

Abstract
Cellvibrio japonicus is a Gram-negative soil bacterium that is primarily known for its ability to degrade plant cell wall polysaccharides through utilization of an extensive repertoire of carbohydrate-active enzymes. Several putative chitin-degrading enzymes are also found among these carbohydrate-active enzymes, such as chitinases, chitobiases, and lytic polysaccharide monooxygenases (LPMOs). In this study, we have characterized the chitin-active LPMO, CjLPMO10A, a tri-modular enzyme containing a catalytic family AA10 LPMO module, a family 5 chitin-binding module, and a C-terminal unclassified module of unknown function. Characterization of the latter module revealed tight and specific binding to chitin, thereby unraveling a new family of chitin-binding modules (classified as CBM73). X-ray crystallographic elucidation of the CjLPMO10A catalytic module revealed that the active site of the enzyme combines structural features previously only observed in either cellulose or chitin-active LPMO10s. Analysis of the copper-binding site by EPR showed a signal signature more similar to those observed for cellulose-cleaving LPMOs. The full-length LPMO shows no activity toward cellulose but is able to bind and cleave both α- and β-chitin. Removal of the chitin-binding modules reduced LPMO activity toward α-chitin compared with the full-length enzyme. Interestingly, the full-length enzyme and the individual catalytic LPMO module boosted the activity of an endochitinase equally well, also yielding similar amounts of oxidized products. Finally, gene deletion studies show that CjLPMO10A is needed by C. japonicus to obtain efficient growth on both purified chitin and crab shell particles.
Keywords: cellulose, chitin, chitinase, electron paramagnetic resonance (EPR), gene knockout, Cellvibrio japonicus, x-ray crystallography, carbohydrate-binding module (CBM), lytic polysaccharide monooxygenase (LPMO)
Introduction
Lytic polysaccharide monooxygenases (LPMOs)2 are copper-dependent enzymes that cleave glycosidic bonds of polysaccharides organized in crystalline macrostructures. After the discovery of this enzyme activity in 2010 (1), high diversity in sequence, modularity, and substrate preference has been unraveled. LPMOs are currently classified in families 9, 10, 11, and 13 of the auxiliary activities (AA) in the CAZy database (2, 3). Families AA9, AA11, and AA13 are strictly fungal, whereas family AA10 includes enzymes from bacteria, viruses, and some eukaryotic organisms. Family AA9 LPMOs (LPMO9s) have been shown to target cellulose (4–7) and some hemicelluloses (8–10), whereas LPMO11s and LPMO13s have been demonstrated to cleave chitin (11) and starch (12, 13), respectively. LPMO10s have been shown to act on cellulose, chitin, or both (1, 14, 15). Although the majority of family AA10 LPMOs exist as single domain enzymes, several enzymes also contain one or more additional carbohydrate-binding modules (CBMs) that may reflect substrate preferences (16). One example is the bi-modular Streptomyces coelicolor LPMO, ScLPMO10C (CelS2), which contains a cellulose-binding CBM2 that contributes to activity (14, 15).
So far, LPMOs have been reported to either oxidize the C1 or C4 carbon of the polysaccharide, yielding either a lactone (C1 oxidation) or a 4-ketoaldose (C4 oxidation) product (1, 17, 18). Enzymes having mixed C1/C4 activity have also been identified (6, 15). When combined with glycoside hydrolases (GHs), LPMOs contribute synergistically to overall substrate solubilization, a property that has been documented for both cellulose and chitin as substrates (19–21). This synergy is thought to result from a combination of several factors. First, LPMO activity results in an increase in available chain ends in regions of the polysaccharide that otherwise are inaccessible to GHs, yielding an increase in the number of GH attachment points (1, 19). Second, the LPMO activity appears to render the crystalline regions of the substrate more amorphous, increasing susceptibility to enzymatic hydrolysis (1, 19, 22–25). Furthermore, the decrystallization work required by GHs to extract polysaccharide chains with oxidized termini is lower than for chains having non-oxidized termini, thereby increasing GH activity on oxidized substrates (23). Notably, LPMO functionality is likely to depend on the crystalline nature of the substrate (19). Chitin exists predominantly in two naturally occurring polymorphic forms, the highly recalcitrant α-form, with anti-parallel chain arrangement, and the less recalcitrant β-form, with a parallel chain arrangement (26, 27). Synergy between different LPMOs has been observed, indicating that LPMOs may target different regions within the same substrate (15).
From a microbiological perspective, expression and secretion of LPMOs as a response to growth on recalcitrant substrates is well documented. LPMOs are often detected in large amounts in the culture supernatants of bacteria growing on insoluble substrates like chitin (28, 29) and cellulose (30). Quantitative proteomic approaches have reported similar findings for well known cellulose degraders like Thermobifida fusca (31), Streptomyces sp. SirexAA–E (32) and, as a fungal example, Neurospora crassa (33). Furthermore, studies of the transcription of LPMO-encoding genes corroborate the notion that these enzymes are abundantly produced during microbial decomposition of recalcitrant biomass (34–37). Conversely, the functional significance of LPMOs in vivo is hitherto relatively unexplored. The only study so far addressing LPMO function in an in vivo context reports that the cellulose-active LPMO10B of Cellvibrio japonicus (CjLPMO10B) is important for the ability of this bacterium to grow on cellulose (36).
Despite the relatively short existence of the LPMO field, a wealth of structural, biochemical, and bioinorganic data has been reported for these enzymes. Structural studies show that LPMOs have a characteristic pyramidal shape that arises from a central skewed β-sandwich decorated by loops and helices (4, 38–41). A large, relatively flat surface represents the substrate-binding region of the enzyme, which is dominated by a network of mainly polar residues (39, 42). Whereas most LPMO10s have a single solvent-exposed aromatic amino acid on the substrate-binding surface, LPMO9s can have up to three. The active site of LPMOs is highly conserved and includes a copper ion that, in its reduced form, is bound in a T-shaped geometry by three nitrogen ligands provided by two fully conserved histidine residues (4, 41, 43).
In this study, we have investigated the role of LPMOs in chitin degradation by C. japonicus. Next to cellulose-active CjLPMO10B, this microbe produces an additional LPMO, CjLPMO10A, that comprises three domains, including a CBM5 likely to bind chitin and a domain with unknown function. The impact of the LPMOs on the ability to grow on chitin was probed by comparing bacterial growth rates of wild type and lpmo gene-disrupted strains cultivated on a range of chitinous substrates. Furthermore, full-length CjLPMO10A as well as truncated variants were recombinantly expressed and biochemically characterized using x-ray crystallography, EPR, and/or enzyme activity assays. In addition to unraveling the functionality of the first three-domain LPMO ever characterized at this level of detail, these studies also revealed a novel CBM that binds with high affinity to crystalline chitin. This chitin-binding module is the first representative of the new CAZy CBM73 family.
Experimental Procedures
Cloning, Expression, and Purification of Recombinant CjLPMO10A
A codon-optimized gene for Escherichia coli expression (GenScript) encoding CjLPMO10A (residue 1–397, UniProt ID; B3PJ79), including the native signal sequence (residues 1–36), was amplified using primers with 15-base overhangs for In-Fusion cloning into the pRSET B (Invitrogen) expression vector (Table 1). Two different reverse primers were used, one to clone the full-length cjlpmo10A gene (residues 1–397) and one to clone the catalytic LPMO domain only (residues 1–216). The amplified genes were fused into the pRSET B expression vector (linearized by the restriction endonucleases NdeI and HindIII (New England Biolabs)) using the In-Fusion HD cloning kit (Clontech). Sequenced vectors were transformed by heat shock into chemically competent One Shot® BL21 StarTM (DE3) cells (Invitrogen). Fresh colonies were inoculated in lysogenic broth (LB) media containing 50 μg/ml ampicillin. The full-length enzyme was expressed by growing the transformant at 37 °C for 8 h followed by incubation for 16 h at 25 °C, whereas the truncated version was expressed by growing at 30 °C for 20 h prior to harvest. All cultures were shaken at 180 rpm during cultivation. Cells were harvested by centrifugation for 10 min at 5500 × g. The periplasmic fraction containing the mature protein (lacking the signal peptide that encodes export to the periplasm; residues 1–36) was extracted from the harvested cells using an osmotic shock method (44), and the resulting extracts were sterilized by filtration through a 0.22-μm filter and adjusted to 50 mm Tris/HCl, pH 9.0, prior to protein purification.
TABLE 1.
