oic acid (3OChA). Cholesterol is practically insoluble in water, so the extent to which these results are affected by the relative solubility of these compounds is unclear. Curiously, M. smegmatis KstR appears unaffected by 4-androstene-3,17-dione and 1,4-androstadiene-3,17-dione, which are further metabolized sterol compounds that lack an alkyl chain.
FIGURE 1.
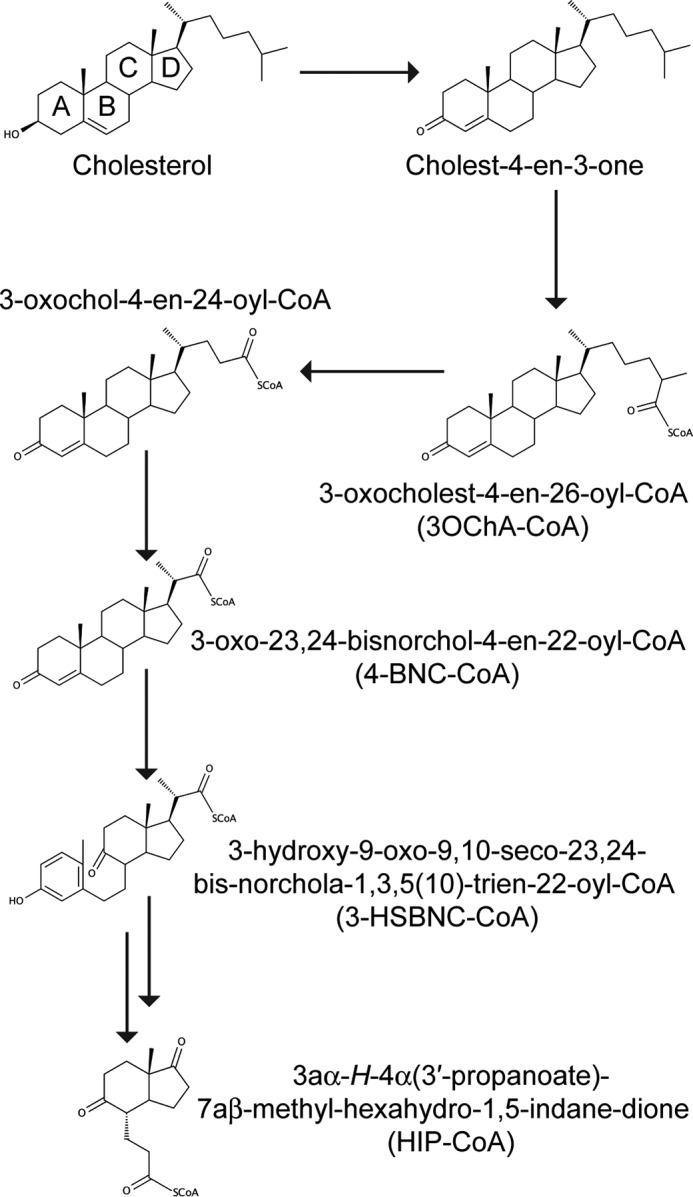
Schematic of cholesterol degradation in M. tuberculosis. A–D refer to rings A–D.
Casabon et al. (15) established that the cognate ligand for M. tuberculosis KstR2 is the coenzyme A (CoA) thioester of the two-ring sterol metabolite 3aα-H-4α(3-propanoate)-7aβ-methylhexahydro-1,5-indanedione (HIP). The molecular determinants of this interaction were delineated in a subsequent study by Crowe et al. (20). We therefore set out to establish whether the CoA thioester adducts of early metabolites of cholesterol would be functional inducers of KstR regulation and to understand the molecular details of their mode of action.
Experimental Procedures
Chemical and Reagents
ATP, CoASH, and 2-hydroxypropyl-β-cyclodextrin were purchased from Sigma-Aldrich. 3OChA, 3-oxo-23,24-bisnorchol-4-en-22-oic acid (4-BNC), and 3-hydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oic acid were purchased from Steraloids, Inc. (Newport, RI). Restriction enzymes and the Expand High Fidelity PCR System were purchased from New England Biolabs (Ipswich, MA) and Roche Applied Science, respectively. Oligonucleotides for amplifying KstR were purchased from Integrated DNA Technologies (San Diego, CA). All other reagents were of HPLC or analytical grade. Water for buffers was purified using a Barnstead Nanopure Diamond system (Dubuque, IA) to a resistivity of at least 18 megaohms·cm−1. Recombinant tobacco etch virus protease was expressed and purified in house. HiTrap chelating affinity columns were purchased from Amersham Biosciences. Amicon® Ultra centrifugal filters were purchased from Merck Millipore. Size exclusion columns and surface plasmon resonance (SPR) consumables were purchased from GE Healthcare.
Preparation of CoA Thioesters
3-Oxo-23,24-bisnorchol-4-en-22-oyl-CoA (4-BNC-CoA), 3-oxocholest-4-en-26-oyl-CoA (3OCh-CoA), and 3-hydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oyl-CoA (3-HSBNC-CoA) were prepared essentially as described previously (21–23). M. tuberculosis His6-FadD19 was obtained and used to synthesize 3OCh-CoA from 3OChA as described previously (23). Briefly, 0.6 mg of 3OCh dissolved in 25% low molecular weight 2-hydroxypropyl-β-cyclodextrin was mixed with 2 mg of M. tuberculosis FadD19 for 13 h at room temperature. Proteins were removed by precipitation with methanol at 4 °C for an hour. This mixture was filtered before being deposited onto a 60 mg/3 ml Strata-X 33-μm polymeric reversed phase solid phase extraction column (Phenomenex, Torrance, CA) conditioned with 2 ml of methanol and equilibrated with 2 ml of Nanopure water. The column was washed with 1.5 ml of 5% methanol and then with 1 ml of 90% methanol. Unreacted 3ChOA was removed using 3 ml of 2% formic acid in acetonitrile followed by 1 ml of acetonitrile. 3ChO-CoA was eluted using 4 ml of 2% formic acid in methanol, 1 ml of methanol, and 2.5 ml of 2% formic acid in methanol in succession. The eluate was dried under N2 and suspended in 25% β-cyclodextrin. The purity of the CoA thioester was verified by HPLC, eluting as a single peak from a Luna 3-μm PFP(2) 50 × 4.6-mm column (Phenomenex) equilibrated in 0.1 m ammonium acetate, pH 4.5, and run with a linear gradient to 98% methanol. 3ChO-CoA was quantified by recording its spectrum in water and assuming the same extinction coefficient as for 4-BNC-CoA (ϵ248 = 17,200 m−1 cm−1 (21)). 3aα-H-4α(3′-propanoyl-CoA)-7aβ-methylhexahydro-1,5-indanedione (HIP-CoA) was prepared as reported previously (15, 20).