Primers used to amplify the gene encoding CjLPMO10A
The full-length protein and the truncated catalytic domain were cloned into the pRSET B expression vector using In-Fusion cloning, whereas the CBM5 and the C-terminal domain were cloned into the pNIC-CH expression vector using ligation-independent cloning. Vector overhang sequences are shown as boldface letters; the underlined sequences shows the NdeI (forward) and HindIII (reverse) restriction sites used to linearize the pRSET B vector prior to In-Fusion cloning.
| InFusion cloning primers | 5′ to 3′ |
|---|---|
| pRSET B_cjlpmo10A forward primer | GAAGGAGATATACATATGTTCAATACCCGT |
| pRSET B_cjlpmo10A reverse primer (full-length) | CAGCCGGATCAAGCTTTTATTGACACGAGC |
| pRSET B_cjlpmo10A reverse primer (catalytic domain) | CAGCCGGATCAAGCTTTTAGCCGAAATCAAC |
| pNIC-CH_cjcbm5 forward primer | TTAAGAAGGAGATATACTATGGACACCTGTGCTACGCTG |
| pNIC-CH_cjcbm5 reverse primer | AATGGTGGTGATGATGGTGCGCGCCGCTGGTCACGCAGTTA |
| pNIC-CH_cjcbm73 forward primer | TTAAGAAGGAGATATACTATGGGCAATTGTATCAGTCCG |
| pNIC-CH_cjcbm73 reverse primer | AATGGTGGTGATGATGGTGCGCTTGACACGAGCGGACCAG |
Periplasmic extracts were loaded onto a 5-ml Q-Sepharose FF column (GE Healthcare) connected to an ÄKTA purifier FPLC system (GE Healthcare). Elution of bound proteins was achieved by applying a linear salt gradient (0–500 mm NaCl) over 100 min at a flow rate of 3.0 ml/min. LPMO-containing fractions were pooled and concentrated to 1 ml before being loaded onto a HiLoad 16/60 Superdex 75 size exclusion column (GE Healthcare) operated with a running buffer consisting of 20 mm Tris/HCl, pH 8.0, and 250 mm NaCl, at a flow rate of 1 ml/min. Fractions containing pure LPMO were identified by SDS-PAGE and subsequently pooled and concentrated using Amicon ultracentrifugal filters (Millipore) with a molecular mass cutoff of 10 kDa. Purified LPMOs were saturated with copper by incubation with a 3-fold molar excess of Cu(II)SO4 for 30 min at room temperature as described previously (15, 45). Excess copper was removed by desalting the proteins using a PD MidiTrap G-25 desalting column (GE Healthcare) equilibrated with 20 mm Tris/HCl, pH 8.0. The concentrations of the desalted copper-saturated enzymes were measured with the Bradford assay (Bio-Rad), and the protein solutions were stored at 4 °C until further use.
The CBM5 (residues 251–309) and the C-terminal domain (residues 338–397) were cloned separately into the pNIC-CH (Addgene) expression vector by ligation-independent cloning (46) using the primers listed in Table 1. This cloning procedure adds a poly-histidine tag (His6 tag) to the C terminus of the protein. After verification of the sequences, the expression vectors were transformed into chemically competent One Shot® BL21 StarTM (DE3) cells (Invitrogen). Cells harboring the plasmids were pre-cultured for 8 h in 5 ml of LB medium containing kanamycin (50 μg/ml) and subsequently used to inoculate 500 ml of kanamycin containing TB medium (Terrific Broth) that was then incubated overnight at 20 °C in an LEX-24 Bioreactor (Harbinger Biotechnology, Canada) using compressed air for aeration and mixing. Expression was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 0.1 mm at an absorbance at 600 nm (A600) of 0.8, followed by incubation for 24 h at 20 °C. Cells were harvested by centrifugation (5500 × g, 10 min) and resuspended in lysis buffer (50 mm Tris/HCl, pH 8.0, 500 mm NaCl, and 5 mm imidazole). Cells were disrupted by pulsed sonication two times for 2 min (3 s on and 2 s off), and cell debris was removed by centrifugation (75,000 × g, 30 min). The supernatant was sterilized by filtration using a 0.22-μm syringe filter and directly loaded onto a 5-ml HisTrap HP nickel-Sepharose column (GE Healthcare) equilibrated with Buffer A (50 mm Tris/HCl, pH 8.0, 500 mm NaCl, 5 mm imidazole). Protein was eluted by applying a 25-column volume linear gradient of a buffer containing 50 mm Tris/HCl, pH 8.0, 500 mm NaCl, and 500 mm imidazole, at a flow rate of 2.5 ml/min. Protein-containing fractions were analyzed by SDS-PAGE and subsequently concentrated, with concomitant buffer exchange to 20 mm Tris/HCl, pH 8.0, using an Amicon® ultracentrifugal filter (Millipore) with a 3-kDa cutoff. The protein concentration was measured using A280 and the protein's calculated molar extinction coefficients (ϵCBM5 = 22,585 m−1 cm−1 and ϵC-terminal domain = 28210 m−1 cm−1).
Crystallization, Diffraction Data Collection, Structure Determination, and Model Refinement
Crystals of the mature catalytic LPMO10 module (CjLPMO10Acd; residues 37–216) were obtained by hanging drop vapor diffusion at room temperature by mixing equal volumes of the Cu(II)-saturated LPMO solution (in 20 mm Tris/HCl, pH 8.0) and the reservoir solution. Crystals were obtained in 0.1 m sodium acetate, pH 5.2, and 22% w/v PEG4000 at a protein concentration of 9 mg/ml. Prior to freezing, crystals were soaked in a cryo-protectant consisting of the reservoir solution adjusted to 20% PEG400. X-ray diffraction images were collected at the MASSIF ID30A beamline at the European Synchrotron Radiation Facility. The crystals generally diffracted to ∼2 Å. The collected data set was integrated, scaled, and analyzed using XDS (47), Aimless (48), and CCP4i (49). The structure was solved by molecular replacement using Phaser (50) using a polyalanine model of SmLPMO10A (CBP21; PDB 2BEM (39)). The model was rebuilt into the CjLPMO10Acd sequence using PHENIX (51), and the final refinement and model adjustments were carried out using RefMac5 (52, 53) and Coot (54).
Binding of Individual CjLPMO10A Domains to Insoluble Substrates
Binding studies were performed using a variant of the protocol described by Vaaje-Kolstad et al. (39), using shrimp shell α-chitin (Seagarden AS, Avaldsnes, Norway), β-chitin (extracted from squid pen, Batch 20140101, France Chitin, Orange, France), Avicel® PH-101 (Fluka), or Whatman No. 1 filter paper (95% pure, 0.5-μm particle size) as substrates. Each binding reaction contained 10 mg/ml substrate and 0.08 mg/ml protein in 50 mm sodium phosphate buffer, pH 7.0, and was carried out at 22 °C in an Eppendorf Comfort Thermomixer set to 1000 rpm. At various time points (2.5, 5, 15, 30, 60, 120, and 240 min), a sample was taken and filtered, using a 96-well filter plate (Millipore) operated by a Millipore vacuum manifold, to remove insoluble substrate and substrate-bound protein. The concentration of protein in the supernatant was determined by A280 (Eppendorf Biophotometer, Eppendorf, Hamburg, Germany).
The equilibrium binding constants (Kd) and binding capacity (Bmax) were determined for the CBM5 and the C-terminal domain by mixing protein solutions with varying concentrations (0, 10, 20, 50, 75, 100, 150, and 300 μg/ml) with 10 mg/ml α-chitin. Before adding the chitin, A280 was measured for each of the prepared protein solutions (in 50 mm sodium phosphate buffer, pH 7.0), to create individual standard curves for each protein. After addition of α-chitin, the solutions were placed at 22 °C in an Eppendorf Comfort Thermomixer set to 1000 rpm for 2 h. Subsequently, samples were filtered using a 96-well filter plate (Millipore), and the concentration of free protein in the supernatant was determined by measuring A280. All assays were performed in triplicate and with blanks (buffer and 10 mg/ml α-chitin). The equilibrium dissociation constants, Kd (μm), and substrate binding capacities, Bmax (μmol/g α-chitin), were determined by fitting the binding isotherms to the one-site binding equation where P represents protein: [Pbound] = Bmax [Pfree]/Kd + [Pfree], by nonlinear regression using the Prism 6 software (GraphPad, La Jolla, CA).
Electron Paramagnetic Resonance
Metal-free truncated CjLPMO10A was generated by treating the LPMO with a 1:10-fold molar surplus of EDTA overnight at 4 °C. The excess EDTA was removed by desalting the protein using a PD MidiTrap G-25 desalting column (GE Healthcare) equilibrated with 20 mm Chelex-treated (Bio-Rad) Pipes buffer, pH 6.5. To avoid excess copper, 100 μm Cu(II)SO4 was added to 110 μm apo-CjLPMO10Acd before freezing the protein in liquid nitrogen. EPR spectra were recorded as described previously (55) using a BRUKER EleXsys 560 SuperX instrument equipped with an ER 4122 SHEQ SuperX high sensitivity cavity and a liquid helium-cooled Oxford ESR900 cryostat. The microwave power was set to 0.5 milliwatts, the modulation amplitude was 5 G, and the spectra were recorded at 30 K. The EasySpin toolbox developed for Matlab was used to simulate and fit EPR spectra (56).
Enzyme Activity Assays
Reaction mixtures contained full-length or truncated CjLPMO10A at a concentration of 0.5 μm and 10 mg/ml purified shrimp shell α-chitin particles or purified squid pen β-chitin particles and 1 mm ascorbate, in 20 mm Bistris propane/HCl, pH 7.2. The reactions were incubated at 37 °C in an Eppendorf Comfort Thermomixer at 1000 rpm for up to 24 h. Samples were taken at 2, 4, 6, 8, and 24 h, and the soluble fractions were immediately separated from the insoluble substrate particles by filtration using a 96-well filter plate (Millipore) operated by a Millipore vacuum manifold. By separating soluble and insoluble fractions, LPMO activity is stopped, as CjLPMO10A does not oxidize soluble chito-oligosaccharides (results not shown). A part of the soluble fraction from the 24-h samples was analyzed by MALDI-TOF MS and UPLC (see below), and the remaining parts, as well as the samples from the earlier time points, were further degraded by the S. marcescens GH20 β-N-acetylhexosaminidase, also known as chitobiase (SmCHB), for quantification of chitobionic acid (GlcNAcGlcNAc1A) as described previously (45). Before adding chitobiase to a final concentration of 1.6 μm, the pH was lowered by adding BisTris buffer, pH 6.0, to a concentration of 50 mm. The reactions were incubated at 37 °C for 2 h and thereafter analyzed by UPLC (see below for details).
Synergy experiments for degradation of α-chitin (10 mg/ml) were performed with 0.5 μm full-length or truncated CjLPMO10A (the latter variant is named CjLPMO10Acd) and 0.5 μm the S. marcescens GH18 endochitinase C (SmChi18C). The reactions were performed in triplicate, and the time points and conditions were identical to those described above. The soluble fraction obtained at each sampled time point was further degraded by 1.6 μm chitobiase for quantification of GlcNAc (native monomer) and chitobionic acid (oxidized dimer). To quantify the oxidized products that remain in the insoluble fraction (i.e. long oxidized chitin chains that remain bound to the chitin particles), half of the enzyme reaction after 24 h was boiled for 10 min to ensure complete inactivation of the LPMO. Thereafter, 5 μm SmChi18A, an exochitinase, and 3 μm SmChi18C were added, and the samples were incubated for another 24 h to totally solubilize chitin particles and thereby release all oxidized products in the sample. The samples were then treated with SmCHB to enable quantification of chitobionic acid.
Qualitative and Quantitative Analysis of (GlcNAc)nGlcNAc1A and GlcNAc
Qualitative analysis of oxidized chito-oligosaccharides was carried out using a 150 × 2.1-mm HILIC column (Acquity UPLC BEH Amide) and the protocol described by Loose et al. (45). The sample injection volume was set to 10.0 μl, and the UPLC was operated with a flow rate of 0.4 ml/min and a gradient combining 100% acetonitrile (solvent A) and 15 mm Tris/HCl, pH 8.0 (solvent B), as follows: 80% A, 20% B was run for 3.5 min, followed by a 5.5 min gradient to 70% A, 30% B and a 0.5 min gradient to 55% A, 45% B. The latter condition was held for 1 min, followed by column reconditioning obtained by a 1-min gradient back to initial conditions (80% A and 20% B) and subsequent running at these conditions for 4 min. Products were detected by monitoring absorbance at 205 nm (45).
Quantification of GlcNAc and GlcNAcGlcNAc1A was accomplished using an RSLC system (Dionex) equipped with a 100 × 7.8-mm Rezex RFQ-Fast Acid H+ (8%) (Phenomenex, Torrance, CA) column operated at 85 °C. Eight-μl samples were injected into the column, and solutes were eluted isocratically using 5 mm sulfuric acid as mobile phase with a flow rate of 1 ml/min.
Standards of GlcNAcGlcNAc1A (10–3000 μm) and GlcNAc (50–8000 μm) were used for quantification; GlcNAc (99% purity) was purchased from Sigma, and GlcNAcGlcNAc1A was generated in-house by complete oxidation of N-acetyl-chitobiose (Megazyme; 95% purity) by the Fusarium graminearum chitooligosaccharide oxidase (ChitO) as described previously (45, 57).
C. japonicus Gene Disruption
Insertional inactivation of C. japonicus genes was performed using vector integration mutagenesis (58). Briefly, a 500-bp internal region of the gene to be disrupted was cloned into the plasmid pK18mobsacB (59) at the EcoRI and XbaI sites. This plasmid was introduced into E. coli strain S17 λPIR via electroporation. Conjugation of the plasmid into C. japonicus was performed by tri-parental mating as done previously (60) using an additional strain of E. coli containing plasmid pRK2013 (61). Selection of C. japonicus mutants was on minimal medium supplemented with 50 μg/ml kanamycin. Mutants were confirmed via PCR screening.
Growth Analysis of C. japonicus Strains
All strains were grown using MOPS-defined growth medium. Glucose, N-acetylglucosamine (GlcNAc), and β-chitin (squid pen) were used as sole carbon sources at a concentration of 0.2% (w/v). Shells from Chesapeake blue crabs (Callinectes sapidus) were crushed to small sized pieces (∼4 mm2) and used as a sole carbon source at a concentration of 1% (w/v). All cultures were incubated at 30 °C with a high level of aeration (225 rpm). Growth was measured using absorbance (A600) with a Spec20D+ spectrophotometer (Fisher) with 18-mm culture tubes. After removal from a shaking incubator, the chitin and crab shell-containing tubes sat on the bench top for 5 min before A600 reading to allow insoluble chitin material to settle to the bottom of the tube. All growth experiments were performed in biological triplicates. Growth rate calculations were performed with the Prism 6 software package (GraphPad, La Jolla, CA).
Results
Sequence Analysis of CjLPMO10A
CjLPMO10A consists of 397 amino acids comprising a signal peptide (residues 1–36) that is cleaved off during secretion, followed by an N-terminal family AA10 LPMO domain (residues 37–216; CjLPMO10Acd),a CBM5 substrate binding domain (residues 251–309), and a C-terminal uncharacterized domain (residues 338–397) (see Fig. 1A). Phylogenetic analysis of selected AA10 LPMO sequences (catalytic domains only) shows that CjLPMO10Acd does not cluster with any of the established clades or subclades (Fig. 1B), as also observed by Book et al. (62). The most similar characterized chitin- and cellulose-oxidizing LPMO10s (SmLPMO10A (CBP21) and ScLPMO10C) show 33 and 36% sequence identity to the CjLPMO10Acd sequence, respectively. The CBM5 sequence is typical for chitin-active enzymes and is 45% identical to a well described ortholog present in a Streptomyces griseus chitinase (SgChi18C), for which the three-dimensional structure is known (63). The sizes of the C-terminal domain and the CBM5 are similar (∼60 residues), and despite low overall sequence similarity, conserved aromatic amino acids of the CBM5s align with similar residues in the C-terminal domain (Fig. 2). The C-terminal domain is rich in glycine (seven), cysteine (four), tryptophan (four), and tyrosine (four) residues. This domain is also found in three of the four C. japonicus GH18 chitinases. Analysis of the sequence by Pfam and dbCAN did not yield any significant results. The sequence analysis in combination with binding data (see below) show that the C-terminal domain is a chitin-specific CBM and is the first representative in a new CAZy family called CBM73.
FIGURE 1.