Gene Cloning
Two plasmid constructs were used in this study to produce recombinant KstR protein. To produce His6-tagged KstR, the kstR open reading frame from M. tuberculosis (Rv3574) was cloned into pET30a, resulting in the pSK35 plasmid as described elsewhere (13). To produce KstR fused with N-terminally His6-tagged maltose-binding protein (MBP), the kstR coding region was amplified using a two-step nested PCR approach using primers listed in Table 1 and cloned into the expression vector pDEST-566 using the Gateway® cloning system (Invitrogen) as described in Moreland et al. (24). Briefly, the first PCR step created a tobacco etch virus cleavage site at the 5′-end of kstR, whereas the second PCR step introduced a recombination site to both ends of the gene. The final PCR product was introduced into the donor vector pDONR221 through a recombination step (BP reaction). The resulting construct pDONR221-kstR was sequence-verified before introducing the gene into the entry vector pDEST-566 via a second recombination event (LR reaction). The final construct was verified by digestion with the restriction enzyme BsrGI.
TABLE 1.
Oligonucleotide sequences
The underlined residues indicate the common sequence in the gene-specific and generic primers used for Gateway cloning.
| Name | Sequence |
|---|---|
| PCR 1 | |
| Forward gene-specific primer | 5′-GGCAGCGGCGCGGTGGCGGTACTTGCCGAGTCCGAG-3′ |
| Reverse gene-specific primer | 5′-AAAGCTGGGTGCTAGGCGCTGTCTTGATCGCCGAT-3′ |
| PCR 2 | |
| Forward generic primer | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAACCTGTATTTTCAGGGCAGCGGCGCG-3′ |
| Reverse generic primer | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTG-3′ |
| EMSA | |
| Forward strand | 5′-TGCCCACTAGAACGTGTTCTAATAGTGCT-3′ |
| Reverse strand | 5′-AGCACTATTAGAACACGTTCTAGTGGGCA-3′ |
| SPR | |
| Forward strand | Biotin-5′-TGCCCACTAGAACGTGTTCTAATAGTGC-3′ |
| Reverse strand | 5′-GCACTATTAGAACACGTTCTAGTGGGCA-3′ |
Expression and Purification of KstR
Both KstR constructs were expressed in Escherichia coli BL21(DE3). The E. coli culture harboring the plasmid was grown in autoinduction medium ZYM-5052 (25) at 37 °C until A600 reached 0.6–0.8 and then transferred to 18 °C for incubation overnight. Induced cells were harvested by centrifugation (4000 × g, 10 min, 4 °C), resuspended in lysis buffer (20 mm HEPES, 150 mm NaCl, pH 7.4) containing 10 mm imidazole and passed through a cell disruptor (ConstantSystem) at 18 kp.s.i. The lysate was centrifuged (16,000 × g, 30 min, 4 °C), and the soluble KstR present in the supernatant was recovered using a Ni2+-charged HiTrap chelating column. Recombinant KstR was eluted from the column in the same buffer containing 250 mm imidazole and mixed with recombinant tobacco etch virus protease to cleave the affinity tag. To remove the imidazole, the mixture was dialyzed three times against 1000 ml of lysis buffer containing 0.5 mm tris(2-carboxyethyl)phosphine. The mixture was then passed through the Ni2+-charged HiTrap column again. The cleaved KstR was eluted in lysis buffer containing 20 mm imidazole, whereas the cleaved His6 tag and tobacco etch virus protease remained bound on the column. KstR protein containing <5% other proteins (as judged by SDS-PAGE) was concentrated and exchanged back into lysis buffer using an Amicon Ultra filter (molecular mass cutoff, 10 kDa), snap frozen in liquid nitrogen, and stored at −80 °C.
Size Exclusion Chromatography with Multiangle Light Scattering (SEC-MALS)
SEC-MALS was used to determine the solution molecular mass of KstR. Peak separation was performed using a Superdex 200 10/300 GL column connected to a Dionex HPLC system. The column was equilibrated with buffer containing 20 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm sodium azide before being loaded with 100 μl of 2.5–5 mg/ml KstR. Multiangle light scattering was measured with an SLD7000 MALS detector and a Shodex RI-101 refractive index detector. Data were analyzed using PSS WinGPC Unichrom software.
Electrophoretic Mobility Shift Assay (EMSA)
A dsDNA probe of the M. tuberculosis KstR operator sequence in the intergenic region between Rv3573c and Rv3574 (Table 1) was prepared by heating complementary single-stranded DNA oligomers to 95 °C and annealing at room temperature in 20 mm Tris-HCl, 10 mm MgCl2, pH 8.0, 75 mm NaCl as described previously (20). Binding assays were performed in 20 mm HEPES, pH 8.0, 10 mm MgCl2, 75 mm NaCl, 10% glycerol and contained 6 pmol of DNA probe, 12 pmol of M. tuberculosis KstR, and 0.2 μmol of each potential inducer. Assays were incubated at 37 °C for 30 min prior to being loaded onto a 9% polyacrylamide gel containing 0.5× TBE (50 mm Tris base, 50 mm boric acid, 1 mm EDTA, pH 8.3) and then run at 105 V for 45 min. Gels were stained with ethidium bromide (EtBr) and visualized under UV light using an AlphaimagerTM 2200 (Alpha Innotech Corp.) gel documentation system.
Intrinsic Fluorescence Quenching Assay
The intrinsic tryptophan fluorescence of KstR was measured using a Pistar-180 spectrometer (Applied Photophysics). Protein was excited at 280 nm (bandwidth of 7 nm), and emission spectra were scanned from 300 to 360 nm. A spectral maximum was found at 328 nm, and emission at this wavelength was therefore used for fitting the quenching curve. KstR concentrations in this assay refer to the monomer. The fluorescence intensities of samples containing 2 μm KstR and ligand at a series of concentrations in buffer (20 mm HEPES, 150 mm NaCl, pH 7.4) were measured using 10 × 2-mm Quartz Suprasil® cuvettes (Hellma). A solution of the ligand dissolved in buffer at the highest concentration used has an absorbance of <0.02 at the excitation and emission wavelengths, thus the inner filter effect was negligible. The measured fluorescence intensities were fitted to the following equation developed from a simple 1:1 binding model using GraphPad Prism 6.0.
 |
where F is the fluorescence in the presence of ligand, Fmax is the fluorescence in absence of ligand, Fmin is the fluorescence of ligand-saturated protein, [L] is ligand total concentration (μm), [M]0 is protein concentration (μm), and Kd is the dissociation constant (μm).
SPR
SPR was performed using a Biacore T200 (GE Healthcare). To assay KstR-DNA interactions, 28-bp double-stranded DNA similar to that used in the EMSA was biotinylated at the 5′-end of one strand (Table 1) and immobilized on a streptavidin sensor chip. The density of DNA on the chip surface was kept low (less than 100 response units). KstR concentrations in this assay refer to the dimer. KstR in HBS-EP+ buffer (10 mm HEPES, 150 mm NaCl, 3 mm EDTA, 0.05% Surfactant P20, pH 7.4) at a range of concentrations up to 500 μm was injected over the immobilized DNA surface at a flow rate of 30 μl/min for 180 s to collect data for the association phase followed by a 120-s buffer injection at the same flow rate for the dissociation phase. The surface was regenerated by injecting 0.03% SDS at 50 μl/min for 20 s and then washed with a brief injection of buffer. The equilibrium response was measured 4 s before the end of the association phase and used to calculate the dissociation constant Kd. In the ligand inhibition assay, a mixture of 15 nm KstR and ligand at various concentrations was used for the association phase with the same injection procedure. Curve fitting to SPR data was performed using a one-site binding equation with specific and nonspecific interaction components using GraphPad Prism 6.0.