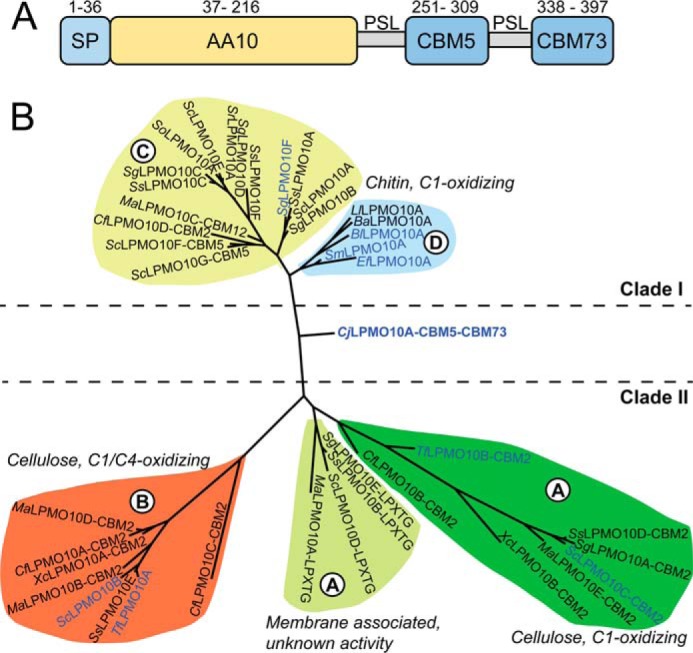
Sequence analysis of CjLPMO10A. A, domain architecture of CjLPMO10A. The full-length enzyme contains a signal peptide (SP: residues 1–36) that is cleaved off during secretion resulting in the mature enzyme that possesses an N-terminal AA10-type LPMO domain followed by a family 5 chitin-binding module (CBM5) and a C-terminal CBM classified as CBM73 (see text). The three domains are separated by two poly-serine linkers (PSL). The modules and linkers are scaled according to the number of amino acids they contain. B, phylogeny of selected LPMOs from auxiliary activities family 10 built on a previous classification by Book et al. (62), using Phylogeny.fr (64). Enzyme names include additional domains and other relevant sequence motifs, but the phylogeny is based on the catalytic domains only. The indicated clades are as defined in the study by Book et al. (62). Clade I (subclade C and D) contains chitin-oxidizing LPMOs and Clade II (subclade A and B) contains cellulose-oxidizing LPMOs as well as membrane-associated LPMOs with unknown function. Blue colored protein names represent enzymes that have been characterized.
FIGURE 2.

Multiple sequence alignment of CBM5s and CBM73s. The five CBM5 sequences originate from C. japonicus LPMO10A, Chi18C, and Chi18D, Cellvibrio mixus LPMO10, and S. griseus Chi18C. The five CBM73 sequences originate from C. japonicus LPMO10A, Chi18B, Chi18C, Chi18D and C. mixus LPMO10. The pink stars indicate two aromatic residues (YW or WW) for which experiments have shown that they are important for substrate binding by CBM5s (63).
Three-dimensional Structure of CjLPMO10Acd
The structure of the catalytic domain of CjLPMO10A containing copper in the active site was determined to 1.85-Å resolution with three molecules in the asymmetric unit (Table 2). The overall structure of CjLPMO10Acd has a pyramid-like shape that is characteristic for all LPMOs, displaying an Ig-like β-sandwich fold with a large protrusion that contains several small helices and loops (Fig. 3). The β-sandwich is formed by two distorted β-sheets, one containing three antiparallel β-strands (S1, S4, and S7) and one containing four antiparallel β-strands (S5, S6, S8, and S9). The two short β-strands S2 and S3 are oriented antiparallel or parallel, respectively, to the four-stranded β-sheet (Fig. 3A). The large protrusion, between β-strands S1 and S3, faces β-strands S1 and S9 and contains five short α-helices and several loops and contributes to approximately half of the putative substrate binding surface. Notably, those protrusions contain most of the structural diversity in all LPMO families. Structural alignment of CjLPMO10Acd and other structurally characterized LPMO10s shows that the C. japonicus enzyme shares features with both chitin- and cellulose-oxidizing LPMO10s (Fig. 4). Similar to ScLPMO10C (CelS2), ScLPMO10B, and BaLPMO10A, in CjLPMO10Acd the planar face of the substrate-binding surface protrusion is extended by one amino acid (Glu-56) compared with SmLPMO10A (CBP21) and EfLPMO10A.
TABLE 2.
Diffraction data and refinement statistics for the CjLPMO10Acd structure (PDB code 5FJQ)
| Data collection | Refinement statistics | ||
|---|---|---|---|
| Beamline | ID30A (ESRF, Grenoble) | Rcryst/Rfree (%)a | 22.1/28.4 |
| Wavelength (Å) | 0.965 | Root mean square deviation bond lengths (Å) | 0.02 |
| Space group | C2 | Root mean square deviation bond length angles (°) | 1.9 |
| Unit cell dimensions | No. of atoms/B-factor | ||
| a, b, c (Å) | 91.50, 50.17, 114.53 | Protein | 4320/20.5 |
| α, β, γ | 90.0, 90.0, 90.0 | Solvent | 541/27.5 |
| Resolution (Å) | 45.75 to 1.85 (1.90 to 1.85) | Metal | 3/22.7 |
| Unique reflections | 43,624 (3175) b | Ramachandran plot (%)c | |
| Multiplicity | 2.25 (2.35) | Most favored | 95.5 |
| Completeness (%) | 96.9 (96.5) | Additionally allowed | 3.9 |
| Mean I/σI | 13.1 (5.4) | Outliers | 0.6 |
| Rmeasd | 0.063 (0.196) | ||
a Rcryst = Σhkl‖Fo‖Fc‖/Σhkl‖Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated from a randomly chosen 5% sample of all unique reflections not used in refinement.
b Values in parentheses are for the highest resolution shells.
c Data were defined using MolProbity (66).
d Rmeas was defined by Diederichs and Karplus (65).
FIGURE 3.

Three-dimensional structure of CjLPMO10Acd. A, cartoon representation of the CjLPMO10Acd secondary structure. α-Helices are shown in blue, and β-strands are shown in yellow. The copper ion is shown as an orange sphere, and the coordinating histidine side chains (His-37 and His-136) are shown as gray sticks. The dotted circle shows the protrusion that includes most of the structural diversity in LPMOs. B, surface projection of the substrate binding surface in CjLPMO10Acd, chain B. The main chain is shown in cartoon representation, and side chains are shown as sticks. The copper ion and selected solvent water molecules are shown as orange and red spheres, respectively. Surface-exposed residues are labeled using the single letter amino acid code, and those discussed in the main text are marked with bold letters (E56, Q78, E79, T133, R197, D202, and E205). Glu-79 and Thr-76 show different rotamers in chain A (yellow sticks) compared with chain B and C (chain B; gray sticks).
FIGURE 4.

Copper active sites of C1-oxidizing LPMO10s. A, superposition of the CjLPMO10A copper active site with chitin-active Enterococcus faecalis LPMO10A (PDB 4ALC (67)) and cellulose-active S. coelicolor LPMO10C (PDB 4OY7 (15)). Carbon atoms in the three enzymes are colored yellow (EfLPMO10A), green (ScLPMO10C), and gray (CjLPMO10A). Side chains are colored according to the following properties: residues fully conserved in all LPMO10s (purple), residues highly conserved among chitin- and cellulose-specific LPMO10s (yellow and green, respectively), and residues that are not conserved (gray). Residues that are either fully or not conserved are numbered according to the CjLPMO10A sequence. Note the similar spatial location of Glu-64 in EfLPMO10A and Glu-205/Glu-217 in the other two enzymes. B, sections of a structural sequence alignment related to the active sites in four chitin-oxidizing LPMO10s (SmLPMO10A, PDB 2BEM; BaLPMO10A, PDB 2YOY; EfLPMO10A, PDB 4ALC; and CjLPMO10A, PDB 5FJQ) and one cellulose-active LPMO10 (ScLPMO10C, PDB 4OY7); the sequence of cellulose-oxidizing T. fusca LPMO10B, for which no structure is known, was added to the structure-based alignment. The top arrows illustrates on which β-strands the residues are found. The green and yellow color indicates similarity to cellulose-active ScLPMO10C and chitin-active EfLPMO10A, respectively. The WDR insertion in CjLPMO10A is located ∼15 Å from the catalytic center, and its side chains are not part of the presumed substrate-binding surface shown in Figs. 3B and 4A. C, active site of CjLPMO10A (chain B). D, active site of the copper-soaked AA10 from Melolontha melolontha entomopoxvirus (PDB 4X27 (68)) which shows 27% overall sequence identity with CjLPMO10A.
The active site of CjLPMO10A is similar to other LPMO10s, containing a copper ion coordinated by two histidines (His-37 and His-136) in a T-shaped histidine brace (Fig. 4). Of the three protein chains in the asymmetric unit, two (chain A and C) show copper coordinating protein ligands only, whereas one (chain B) also shows copper coordinating two water molecules (Fig. 3B). The variation in copper coordination in the three chains may reflect variation in the oxidation state of the copper (43, 67).