Crystallization and X-ray Data Collection
All crystals in this study were grown at 18 °C using the vapor diffusion method in hanging drop (free KstR) or sitting drop (KstR·ligand complex) format. Purified KstR at ∼5 mg/ml was mixed with crystallization mother liquor at a 1:1 volume ratio to make a 4-μl (His6-KstR construct) or 2-μl drop (MBP-fused construct). For protein·ligand complexes, KstR was mixed with its ligand at a final concentration of 220 μm KstR monomer and 250 μm ligand, and the mixture was left at room temperature for 3 h before being dispensed with crystallization mother liquor in a 1:1 volumetric ratio to make 0.4-μl drops using a Cartesian Honeybee nanoliter dispensing robot. Except for crystals of apoprotein, which were cryoprotected with an addition of 25% glycerol to its mother liquor, crystals were mounted and flash cooled in their mother liquor. X-ray diffraction data from all crystals were collected using beamline MX2 at the Australian Synchrotron on an ADSC QUANTUM 315R CCD detector. Data were indexed and integrated using iMosflm (26) or XDS (27) and then scaled and merged using Aimless in the CCP4 program suite (28).
Structure Determination and Refinement
The initial structure of KstR (from the MBP-KstR construct) was solved by molecular replacement using BALBES (29) (CCP4 program suite). Chain A of a TetR transcriptional regulator from Rhodococcus sp. RHA1 (Protein Data Bank code 3BJB) was used as the search model. Model building was done using ARP/wARP (30), and refinement was initially carried out using Refmac5 (31). The structure was further refined with diffraction data obtained at 2 and 1.85 Å using phenix.refine (32) and AutoBuster (33). For the ligand-bound structures, the apoprotein structure was used for molecular replacement, and the obtained solutions were then input to PHENIX AutoBuild (34) to improve model quality and refined using phenix.refine. Initial ligand geometry was generated using the Coot Ligand Builder (35), ligand geometrical restraints were generated using eLBOW (36), and manual rebuilding was carried out using Coot (35). Structures were visualized and illustrated using PyMOL (37).
Results
Analysis of KstR-DNA Interactions
We reported previously that M. tuberculosis KstR forms a dimer in solution (13), whereas it has been reported that a small fraction of monomer might also exist in a solution of purified M. smegmatis KstR (19). SEC-MALS analysis of purified M. tuberculosis KstR showed a single, homogeneous peak of molecular mass 44.5 kDa (Fig. 2), which is very close to the theoretical mass of the KstR dimer (44.6 kDa). No peak consistent with the predicted size of the protein monomer was observed, demonstrating that M. tuberculosis KstR is an obligatory homodimer.
FIGURE 2.
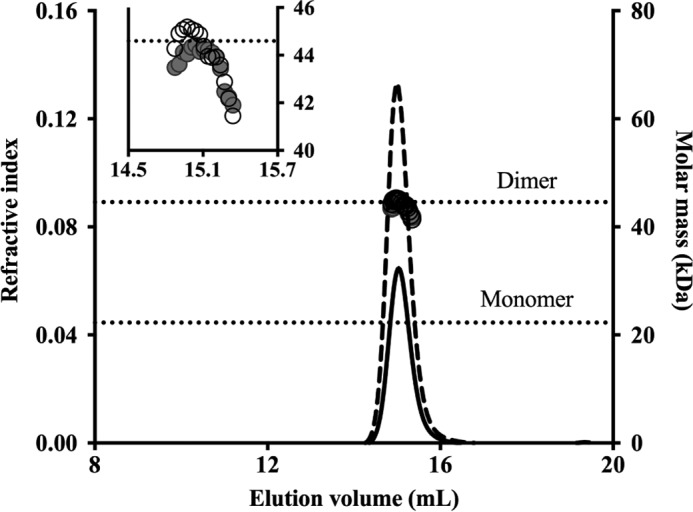
SEC-MALS analysis of KstR. KstR was eluted in a single peak as detected by refractive index detection (solid and dashed lines represent samples loaded at 2.5 and 5.0 mg/ml, respectively). Calculated molar mass across the peaks (filled and empty circles at 2.5 and 5.0 mg/ml, respectively) indicates a dimeric form of KstR in solution. The graph shows the complete elution range of the Superdex 200 10/300 GL column, and the inset shows an enlarged region around the peak.
In our previous study, KstR bound as a dimer to its target DNA, a 28-bp fragment from the intergenic region between the open reading frames Rv3573c and Rv3574 (kstR) in the M. tuberculosis genome (13). In this study, the affinity of KstR for its target DNA was quantitated by SPR using a biotinylated version of the same 28-bp (17.2-kDa) DNA fragment immobilized on the surface of a streptavidin sensor chip. Under these conditions, M. tuberculosis KstR showed relatively fast association and dissociation kinetics with a global Kd of 9.9 ± 0.6 nm (Fig. 3A). However, the kinetic data could not be fitted to a simple Langmuir model, indicating a more complex mode of interaction. The maximum response at saturated binding (i.e. at 500 nm KstR dimer) correlates with the amount of immobilized DNA in a 1:1 stoichiometry of DNA duplex:KstR dimer (i.e. 37 response units of KstR dimer and 18 response units of DNA versus 45 kDa of KstR dimer and 18 kDa of DNA).
FIGURE 3.

Analysis of the interaction of KstR with DNA and ligands. A, global fitting of SPR equilibrium response of the KstR-DNA interaction. Change in response units (RU) at equilibrium is plotted against concentration of KstR dimer. B, EMSA to identify KstR ligands. All lanes contain 0.6 μm DNA probe and 1.2 μm KstR monomer plus either 20 μm possible effector or 600 ng of nonspecific (NS) (salmon sperm) DNA. C, intrinsic fluorescence of KstR in the presence of 3OCh-CoA (circles, solid line) or 4-BNC-CoA (squares, dashed line). Samples contain 2 μm KstR monomer and 0–8 μm ligand. D, SPR analysis of 15 nm KstR dimer and immobilized DNA in the presence of 3OCh-CoA (circles, solid line) or 4-BNC-CoA (squares, dashed line). Residual binding is normalized against the binding of KstR in the absence of ligand. Shown values are the average of triplicate experiment with two readings for each repeat. Error bars indicate standard deviation for all readings.
The Endogenous Ligands of KstR Are Thioesters of Cholesterol Side-chain Degradation Products
To identify the likely endogenous effectors of M. tuberculosis KstR, EMSAs of KstR-DNA binding were performed in the presence of a selection of six potential effector molecules. Among these, only 4-BNC-CoA and 3OCh-CoA released KstR from its target DNA (Fig. 3B). Curiously, neither 4-BNC nor 3OChA induced DNA release despite the previous report that the latter is able to bind to M. smegmatis KstR (19). Moreover, neither CoASH nor HIP-CoA, a downstream metabolite in which the side chain and ring A are completely degraded (15, 20), acted as KstR effectors. Finally, 3-HSBNC-CoA, the equivalent of 4-BNC-CoA with a cleaved ring B, also did not act as an effector (Fig. 3B). Together, these results suggest that the four steroid rings and the CoA esterified side-chain moiety are required for a functional interaction with 3OCh-CoA having a greater effect than 4-BNC-CoA.
To determine the affinity of the interactions, the intrinsic tryptophan fluorescence of KstR was monitored in the presence of various concentrations of both identified cognate ligands (Fig. 3C). KstR has two tryptophan residues in its sequence, one of which (Trp-164) flanks the potential ligand-binding pocket and thus is positioned to have its fluorescence modified when the pocket is occupied. A strong quenching effect (up to 70% of the fluorescence intensity at 328 nm) was observed in the presence of both ligands. Fitting quenching curves to a one-site binding model gave calculated Kd values of 60 ± 20 and 280 ± 50 nm for 3OCh-CoA and 4-BNC-CoA, respectively. No cooperativity was observed, suggesting that the two binding sites of the dimer have equal and independent affinity toward both ligands. The assay was in agreement with the EMSA results as the stronger affinity of KstR for 3OCh-CoA is consistent with the lower concentration of that ligand required to cause DNA release.
The effect of ligand binding on the interaction of KstR with its target DNA was investigated using an SPR-based release assay (Fig. 3D). The same biotinylated 28-bp DNA fragment was used as a target in measuring the inhibitory effect of ligands on the ability of KstR to bind to DNA. At a fixed concentration of 30 nm KstR, 3OCh-CoA and 4-BNC-CoA showed IC50 values of 36 ± 1 and 143 ± 1 nm, respectively. In both cases, strong inhibition (∼95% of KstR-DNA binding activity) was obtained, demonstrating the effective inactivation that these ligands can impose on KstR. The results confirm that 3OCh-CoA and 4-BNC-CoA are both functionally effective, high affinity ligands for KstR with 3OCh-CoA exhibiting both a stronger binding affinity and a more effective induction of DNA release.
The Structure of Ligand-free KstR
The structure of ligand-free KstR was determined in two different crystal forms (Table 2). In form I (Protein Data Bank code 3MNL), tag-free protein crystallized in space group P212121 with the asymmetric unit containing two protein chains. As expected, the global structure of KstR shows an all-helical protein with nine α-helices arranged into a two-domain architecture (Fig. 4A). The DBD is located at the N terminus and comprises helices α1–3 of which helices α2 and α3 form the typical helix-turn-helix motif characteristic of TetR family repressors (37). The rest of the protein forms a LBD that primarily contacts the DBD via helix α4. A four-helix bundle, comprising helices α8 and α9 from each monomer, forms the dimer interface. The buried surface between the two chains is 1099 Å2, or almost 12% of the ∼9500-Å2 total surface area of each monomer, as measured by PISA (38), indicating a strong dimer. This is in agreement with the dimeric arrangement of KstR seen in solution as well as with other known TetR family structures (e.g. Refs. 39 and 40). Each LBD in the dimer contains a potential ligand-binding pocket that is closed by the side chain of Met-104 from helix α5 at one end and is open to the dimer interface at the opposite end. The pocket is formed by the α4 helix, the triangle of helices α5–7, and helix α8 and is extensively lined with hydrophobic residues, indicating a propensity for binding hydrophobic ligands. The distance between the two DBDs in the dimer is 42.6 Å (calculated from Cα of Tyr-54 in DNA-binding helix α3 in each subunit), which far exceeds the 34-Å distance of two consecutive major grooves in DNA, suggesting that the conformation seen in the crystals would be unfavorable for DNA binding. Form II crystallized in space group I121 with two dimers in the asymmetric unit. The dimers have high structural similarity to each other (0.46-Å r.m.s.d. over all main-chain atoms) but differ somewhat from the dimer seen in form I (1.51- and 1.63-Å r.m.s.d. between form I and dimer 1 or 2 from form II, respectively). A molecule of PEG is found bound to the protein in form II, causing helices α4 and α5 to move apart slightly and altering the conformation of Met-104, thus opening the mouth of the ligand-binding pocket. The distances between the two DBDs of both dimers are also substantially smaller than that found in form I (38.4 and 37.7 Å), indicating inherent flexibility in the α3-α4 loop and that form II may more closely resemble a conformation of KstR able to bind DNA (Fig. 4B).
TABLE 2.
Crystallographic statistics for the KstR structures
Values in parentheses represent the outermost shell of data. CC, correlation coefficient.
| Protein Data Bank code | 3MNL | 5CXG | 5CW8 | 5CXI |
| Description | Apo-KstR | KstR in complex with PEG | KstR in complex with 3OCh-CoA | KstR in complex with 4-BNC-CoA |
| Crystallization | ||||
| Expression construct | His6-MBP-KstR | His6-KstR | His6-KstR | His6-KstR |
| Condition | 0.2 m ammonium fluoride, 17.5% PEG 3350 | 8% PEG 3350, 25% PEG 400, 0.1 m MgCl2, 0.1 m Tris-HCl, pH 8.5 | 10 mm NiCl2, 0.1 m Tris, pH 8.5, 1.0 m Li2SO4 | 0.1 m MES/imidazole, pH 6.5, 10% PEG 4000, 20% glycerol, 20 mm carboxylic acid mixturea |
| Data collection | ||||
| Space group | P212121 | I121 | I121 | I121 |
| Cell dimension | ||||
| a, b, c (Å) | 4.02, 54.57, 166.0 | 126.03, 38.38, 154.72 | 49.69, 67.10, 124.23 | 49.78, 67.26, 124.23 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 111.69, 90.00 | 9.00, 96.74, 90.00 | 90.00, 96.79, 90.00 |
| Resolution (Å) | 83.08-1.80 | 37.41-2.10 | 47.88-2.60 | 47.88-2.00 |
| No. of unique reflections | 35,558 | 40,309 | 12,603 | 27,478 |
| Rmerge | 0.056 (0.606) | 0.140 (0.805) | 0.197 (0.884) | 0.198 (1.095) |
| I/σ(I) | 26.25 (3.2) | 4.6 (1.4) | 6.7 (2.3) | 9.5 (3.4) |
| CC1/2 | 0.999 (0.801) | 0.990 (0.669) | 0.985 (0.706) | 0.993 (0.731) |
| Completeness (%) | 99.9 (99.9) | 98.5 (94.6) | 100.0 (100.0) | 99.6 (99.0) |
| Redundancy | 6.8 (6.4) | 3.6 (3.5) | 6.4 (6.4) | 8.3 (8.3) |
| Refinement | ||||
| Resolution (Å) | 24.68-1.80 | 37.41-2.10 | 47.80-2.60 | 39.83-2.00 |
| No. of reflections | 35,467 | 40,305 | 12,603 | 27,469 |
| Rwork/Rfree | 19.1/20.5 | 21.0/25.8 | 19.0/23.1 | 23.8/28.0 |
| No. of atoms | ||||
| Protein | 2,758 | 5,129 | 2,635 | 2,661 |
| Ligand | 31 | 69 | 63 | |
| Water | 117 | 208 | 72 | 234 |
| B-factors (Å2) | ||||
| Protein | 30.63 | 48.68 | 33.65 | 26.25 |
| Ligand | 49.86 | 33.05 | 21.59 | |
| Water | 37.39 | 41.97 | 35.56 | 32.35 |
| r.m.s.d. from ideal | ||||
| Bond lengths (Å) | 0.009 | 0.012 | 0.004 | 0.011 |
| Bond angles (°) | 0.85 | 0.655 | 0.75 | 0.625 |
a Carboxylic acid mixture: sodium formate, ammonium acetate, trisodium citrate, sodium potassium l-tartrate, sodium oxamate.