Interestingly, the direct environment of the copper coordination sphere shows features of both cellulose and chitin-active LPMO10s, even though CjLPMO10A is active toward chitin substrates only (see below for activity data). The cavity positioned in close proximity to the active site in all chitin-specific LPMOs with known structure (members from family AA10 and one AA11) is in CjLPMO10A filled by Arg-197, Asp-202, and Glu-205, similar to what is observed in cellulose-oxidizing ScLPMO10C (Fig. 4A). Arg-197 is positioned in proximity to His-136 and shares an H-bond with Glu-205 that coordinates one of the water molecules interacting with the copper ion in chain B. The positioning of Glu-205 in relation to the active site is similar to what is observed for cellulose C1-oxidizing LPMO10s (15), viral AA10s with unknown substrate specificity (68), chitin-active AoLPMO11 (11), and starch-active AoLPMO13 (13) but different from previously characterized chitin-oxidizing LPMO10s. Whereas this glutamate in other chitin-oxidizing LPMO10s is located on the second β-strand (S2), the similarly positioned glutamate in CjLPMO10A is found on β-strand nine (S9), as in cellulose-active LPMO10s. Gln-78 and Thr-133 in CjLPMO10A are highly conserved in chitin-active LPMOs (family AA10 and AA11) and tend to be Phe and Trp in the cellulose-active family AA10 LPMOs (Fig. 4B). Thus, based on currently available structure-function data, this particular feature, i.e. the Gln-Thr pair, stands out as a possible determinant of chitin activity. A special characteristic of CjLPMO10A is the presence of a non-conserved glutamate (Glu-79), which directly interacts with the N-terminal histidine (His-37) in chain B and C but shows another rotamer in chain A (Fig. 3B). Noteworthy, the active site of CjLPMO10A bears resemblance to the active site of the recently described entomopoxvirus AA10 structures (Fig. 4D), whose activity has not been demonstrated yet (68). It should be noted that a cavity formed by the non-conserved residues Thr-76, Glu-79, Arg-103, Ala-104, and Asn-105 is present on the putative binding surface (Fig. 3B), but the significance of this topological feature is unknown.
EPR Spectroscopy
The immediate environment of the active site copper ion in CjLPMO10Acd was analyzed by EPR spectroscopy, as described previously (55), and the EPR spectra were simulated (Fig. 5). The estimated spin Hamiltonian parameters are summarized in Table 3. The g and ACu tensors reflect the copper coordination in the LPMO active site, and of these the gz and AzCu could be calculated with high accuracy. The gz and AzCu tensors for CjLPMO10A (gz = 2.267; AzCu = 153·10−4 cm−1) are different compared with other chitin-oxidizing LPMO10s, which yield values that fall between type 1 and type 2 copper enzymes (43, 55). With a higher AzCu value, the EPR spectrum of CjLPMO10A resembles that of cellulose-oxidizing LPMOs that yield EPR signals typical for type 2 copper centers (4, 11, 13, 15, 41, 55) based on the Peisach-Blumberg classification of type 1 and type 2 copper enzymes (69).
FIGURE 5.

Electron paramagnetic resonance spectroscopy of Cu(II)-saturated CjLPMO10Acd. A, X-band EPR spectra (—) with simulation (- - -) for Cu(II)-saturated CjLPMO10A (top) and Cu(II) in Pipes buffer, pH 6.5. B, comparison of EPR signals of CjLPMO10A, ScLPMO10C (cellulose-active), and SmLPMO10A (chitin-active). The EPR spectra were recorded at 30 K using a microwave power of 0.5 milliwatts.
TABLE 3.
Spin Hamiltonian parameters
Data assume co-linear g and ACu tensors in all simulations.
| Parameter | Cu(II) buffer | CjLPMO10A | ScLPMO10Ca | SmLPMO10Aa |
|---|---|---|---|---|
| gx | 2.059 | 2.036 | 2.015 | 2.039 |
| gy | 2.059 | 2.091 | 2.102 | 2.116 |
| gz | 2.270 | 2.267 | 2.267 | 2.260 |
| AxCub | 12.3 | 31.8 | 11.7 | 42.3 |
| AyCub | 12.3 | 26.1 | 17.0 | 50.3 |
| AzCub | 165 | 154 | 153 | 116 |
a Data were from a previous study by Forsberg et al. (55). ScLPMO10C acts on cellulose; SmLPMO10A acts on chitin.
b The unit used is 10−4 cm−1.
CjLPMO10A Domains Bind to Chitin
The purified LPMO and CBM domains were used in binding assays to determine their substrate specificities. The protein yields (pure protein) were 1.2, 18, 5, and 6.3 mg/liter culture for CjLPMO10A, CjLPMO10Acd, CBM5, and the C-terminal domain, respectively. Reduction of the protein concentration measured as A280 over time showed that the LPMO domain (CjLPMO10Acd) binds weakly and slowly to α- and β-chitin and not to cellulose (Fig. 6A). The CBM5 (Fig. 6B) showed strong binding to the two chitin substrates and weak binding to the cellulose substrates (Avicel and filter paper). The data for the C-terminal domain showed even stronger binding, with 90% of the binding to both α- and β-chitin within 15 min of incubation (Fig. 6C). Also, this domain seemed more specific than the CBM5 because no binding to cellulose was observed. All in all, the data indicate that the C-terminal domain is a novel chitin-binding module, which has been given the family name CBM73 by the curators of CAZy. To obtain further insight into the binding of the CBMs, binding dissociation constants (Kd) and binding capacities (Bmax) were determined (Fig. 6D) using α-chitin as a substrate. The Kd and Bmax values obtained were 5.3 μm and 4.8 μmol/g α-chitin for the CBM5 module and 4.3 μm and 6.4 μmol/g α-chitin for the CBM73 module. These values are in the same range as dissociation constants and binding capacities reported for other CBMs (39, 70, 71).
FIGURE 6.

Binding of the CjLPMO10A domains to chitin and cellulose. A–C show binding of the catalytic LPMO domain (A), the CBM5 domain (B), and the C-terminal domain (C) to chitin and cellulose substrates. The percentage of free protein was determined by measuring the reduction in protein concentration (A280) over time, in the absence of an electron donor. The experiments were carried out at 22 °C using 10 mg/ml substrate (α-chitin, β-chitin, Avicel, or filter paper) in 50 mm sodium phosphate buffer, pH 7.0. D, plots of binding data for the CBM5 (■) and the C-terminal (CBM73) domain (●) incubated with α-chitin. Pbound corresponds to bound protein (μmol/g substrate), and Pfree corresponds to non-bound protein (μm). Each point represents the average of values obtained in three independent experiments.
CjLPMO10A Is a Chitin-targeting LPMO
Chromatographic and mass spectrometry analysis of soluble products generated upon incubation of CjLPMO10A with a range of polysaccharide substrates (cellulose, mannans, xyloglucan, xylans, starch, soluble chito-oligosaccharides, and chitin) revealed activity toward α- and β-chitin (see below), whereas activity toward other substrates was not detected (results not shown). The products detected upon incubation with chitin were chito-oligosaccharide aldonic acids (i.e. products oxidized at the C1 carbon), ranging in degree of polymerization from three (DP3) to eight (DP8), with DP6 being the most abundant product after 24 h of incubation (Fig. 7). Product profiles were similar for α- and β-chitin. The full-length enzyme produced significantly higher amounts of product than the truncated enzyme (CjLPMO10Acd) on both α- and β-chitin, indicating that one or both of the additional domains (CBM5 and the C-terminal domain) are important for the efficiency of the enzyme.
FIGURE 7.

Activity of full-length and truncated CjLPMO10A. A, chromatographic analysis of oxidized chito-oligosaccharides from degradation reactions containing 10 mg/ml α-chitin (top chromatograms) or β-chitin (middle chromatograms) after 24 h of incubation with full-length (solid lines) or truncated (dotted lines) CjLPMO10A, with standards for oxidized chito-tetraose (DP4ox) and chito-pentaose (DP5ox). B, MALDI-TOF MS analysis of products generated by the full-length LPMO acting on α-chitin. Prior to the analysis, the samples were saturated with sodium, to simplify the spectra. For each oligomeric product, two major adducts are observed: [DP4–8ox + Na]+ and [DP4−8ox−H + 2Na]+. All the reactions were carried out in 20 mm Bistris propane buffer, pH 7.2, with 0.5 μm Cu2+-saturated LPMO and 1 mm ascorbate as external electron donor.