FIGURE 4.
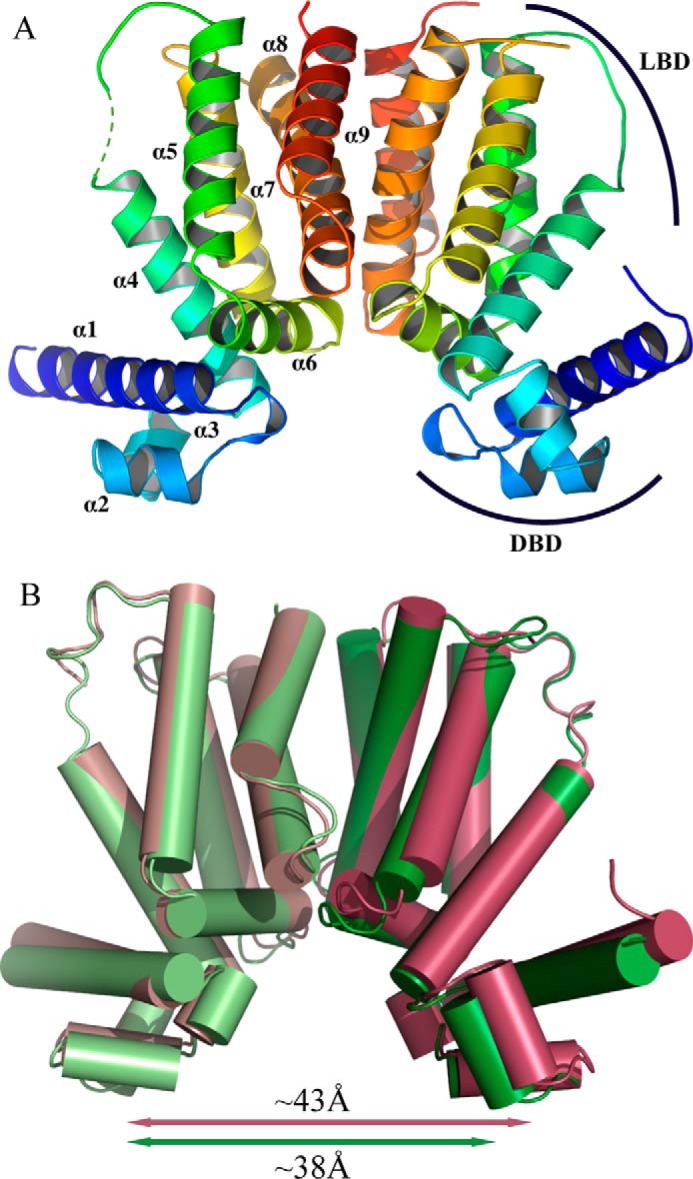
Structure of ligand-free KstR. A, the dimer of the form I structure with each monomer colored from blue (N terminus) to red (C terminus) and helices labeled. The LBD and DBD are indicated. B, superposition of form I (maroon) and form II (dimer 1; green) overlaid at the central four-helix bundle composed of helices α8 and α9 from each subunit.
The Structures of KstR in Complex with Its Endogenous Ligands
The co-crystal structures of KstR in complex with 3OCh-CoA and 4-BNC-CoA were both determined by molecular replacement. Both crystallized in space group I121 with one KstR dimer in the asymmetric unit. The structures are highly similar to each other with a r.m.s.d. of 0.24 Å over the whole dimer. As expected from structures of other TetR family members (39, 40), the DBDs are widely spaced in the complexes with 3OCh-CoA and 4-BNC-CoA, 43.4 and 43.8 Å, respectively. The SPR results described above confirm that these ligand-bound forms of KstR are incompetent for DNA binding, and these structures therefore represent KstR in its inactive form.
Both steroids are positioned in the predicted ligand-binding pocket of KstR (Fig. 5A), which adopts an open conformation compared with the ligand-free structures to accommodate the ligand. The four steroid rings of the 3-oxo-cholest-4-enoyl moiety of both 3OCh-CoA and 4-BNC-CoA make almost entirely hydrophobic interactions in the pocket and are sandwiched among helices α4, α5, α7, and α8 (Fig. 5B). The notable exception is the 3-oxo group, which forms hydrogen bonds with Arg-158 and a nearby highly coordinated water molecule (Fig. 5C). As the side chain of Arg-158 also makes a hydrogen bond with the side-chain carboxyl group of another conserved residues, Asp-137, the organization of this area is likely to be key to ligand recognition. Although both the steroidal ring and its aliphatic side chain are clearly visible and well ordered (the refined atomic B-factors are less than 20 Å2), there is no visible electron density for the CoA moiety beyond the sulfur atom. The lack of electron density for this part of the ligand raises a question of whether the CoA arm makes contact with the protein and is required for protein-ligand recognition or whether it is free and hence functions primarily as a solubilizing group for the steroid. The entrance to the pocket is flanked by α4, α5, and part of the α1 helix where there are a number of positively charged residues (Arg-102, Arg-106, Lys-99, Arg-70, Arg-74, and Lys-78) that are potential binding sites for the phosphate groups that are part of the phosphopantothenate component of CoA. There is weak electron density visible around this area, and although none of it is strong enough to be assigned with any certainty to the CoA tail, it seems possible that the tail is localized in this area in a statistically disordered state. Apart from the interaction made by the 3-oxo group, no other specific polar interactions were found at the keto or sulfide group of the tail of 3OCh-CoA, suggesting that these points do not contribute to ligand binding.
FIGURE 5.

Structure of ligand-bound KstR. A, difference electron density in the ligand pocket of KstR indicates the presence of bound ligands. Fo − Fc electron density maps (green mesh; contoured at 2σ) were calculated prior to modeling the ligands (gray stick model) in structures of KstR determined in complex with 3OCh-CoA and 4-BNC-CoA. B, binding of 4-BNC-CoA (shown as a stick model) into the ligand-binding pocket of KstR (shown as schematic and surface representations). C, the contacts made by KstR to both 3OCh-CoA and 4-BNC-CoA. Hydrophobic contacts shown in red; hydrogen bonds are shown as green dashed lines.