CjLPMO10A Acts in Synergy with an Endochitinase
Synergy experiments showed that the degradation of α-chitin by S. marcescens endochitinase (SmChi18C) was substantially increased in the presence of either full-length or truncated CjLPMO10A (Fig. 8A). The level of synergy obtained was equal for both CjLPMO10A variants. In both cases, after 24 h 19.2 and 5.0% of the chitin were solubilized in the presence and absence of the LPMO, respectively. In the absence of a chitinase, CjLPMO10A and CjLPMO10Acd dissolved 5.2 and 1.4% of the chitin after 24 h of incubation, respectively. Quantification of solubilized oxidized products showed that the two CjLPMO10A variants are equally active when SmChi18C is present in the reaction mixture (Fig. 8B). In the absence of the chitinase, the full-length LPMO showed higher activity, whereas the truncated LPMO variant appeared unaffected except for the final sampling time point of the reaction (Fig. 8B). The data thus show that, in the absence of a chitinase, the activity of the truncated variant (CjLPMO10Acd) ceases earlier compared with the full-length enzyme (Fig. 8B).
FIGURE 8.

Synergy between CjLPMO10A and the SmChi18C endochitinase in the degradation of α-chitin. A and B show quantification of GlcNAc and chitobionic acid (GlcNAcGlcNAc1A), respectively, obtained after chitobiase digestion of soluble products from the degradation of 10 mg/ml α-chitin. C, peak areas for chitobionic acid from solubilized material only (gray) and from the complete reaction mixture (black) products after degradation of 2 mg/ml α-chitin using full-length or truncated CjLPMO10A. The total oxidized sugar content was measured after full solubilization of the LPMO-pretreated chitin substrate by the S. marcescens chitinases Chi18A, Chi18C, and chitobiase (CHB). All reactions were carried out at 37 °C in Bistris propane buffer, pH 7.2, in triplicate with 0.5 μm Cu2+-saturated LPMO and/or 0.5 μm SmChi18C and in the presence of 1 mm ascorbate as external electron donor.
The quantity of soluble products formed by full-length and truncated CjLPMO10A was then compared with the quantity of oxidized products remaining bound to the insoluble chitin fraction. After incubation of the enzyme with the substrate for 24 h, essentially all products generated by the full-length enzyme were soluble, whereas ∼75% of the products formed by the truncated enzyme remained in the insoluble fraction of the substrate (Fig. 8C).
CjLPMO10A Is Required for Efficient Utilization of Insoluble Chitinous Substrates
Growth analysis was performed on wild type C. japonicus and gene disruption mutants of CjLPMO10A and CjLPMO10B. Wild type and mutant strains all grew well in defined media that had either glucose or GlcNAc as a sole carbon source (Fig. 9, A and B). However, when grown on insoluble chitin substrates, the CjLPMO10A mutant showed a reproducible growth defect on β-chitin leading to an ∼2-fold slower growth rate compared with the wild type strain (Fig. 9C). The CjLPMO10B mutant also showed a slight growth defect. A mutant with a defect in the type II secretion system (general secretory protein) was completely unable to grow using insoluble chitinous substrates (Fig. 9, C and D). The type II secretion system has been shown to be the sole mechanism responsible for secretion of carbohydrate-active enzymes into the environment by C. japonicus (60). Thus, growth of the general secretory protein mutant reflects chitin-independent growth of C. japonicus, which, as expected, did not occur. When grown on an unprocessed chitinous substrate, crab shells, the CjLPMO10A mutation led to an extended lag phase of about 100 h, whereas the growth rate was only slightly reduced (Fig. 9D).
FIGURE 9.

Growth of C. japonicus wild type and mutants on various substrates. Wild type and mutant strains were grown in MOPS defined medium with glucose (A), N-acetylglucosamine (B), squid pen β-chitin (C), or unprocessed crab shell (α-chitin) (D) as the sole carbon source. Measurement of growth used optical density (OD) at 600 nm. All experiments were run in biological triplicate with error bars showing standard deviation. Strains presented are wild type (closed circles), general secretory protein mutant (closed squares), CjLPMO10A mutant (open diamonds), and CjLPMO10B (open inverted triangles).
Discussion
C. japonicus is a well studied plant cell wall-degrading bacterium possessing an extensive portfolio of carbohydrate-active enzymes targeting cellulose and other complex plant cell wall polysaccharides (60, 72, 73). However, the genome also contains a system for chitin utilization that has not been studied previously. Apart from the chitin-oxidizing CjLPMO10A described here, the putative chitinolytic machinery includes four GH18 chitinases, one GH19 chitinase, two GH20 N-acetyl-β-hexosaminidases, and one GH46 chitosanase (72). Indeed, our results demonstrate that the bacterium can grow on both pure chitin and an environmentally relevant chitinous substrate (crab shell). The growth data largely corroborate the enzymology for the two LPMOs produced by C. japonicus, showing that CjLPMO10B is specific for cellulose, and CjLPMO10A is specific for chitin. The growth data show that the absence of CjLPMO10A prevents C. japonicus from degrading chitin efficiently, although it is still able to reach growth levels obtained by the wild type. This suggests that the complement of chitinases harbored by the bacterium is able to degrade the insoluble substrate, although not as effectively as in the presence of LPMO activity. This arrangement mirrors the enzyme synergy observed for cellulose degradation by the bacterium, where the cellulose-active LPMO, CjLPMO10B, was indicated to be important for efficient cellulose utilization (36). Somewhat surprisingly, the CjLPMO10B mutant showed reduced growth on chitin, albeit to a much lesser extent than the CjLPMO10A mutant. We do not currently have an explanation for this observation, which may be taken to suggest the presence of previously undiscovered LPMO functionalities.
The ability of C. japonicus to grow on crab shells is not surprising because these shells represent a growth medium rich in protein and carbohydrates (chitin). The crab exoskeleton is constructed of a rigid composite material containing protein-coated α-chitin fibers embedded in a CaCO3 matrix. In the absence of transcriptomic or proteomic data, it is not possible to determine whether it was the protein or the chitin (or both) that was used as a carbon/nitrogen source in the growth experiment depicted in Fig. 9D. Nevertheless, the 100-h lag in growth observed for the CjLPMO10A mutant indicates that CjLPMO10A has a role in rendering the substrate accessible for the bacterium. It is tempting to speculate that the LPMOs are important in the initial growth phase where the concentration of chitinases is low and the LPMO-chitinase synergy is required to solubilize enough substrate to sustain growth.
Since the discovery of LPMOs in 2010 (1), several studies have been published that describe the biochemical and biophysical aspects of these enzymes (4–6, 8, 13, 14, 18, 41, 42). CjLPMO10A is interesting because it represents a chitinolytic enzyme in the uncharacterized chitin-degrading machinery of C. japonicus, and also because its amino acid sequence is substantially different compared with other LPMO10s studied (Fig. 1) (62). Structurally, the overall fold and shape of CjLPMO10A are similar to other LPMO structures, but the pattern of residues on the substrate-binding surface is different. In general terms, the binding surface seems to represent a “hybrid” of the surfaces of known chitin-active and known cellulose-active LPMO10s. The surface also seems to be more extended, in particular through a glutamate (Glu-56) that is far from the active site (Fig. 3B). Also, CjLPMO10A has a cavity in the non-conserved protrusion of the substrate-binding surface (Fig. 3), which is unlike any other LPMO structure published. Other chitin-active LPMO10s have a similarly sized cavity in close proximity to the active site that has been proposed to accommodate O2 (41) or an acetyl group of a GlcNAc (15) during LPMO catalysis. This active site cavity is not present in cellulose-targeting LPMO10s, where a positively charged side chain (arginine or histidine) fills the space represented by the cavity (Fig. 4A) (15). Interestingly, this is also the case for CjLPMO10A, where Arg-197 and, to a lesser extent, Asp-202 and Glu-205 fill the cavity (Figs. 3 and 4). This is another feature rendering the extended active site architecture of CjLPMO10A more similar to that of LPMO10s that are active on cellulose. The hybrid character of CjLPMO10A is also visible in the alignment of sequence fragments surrounding the catalytic center shown in Fig. 4; Arg-197 and Glu-205 in CjLPMO10A are common to cellulose-active LPMO10s, whereas Gln-78 and Thr-133 are common to chitin-active LPMOs. Furthermore, the EPR signature of CjLPMO10A is more similar to the cellulose-active LPMO10s (like ScLPMO10C) than to chitin-active LPMO10s (like SmLPMO10A). The specificity of CjLPMO10A toward chitin (and not cellulose) is intriguing, and further studies of this enzyme may eventually allow a deeper insight into the determinants for LPMO substrate specificity. Interestingly, comparison of CjLPMO10A with the recently published AA10 structure of viral origin (68) revealed an intriguing similarity between the catalytic centers and substrate-binding surfaces of these enzymes (Fig. 4, C and D), even though the overall sequence similarity is low (27% sequence identity). The viral AA10s are essential for viral penetration of the chitinous insect gut peritrophic matrix (that contains α-chitin), which enables infection by virus particles (68).
An interesting property of CjLPMO10A is the presence of two CBMs, a common CBM5 chitin-binding module and a previously uncharacterized C-terminal domain (Fig. 1A). The present results clearly show that the C-terminal domain is a chitin-specific CBM, which has founded the CBM73 family in CAZy. Homologues of CBM73 are present in chitinases from other bacteria from the phylum of Proteobacteria, including three of the four C. japonicus GH18 chitinases as well as several Vibrio sp. chitinases. Despite low sequence similarity to the CBM5 family, conserved substrate-binding aromatic amino acids of the CBM5s align remarkably well with similar residues in the CBM73s (Fig. 2). This indicates that the mechanism of substrate binding may be similar to the CBM5s and the CBM73s. It should be noted, however, that the presence of two additional cysteines, two additional aromatic residues, and multiple glycines (Fig. 2) may endow the CBM73 with properties that are different from CBM5s. Indeed, the two domains showed differences in the binding studies with α-chitin (Fig. 6D), and in contrast to the CBM5, the CBM73 did not bind to cellulose. A deeper investigation of the CBM73s is needed to unravel their structure and the mechanism of substrate interaction and to unravel the degree of functional similarity and complementarity vis-á-vis the CBM5.
Fig. 8B shows that the presence of the CBMs in CjLPMO10A has a major effect on the solubilization of oxidized products by the LPMO. Solubilization ceases much earlier for the truncated variant than for the full-length enzyme. Because release of soluble oxidized products normally requires two cleavages in the same chitin chain, one possible explanation for the observed differences is that the CBMs target the LPMO to similar regions of the substrate, resulting in lytic events in a limited area, i.e. with a high chance of hitting the same chain twice. For the truncated variant, a higher fraction of oxidized chain ends remains attached to the insoluble substrate (Fig. 8C). However, overall, the truncated variant is clearly less active than the full-length enzyme. Therefore, a more probable explanation for the observed differences is that the CBMs enable the LPMO to bind productively to regions of the substrate that are inaccessible for the LPMO domain alone. This explanation is compatible with the binding data showing that the catalytic module alone binds more weakly to chitin than the full-length enzyme (Fig. 6).
Most interestingly, although the presence or absence of the CBMs has a clear effect on LPMO functionality (binding affinity and oxidative activity) when acting alone, the CBMs did not influence the strong synergy obtained when combining CjLPMO10A with an endochitinase in chitin solubilization experiments (Fig. 8A). One possible explanation is that the amount of LPMO added was saturating and that the activity difference between the two variants thus became invisible in terms of overall substrate solubilization (Fig. 8A; note that both enzymes are endo-acting). Importantly, Fig. 8B shows that in the presence of the chitinase the LPMO variants catalyzed similar degrees of substrate oxidation (in contrast to what was observed for the LPMO variants acting alone). Assuming that the LPMO cannot bind productively to a substrate surface that already has been oxidized by the same LPMO, an attractive explanation for these observations is that after LPMO action, the chitinase removes chitin chains in the oxidized region of the substrate. By doing so, the chitinase could uncover new binding surfaces for the LPMOs that may be equally accessible to both LPMO variants. Thus, it may be that the LPMO and the endo-acting chitinase work closely together, removing the substrate layer by layer by their combined action. Such a scenario would add a new dimension to our perception of how LPMOs and GHs collaborate.
In conclusion, we have shown that C. japonicus is able to metabolize chitin in both its pure and native form. The ability of the bacterium to grow efficiently on this recalcitrant substrate is dependent on the presence of the chitin-specific LPMO, CjLPMO10A, an enzyme with an unusual sequence and active site architecture compared with other chitin-active LPMO10s. The presence of two CBMs coupled to the LPMO catalytic domain enables the enzyme to bind tightly to chitin and likely enables the enzyme to access substrate regions that are not accessible for the catalytic domain only.
Author Contributions
Z. F. determined the biochemistry of CjLPMO10A and contributed to the writing of the paper. C. E. N. performed and analyzed the microbiological and genetic experiments in Fig. 9. B. D. determined the crystal structure of CjLPMO10Acd. S. M. designed the REZEX method for quantification of GlcNAc and GlcNAcGlcNAc1A and provided S. marcescens chitinase A and C. J. S. M. L. provided SmCHB and ChitO-oxidized chito-oligosaccharide standards. L. I. C. contributed to experimental design. Å. K. R. designed and analyzed the EPR experiment. J. G. G. and V. G. H. E. contributed to the design and analysis of the experiments and contributed to writing the manuscript. G. V.-K. supervised the work, coordinated the study, and contributed to writing the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank N. Beri for providing the C. sapidus shells and for technical assistance. We are grateful for synchrotron travel support from The Research Council of Norway Grant 216625/F50. We also thank the European Synchrotron Radiation Facility staff for help and beamtime at beamline ID30A Project MX-1468.
This work was supported by The Norwegian Academy of Science and Letters Vista Program Grant 6510, Research Council of Norway Grants 214138, 214613, and 221576, funds provided by University of Maryland at Baltimore County College of Natural and Mathematical Sciences (to the Gardner laboratory), a United States Department of Education Graduate Assistance in Areas of National Need fellowship (to C. N.), NIGMS Initiative for Maximizing Student Development Grant R25-GM55036 from the National Institutes of Health (the IMSD Meyerhoff Graduate Fellows Program), and South-Eastern Norway Regional Health Authority Grants 2012085 and 2015095 (Regional Core Facility for Structural Biology; to B. D.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5FJQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LPMO
- lytic polysaccharide monooxygenase
- AA
- auxiliary activity
- CAZy
- carbohydrate-active enzyme database
- CBM
- carbohydrate-binding module
- cd
- catalytic domain
- GH
- glycoside hydrolase
- gsp
- general secretory protein
- PDB
- Protein Data Bank
- Bistris
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- UPLC
- ultra performanceliquid chromatography.