Comparison of the ligand-free ligand-bound structures of KstR reveals conformational changes in the global structure of KstR that can be attributed to ligand binding. The binding of either ligand induces two changes in the protein: a twist between the α8-α9 pair of helices and its dimeric α8′-α9′ counterpart at the dimer interface and a positional shift of this helical pair relative to the rest of the protein monomer. The former movement translates to a change of the angle of contact between the two subunits in the dimer, whereas the latter results in a rearrangement within each monomer. The alteration of the dimer interface involves a reorientation of ∼4.5° between the α8 helices (Table 3), which causes the DBDs to move in opposite directions, increasing the separation between the domains compared with the form II ligand-free structure (Fig. 6A). In contrast to the variation seen in the ligand-free structures, both ligand complexes exhibit identical dimer interface angles and very similar distances between DBDs, suggesting a rigidification of the ligand-bound structures. The α5–α7 helical bundle, α4 helix, and whole of the DBD in the ligand-bound structure are very similar to those of the form II ligand-free structures (r.m.s.d. of 0.589 Å), indicating that they move together in a concerted manner during the ligand-induced conformational change. In the orientation of the protein that is depicted (Fig. 6A), this translates to an “upward” shift of the DBD relative to the dimer interface helical bundle.
TABLE 3.
Geometrical measurements of the KstR structures
| Form I | Form II |
KstR·3OCh-CoA | KstR·4-BNC-CoA | ||
|---|---|---|---|---|---|
| Dimer 1 | Dimer 2 | ||||
| Protein Data Bank code | 3MNL | 5CXG | 5CXG | 5CW8 | 5CXI |
| Distance between Cα of Tyr-54 and Tyr-54′ (Å) | 42.6 | 38.4 | 37.8 | 43.4 | 43.8 |
| Angle between helix α8 and α8′ (°)a | 85.86 | 86.65 | 86.14 | 81.74 | 81.74 |
| Dihedral angle χ2 of Trp-164 side chain (°)b | |||||
| Chains A/C | 75.9 (75.88%) | 79.3 (63.28%) | 80.7 (79.08%) | −47.3 (1.22%) | −48.1 (1.22%) |
| Chains B/D | 78.3 (75.88%) | 87.5 (90.98%) | 86.7 (90.98%) | −42.0 (1.15%) | −49.6 (1.22%) |
a Orientation vector of each helix is calculated from residues 151 to 172.
b Measured in order of Cα-Cβ-Cγ-Cδ1; percentage of rotamer probability as calculated by MolProbity (48) is shown in parentheses.
FIGURE 6.

Structural changes in KstR due to ligand binding. A, superposition of monomer structures of ligand-free KstR (green; form II, dimer 1) and KstR in complex with 3OCh-CoA (blue). The movement of the DBD relative to the α8-α9 dimer interface is shown by red arrows. B, the ligand-induced movements of the KstR monomer are coordinated with a change in the conformation of Trp-164 (shown as a stick model).
The local dislocation of these helices appears to be linked to a ligand-driven flip of the Trp-164 side chain. In ligand-free KstR, the side chain of Trp-164 is in a relaxed conformation, adopting a common rotamer with a χ2 dihedral angle of 75–80° and its aromatic side chain pointing toward the center of the unoccupied pocket. Upon ligand binding, this side chain rotates by ∼120° around the Cβ–Cγ bond, adopting a rare rotamer with a less energetically favorable χ2 angle of −40 to −50° (Table 3). The rotation of the Trp-164 side chain has the effect of distorting the α5 helix at its C-terminal end, which in turn displaces the α4 and α6 helices and consequently the DBD (Fig. 6B). The contact between the C-terminal end of the α6 helix and N terminus of the α8′ helix in the other monomer remains constant with the result that the ligand-induced shift of the α6 helix alters the angle of contact across the dimer interface. The net result of these displacements is a 6.7-Å shift of the DBD from its position in the ligand-free structure where it would be expected to contact the target DNA.
Discussion
In this study, we have analyzed the interaction of KstR with its cognate DNA and confirmed a 1:1 binding stoichiometry between the DNA duplex and the KstR dimer. This puts KstR in the group of TetR family members that bind to their target DNA sequence motif at a single site (e.g. TetR, SimR, etc. (39, 40)). KstR demonstrates a high affinity for its target DNA with fast binding kinetics. We have also identified two CoA thioester metabolites from early stage cholesterol degradation as ligands for KstR and characterized their binding affinities for the protein and their inhibitory effects on DNA binding using surface plasmon resonance.
KstR regulates most of the enzymes required for side-chain and A/B ring degradation in M. tuberculosis (13). Its identified ligands, 3OCh-CoA and 4-BNC-CoA, occur early in cholesterol catabolism, possessing at least three carbons of the aliphatic side chain. The inability of 3-HSBNC-CoA to act as an effector suggests that it is unlikely that metabolites produced downstream of ring B cleavage act as regulatory ligands. However, it is difficult to definitively conclude which ligand or ligands might function as the physiological effectors of KstR: presumably, all of the three-, five-, and eight-carbon intermediates generated during β-oxidation of the side chain would be competent in this respect. Nevertheless, it is probable that metabolites with a longer side chain are the predominant effectors in vivo, considering that they are produced first. This idea is supported by our observation that 3OCh-CoA binds more tightly to KstR than does 4-BNC-CoA.
The structures of KstR·ligand complexes reveal a ligand pocket that recognizes both the shape of a four-ring steroidal moiety and its highly hydrophobic nature. This is consistent with the inability of 3-HSBNC-CoA and HIP-CoA to act as effectors. However, our structural observations do not exclude the possibility of other four-ring steroids such as cholesterol and cholest-4-en-3-one acting as ligands for KstR.
3OCh-CoA and 4-BNC-CoA are the main substrates of the ChsE4·ChsE5 and ChsE1·ChsE2 complexes that initiate cycles 1 and 3 of cholesterol side-chain β-oxidation (41). Interestingly, both of these cycles produce propionyl-CoA, which can be toxic for bacterial growth due to inhibition of acetyl-CoA production via pyruvate dehydrogenase (42). To alleviate the burden of this toxicity, propionyl-CoA can be fed to the methyl citrate cycle or the methylmalonyl pathway in the presence of vitamin B12 or utilized in the biosynthesis of cell wall lipids (42–44).
TFRs, which are the most abundant family of transcriptional regulators in mycobacteria and other actinomycetes (45), make up a large and structurally variable group of proteins, especially in their LBDs, which have evolved to accommodate a wide variety of ligands. The evolution of ligand-specific binding has progressed in parallel with the allosteric mechanisms that govern protein activity. As a result, these mechanisms are remarkably varied among the TetR-like regulators despite their shared overall architecture. In the majority of TFRs with proposed induction mechanisms, the separation of the DBDs is the result of a pendulum-like movement of helix α4 originating from a displacement of helix α6 upon ligand binding (17, 39, 46, 47). In contrast, SimR from Streptomyces antibioticus has a distinctive rigid body rotation about the dimeric interface center mediated by a dislocation of its α9-α10 helical arm, which occludes the entrance of the ligand pocket of the other subunit in the dimer (40), and in the case of M. tuberculosis KstR2, ligand binding pushes the α4, α6, and α7 helices outward, causing a shift in the DBD in the same direction (20).
For KstR, the distinctive conformational transformation that can be seen when comparing the ligand-free and ligand-bound structures differs from all the mechanisms described previously for other TFRs. The binding of the steroid moiety into the ligand pocket causes changes both within the monomer and across the dimer interface of KstR. The crystallographic evidence presented here suggests an induction mechanism for KstR as follows: the binding of a steroid inside the ligand pocket causes a shift of the α5–α7 helical bundle, α4 helix, and DBD as a whole via a distortion in the α5 helix and the associated flipping of a key tryptophan residue (Trp-164); the concomitant dislocation of the α6 helix shifts the α8 helix of the other subunit in the dimer, causing a small rotation of the contacting angle between subunits at the dimer interface.
Although our EMSA data indicate the essentiality of the CoA adduct of both steroid ligands for effective DNA release, no electron density is observed for the CoA moiety in either structure. As the co-crystal complexes were slow to form, we considered the possibility that the steroid-CoA adducts might have hydrolyzed in situ in the crystallization experiments. Direct analysis of protein·ligand complex crystals using mass spectrometry was confounded by the presence of PEG from the crystallization experiments. However, thioesters are known to be vulnerable to hydrolysis only at the thioester bond, breaking down to form carboxylic acids and thiols. The electron density maps of ligand-bound KstR show clear density for the sulfur atom in all subunits (Fig. 5A), indicating that the thioester bond is still intact. As no other degradation point of CoA has been reported, we interpret this to mean that the ligands retained their integrity in the co-crystal but that the CoA tail is disordered. It is possible that the CoA acts primarily as a solubilizing agent to increase the pool of steroid that is available for KstR binding in the cell. Alternatively, the CoA may play a role in tethering the ligand onto the protein surface via contacts with the flexible and positively charged residues that surround the mouth of the ligand-binding pocket. In either case, it appears not to be contributing any significant specificity to the protein-ligand interaction. However, the length of the alkyl side chain contributes to the differential affinities of the two ligands in a way that is not immediately apparent from inspection of the structures. However, it is possible that the shorter thioester linker in 4-BNC-CoA causes a sterically unfavorable positioning of the CoA portion of the ligand, resulting in weaker binding.
As expected, KstR and KstR2 show overall structural similarity but differ considerably in the details of their interaction with their cognate ligands. Both proteins recognize CoA thioester adducts of cholesterol degradation products, but KstR does not form specific interactions with the CoA moiety, which is disordered in the crystal structures reported. This is in marked contrast to the extensive protein-CoA interaction seen in the complex between KstR2 and the HIP-CoA (19) and highlights the diverse ways in which even TFRs that are closely related in biological function and sequence can interact with and respond to their cognate ligands.
In conclusion, this study further demonstrates the importance of CoA thioesters in the regulation of cholesterol catabolism and provides a framework of structural information that can be used to support the future development of druglike compounds targeting KstR. An antagonist of KstR would be expected to be disadvantageous for M. tuberculosis considering the essentiality of this regulator for the survival of the bacterium in macrophages. Equally, a compound that enables KstR to remain bound to DNA even in the presence of its endogenous ligands would also be harmful for M. tuberculosis persistence due to the requirement of the cholesterol metabolic breakdown pathway during infection. For either purpose, the mechanism of induction by the endogenous ligands provides a guideline for the future design of potential therapeutic compounds targeting KstR.
Author Contributions
N. A. T. H. conducted most of the crystallographic and biophysical experiments supervised by S. S. D. and J. S. L. The ligands were synthesized by I. C., and A. M. C. carried out the EMSA analysis. C. G. determined the structure of the ligand-free protein. S. L. K., E. N. B., and L. D. E. contributed to discussion and analysis of the data. N. A. T. H. and J. S. L. wrote the paper with contributions and approval from all the other authors.
Acknowledgments
We thank Fiona Clow for advice on SPR experiments, David Goldstone for help and advice with SEC-MALS, and Martin Middleditch for mass spectrometry. DNA sequencing was provided by the Centre for Genomics, Proteomics, and Metabolomics (CGPM) within the School of Biological Sciences. X-ray data collection at the MX1 and MX2 beamlines of the Australian Synchrotron was enabled by the New Zealand Synchrotron Access Group. We thank all the beamline staff of the MX beamlines for support.
This work was supported in part by Health Research Council of New Zealand Program Grant 12/1111 (to E. N. B. and J. S. L.) and Canadian Institutes for Health Research (CIHR) Operating Grant MOP-133647 (to L. D. E.). The authors declare that they have no conflicts of interest with the contents of this article.
This paper is dedicated to Israël Casabon, a talented young scientist without whom this work could not have been completed, who sadly passed away before its publication.
The atomic coordinates and structure factors (codes 3MNL, 5CXG, 5CXI, and 5CW8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TFR
- TetR family transcriptional repressor
- DBD
- DNA-binding domain
- LBD
- ligand-binding domain
- 3OChA
- 3-oxocholest-4-en-26-oic acid
- HIP
- 3aα-H-4α(3-propanoate)-7aβ-methylhexahydro-1,5-indanedione
- 4-BNC
- 3-oxo-23,24-bisnorchol-4-en-22-oic acid
- SPR
- surface plasmon resonance
- 4-BNC-CoA
- 3-oxo-23,24-bisnorchol-4-en-22-oyl-CoA
- 3OCh-CoA
- 3-oxocholest-4-en-26-oyl-CoA
- 3-HSBNC-CoA
- 3-hydroxy-9-oxo-9,10-seco-23,24-bisnorchola-1,3,5(10)-trien-22-oyl-CoA
- HIP-CoA
- 3aα-H-4α(3′-propanoyl-CoA)-7aβ-methylhexahydro-1,5-indanedione
- SEC-MALS
- size exclusion chromatography with multiangle light scattering
- r.m.s.d.
- root mean square deviation
- MBP
- maltose-binding protein.
References
- 1. World Health Organization (2015) Global Tuberculosis Report 2015, World Health Organization, Geneva [Google Scholar]
- 2. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 3. Pandey A. K., and Sassetti C. M. (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U.S.A. 105, 4376–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yam K. C., D'Angelo I., Kalscheuer R., Zhu H., Wang J.-X., Snieckus V., Ly L. H., Converse P. J., Jacobs W. R. Jr., Strynadka N., and Eltis L. D. (2009) Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 5, e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brzostek A., Dziadek B., Rumijowska-Galewicz A., Pawelczyk J., and Dziadek J. (2007) Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 275, 106–112 [DOI] [PubMed] [Google Scholar]
- 6. Brzostek A., Pawelczyk J., Rumijowska-Galewicz A., Dziadek B., and Dziadek J. (2009) Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 191, 6584–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubnau E., Fontán P., Manganelli R., Soares-Appel S., and Smith I. (2002) Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect. Immun. 70, 2787–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang J. C., Miner M. D., Pandey A. K., Gill W. P., Harik N. S., Sassetti C. M., and Sherman D. R. (2009) igr genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191, 5232–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nesbitt N. M., Yang X., Fontán P., Kolesnikova I., Smith I., Sampson N. S., and Dubnau E. (2010) A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect. Immun. 78, 275–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu Y., van der Geize R., Besra G. S., Gurcha S. S., Liu A., Rohde M., Singh M., and Coates A. (2010) 3-Ketosteroid 9α-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol. Microbiol. 75, 107–121 [DOI] [PubMed] [Google Scholar]
- 11. Van der Geize R., Yam K., Heuser T., Wilbrink M. H., Hara H., Anderton M. C., Sim E., Dijkhuizen L., Davies J. E., Mohn W. W., and Eltis L. D. (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 1947–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. VanderVen B. C., Fahey R. J., Lee W., Liu Y., Abramovitch R. B., Memmott C., Crowe A. M., Eltis L. D., Perola E., Deininger D. D., Wang T., Locher C. P., and Russell D. G. (2015) Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog. 11, e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kendall S. L., Withers M., Soffair C. N., Moreland N. J., Gurcha S., Sidders B., Frita R., Ten Bokum A., Besra G. S., Lott J. S., and Stoker N. G. (2007) A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 65, 684–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kendall S. L., Burgess P., Balhana R., Withers M., Ten Bokum A., Lott J. S., Gao C., Uhia-Castro I., and Stoker N. G. (2010) Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology 156, 1362–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casabon I., Zhu S.-H., Otani H., Liu J., Mohn W. W., and Eltis L. D. (2013) Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol. Microbiol. 89, 1201–1212 [DOI] [PubMed] [Google Scholar]
- 16. Sassetti C. M., and Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schumacher M. A., Miller M. C., Grkovic S., Brown M. H., Skurray R. A., and Brennan R. G. (2001) Structural mechanisms of QacR induction and multidrug recognition. Science 294, 2158–2163 [DOI] [PubMed] [Google Scholar]
- 18. Grkovic S., Hardie K. M., Brown M. H., and Skurray R. A. (2003) Interactions of the QacR multidrug-binding protein with structurally diverse ligands: implications for the evolution of the binding pocket. Biochemistry 42, 15226–15236 [DOI] [PubMed] [Google Scholar]
- 19. García-Fernández E., Medrano F. J., Galán B., and García J. L. (2014) Deciphering the transcriptional regulation of cholesterol catabolic pathway in mycobacteria: identification of the inducer of KstR repressor. J. Biol. Chem. 289, 17576–17588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crowe A. M., Stogios P. J., Casabon I., Evdokimova E., Savchenko A., and Eltis L. D. (2015) Structural and functional characterization of a ketosteroid transcriptional regulator of Mycobacterium tuberculosis. J. Biol. Chem. 290, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Capyk J. K., Casabon I., Gruninger R., Strynadka N. C., and Eltis L. D. (2011) Activity of 3-ketosteroid 9-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J. Biol. Chem. 286, 40717–40724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Casabon I., Crowe A. M., Liu J., and Eltis L. D. (2013) FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol. Microbiol. 87, 269–283 [DOI] [PubMed] [Google Scholar]
- 23. Casabon I., Swain K., Crowe A. M., Eltis L. D., and Mohn W. W. (2014) Actinobacterial acyl coenzyme A synthetases involved in steroid side-chain catabolism. J. Bacteriol. 196, 579–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreland N., Ashton R., Baker H. M., Ivanovic I., Patterson S., Arcus V. L., Baker E. N., and Lott J. S. (2005) A flexible and economical medium-throughput strategy for protein production and crystallization. Acta Crystallogr. D Biol. Crystallogr. 61, 1378–1385 [DOI] [PubMed] [Google Scholar]
- 25. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 26. Powell H. R., Johnson O., and Leslie A. G. (2013) Autoindexing diffraction images with iMosflm. Acta Crystallogr. D Biol. Crystallogr. 69, 1195–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 32. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., and Womack T. O. (2011) BUSTER, version 2.8.0, Global Phasing, Ltd., Cambridge, UK [Google Scholar]
- 34. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuildwizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D. (2009) Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramos J. L., Martínez-Bueno M., Molina-Henares A. J., Terán W., Watanabe K., Zhang X., Gallegos M. T., Brennan R., and Tobes R. (2005) The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 39. Orth P., Schnappinger D., Hillen W., Saenger W., and Hinrichs W. (2000) Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat. Struct. Biol. 7, 215–219 [DOI] [PubMed] [Google Scholar]
- 40. Le T. B., Stevenson C. E., Fiedler H.-P., Maxwell A., Lawson D. M., and Buttner M. J. (2011) Structures of the TetR-like simocyclinone efflux pump repressor, SimR, and the mechanism of ligand-mediated derepression. J. Mol. Biol. 408, 40–56 [DOI] [PubMed] [Google Scholar]
- 41. Yang M., Lu R., Guja K. E., Wipperman M. F., St Clair J. R., Bonds A. C., Garcia-Diaz M., and Sampson N. S. (2015) Unraveling cholesterol catabolism in Mycobacterium tuberculosis: ChsE4-ChsE5 α2β2 acyl-CoA dehydrogenase initiates β-oxidation of 3-oxo-cholest-4-en-26-oyl CoA. ACS Infect. Dis. 1, 110–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee W., VanderVen B. C., Fahey R. J., and Russell D. G. (2013) Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288, 6788–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Griffin J. E., Gawronski J. D., Dejesus M. A., Ioerger T. R., Akerley B. J., and Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Savvi S., Warner D. F., Kana B. D., McKinney J. D., Mizrahi V., and Dawes S. S. (2008) Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190, 3886–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balhana R. J., Singla A., Sikder M. H., Withers M., and Kendall S. L. (2015) Global analyses of TetR family transcriptional regulators in mycobacteria indicates conservation across species and diversity in regulated functions. BMC Genomics 16, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willems A. R., Tahlan K., Taguchi T., Zhang K., Lee Z. Z., Ichinose K., Junop M. S., and Nodwell J. R. (2008) Crystal structures of the Streptomyces coelicolor TetR-like protein ActR alone and in complex with actinorhodin or the actinorhodin biosynthetic precursor (S)-DNPA. J. Mol. Biol. 376, 1377–1387 [DOI] [PubMed] [Google Scholar]
- 47. Itou H., Watanabe N., Yao M., Shirakihara Y., and Tanaka I. (2010) Crystal structures of the multidrug binding repressor Corynebacterium glutamicum CgmR in complex with inducers and with an operator. J. Mol. Biol. 403, 174–184 [DOI] [PubMed] [Google Scholar]
- 48. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]