References
- 1. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. H. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 2. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Levasseur A., Drula E., Lombard V., Coutinho P. M., and Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., and Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 6. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 7. Westereng B., Ishida T., Vaaje-Kolstad G., Wu M., Eijsink V. G. H., Igarashi K., Samejima M., Ståhlberg J., Horn S. J., and Sandgren M. (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6, e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Agger J. W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W. G., Ludwig R., Horn S. J., Eijsink V. G. H., and Westereng B. (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. U.S.A. 111, 6287–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frommhagen M., Sforza S., Westphal A. H., Visser J., Hinz S. W., Koetsier M. J., van Berkel W. J., Gruppen H., and Kabel M. A. (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol. Biofuels 8, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bennati-Granier C., Garajova S., Champion C., Grisel S., Haon M., Zhou S., Fanuel M., Ropartz D., Rogniaux H., Gimbert I., Record E., and Berrin J. G. (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hemsworth G. R., Henrissat B., Davies G. J., and Walton P. H. (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol. 10, 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vu V. V., Beeson W. T., Span E. A., Farquhar E. R., and Marletta M. A. (2014) A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 13822–13827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lo Leggio L., Simmons T. J., Poulsen J. C., Frandsen K. E., Hemsworth G. R., Stringer M. A., von Freiesleben P., Tovborg M., Johansen K. S., De Maria L., Harris P. V., Soong C. L., Dupree P., Tryfona T., Lenfant N., et al. (2015) Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat. Commun. 6, 5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., and Eijsink V. G. H. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., and Eijsink V. G. H. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horn S. J., Vaaje-Kolstad G., Westereng B., and Eijsink V. G. H. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 [DOI] [PubMed] [Google Scholar]
- 18. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G. H., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose Surface Degradation by a Lytic Polysaccharide Monooxygenase and Its Effect on Cellulase Hydrolytic Efficiency. J. Biol. Chem. 289, 35929–35938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaaje-Kolstad G., Horn S. J., Sørlie M., and Eijsink V. G. H. (2013) The chitinolytic machinery of Serratia marcescens–a model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 280, 3028–3049 [DOI] [PubMed] [Google Scholar]
- 21. Hu J. G., Arantes V., Pribowo A., Gourlay K., and Saddler J. N. (2014) Substrate factors that influence the synergistic interaction of AA9 and cellulases during the enzymatic hydrolysis of biomass. Energy Environ. Sci. 7, 2308–2315 [Google Scholar]
- 22. Nakagawa Y. S., Eijsink V. G. H., Totani K., and Vaaje-Kolstad G. (2013) Conversion of α-chitin substrates with varying particle size and crystallinity reveals substrate preferences of the chitinases and lytic polysaccharide monooxygenase of Serratia marcescens. J. Agric. Food Chem. 61, 11061–11066 [DOI] [PubMed] [Google Scholar]
- 23. Vermaas J. V., Crowley M. F., Beckham G. T., and Payne C. M. (2015) Effects of lytic polysaccharide monooxygenase oxidation on cellulose structure and binding of oxidized cellulose oligomers to cellulases. J. Phys. Chem. B 119, 6129–6143 [DOI] [PubMed] [Google Scholar]
- 24. Vaaje-Kolstad G., Horn S. J., van Aalten D. M., Synstad B., and Eijsink V. G. H. (2005) The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J. Biol. Chem. 280, 28492–28497 [DOI] [PubMed] [Google Scholar]
- 25. Selig M., Vuong T., Gudmundsson M., Forsberg Z., Westereng B., Felby C., and Master E. (2015) Modified cellobiohydrolase–cellulose interactions following treatment with lytic polysaccharide monooxygenase CelS2 (ScLPMO10C) observed by QCM-D. Cellulose 22, 2263–2270 [Google Scholar]
- 26. Gardner K. H., and Blackwell J. (1975) Refinement of the structure of β-chitin. Biopolymers 14, 1581–1595 [DOI] [PubMed] [Google Scholar]
- 27. Minke R., and Blackwell J. (1978) The structure of α-chitin. J. Mol. Biol. 120, 167–181 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki K., Suzuki M., Taiyoji M., Nikaidou N., and Watanabe T. (1998) Chitin binding protein (CBP21) in the culture supernatant of Serratia marcescens 2170. Biosci. Biotechnol. Biochem. 62, 128–135 [DOI] [PubMed] [Google Scholar]
- 29. Schnellmann J., Zeltins A., Blaak H., and Schrempf H. (1994) The novel lectin-like protein CHB1 is encoded by a chitin-inducible Streptomyces olivaceoviridis gene and binds specifically to crystalline α-chitin of fungi and other organisms. Mol. Microbiol. 13, 807–819 [DOI] [PubMed] [Google Scholar]
- 30. Moser F., Irwin D., Chen S., and Wilson D. B. (2008) Regulation and characterization of Thermobifida fusca carbohydrate-binding module proteins E7 and E8. Biotechnol. Bioeng. 100, 1066–1077 [DOI] [PubMed] [Google Scholar]
- 31. Adav S. S., Ng C. S., Arulmani M., and Sze S. K. (2010) Quantitative iTRAQ secretome analysis of cellulolytic Thermobifida fusca. J. Proteome Res. 9, 3016–3024 [DOI] [PubMed] [Google Scholar]
- 32. Takasuka T. E., Book A. J., Lewin G. R., Currie C. R., and Fox B. G. (2013) Aerobic deconstruction of cellulosic biomass by an insect-associated Streptomyces. Sci. Rep. 3, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Phillips C. M., Iavarone A. T., and Marletta M. A. (2011) Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J. Proteome Res. 10, 4177–4185 [DOI] [PubMed] [Google Scholar]
- 34. Yakovlev I., Vaaje-Kolstad G., Hietala A. M., Stefańczyk E., Solheim H., and Fossdal C. G. (2012) Substrate-specific transcription of the enigmatic GH61 family of the pathogenic white-rot fungus Heterobasidion irregulare during growth on lignocellulose. Appl. Microbiol. Biotechnol. 95, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hori C., Igarashi K., Katayama A., and Samejima M. (2011) Effects of xylan and starch on secretome of the basidiomycete Phanerochaete chrysosporium grown on cellulose. FEMS Microbiol. Lett. 321, 14–23 [DOI] [PubMed] [Google Scholar]
- 36. Gardner J. G., Crouch L., Labourel A., Forsberg Z., Bukhman Y. V., Vaaje-Kolstad G., Gilbert H. J., and Keating D. H. (2014) Systems biology defines the biological significance of redox-active proteins during cellulose degradation in an aerobic bacterium. Mol. Microbiol. 94, 1121–1133 [DOI] [PubMed] [Google Scholar]
- 37. Rytioja J., Hildén K., Hatakka A., and Mäkelä M. R. (2014) Transcriptional analysis of selected cellulose-acting enzymes encoding genes of the white-rot fungus Dichomitus squalens on spruce wood and microcrystalline cellulose. Fungal Genet. Biol. 72, 91–98 [DOI] [PubMed] [Google Scholar]
- 38. Karkehabadi S., Hansson H., Kim S., Piens K., Mitchinson C., and Sandgren M. (2008) The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 Å resolution. J. Mol. Biol. 383, 144–154 [DOI] [PubMed] [Google Scholar]
- 39. Vaaje-Kolstad G., Houston D. R., Riemen A. H., Eijsink V. G. H., and van Aalten D. M. F. (2005) Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 280, 11313–11319 [DOI] [PubMed] [Google Scholar]
- 40. Li X., Beeson W. T. 4th, Phillips C. M., Marletta M. A., and Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemsworth G. R., Taylor E. J., Kim R. Q., Gregory R. C., Lewis S. J., Turkenburg J. P., Parkin A., Davies G. J., and Walton P. H. (2013) The copper active site of CBM33 polysaccharide oxygenases. J. Am. Chem. Soc. 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aachmann F. L., Sørlie M., Skjåk-Bræk G., Eijsink V. G. H., and Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hemsworth G. R., Davies G. J., and Walton P. H. (2013) Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 23, 660–668 [DOI] [PubMed] [Google Scholar]
- 44. Manoil C., and Beckwith J. (1986) A genetic approach to analyzing membrane protein topology. Science 233, 1403–1408 [DOI] [PubMed] [Google Scholar]
- 45. Loose J. S., Forsberg Z., Fraaije M. W., Eijsink V. G. H., and Vaaje-Kolstad G. (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 588, 3435–3440 [DOI] [PubMed] [Google Scholar]
- 46. Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 53. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Forsberg Z., Røhr A. K., Mekasha S., Andersson K. K., Eijsink V. G. H., Vaaje-Kolstad G., and Sørlie M. (2014) Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry 53, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 56. Stoll S., and Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 57. Heuts D. P., Winter R. T., Damsma G. E., Janssen D. B., and Fraaije M. W. (2008) The role of double covalent flavin binding in chito-oligosaccharide oxidase from Fusarium graminearum. Biochem. J. 413, 175–183 [DOI] [PubMed] [Google Scholar]
- 58. Gardner J. G., and Keating D. H. (2012) Genetic and functional genomic approaches for the study of plant cell wall degradation in Cellvibrio japonicus. Methods Enzymol. 510, 331–347 [DOI] [PubMed] [Google Scholar]
- 59. Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., and Pühler A. (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 [DOI] [PubMed] [Google Scholar]
- 60. Gardner J. G., and Keating D. H. (2010) Requirement of the type II secretion system for utilization of cellulosic substrates by Cellvibrio japonicus. Appl. Environ. Microbiol. 76, 5079–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Figurski D. H., and Helinski D. R. (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U.S.A. 76, 1648–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Book A. J., Yennamalli R. M., Takasuka T. E., Currie C. R., Phillips G. N. Jr., and Fox B. G. (2014) Evolution of substrate specificity in bacterial AA10 lytic polysaccharide monooxygenases. Biotechnol. Biofuels 7, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Akagi K., Watanabe J., Hara M., Kezuka Y., Chikaishi E., Yamaguchi T., Akutsu H., Nonaka T., Watanabe T., and Ikegami T. (2006) Identification of the substrate interaction region of the chitin-binding domain of Streptomyces griseus chitinase C. J. Biochem. 139, 483–493 [DOI] [PubMed] [Google Scholar]
- 64. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., and Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Diederichs K., and Karplus P. A. (1997) Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 [DOI] [PubMed] [Google Scholar]
- 66. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gudmundsson M., Kim S., Wu M., Ishida T., Momeni M. H., Vaaje-Kolstad G., Lundberg D., Royant A., Ståhlberg J., Eijsink V. G. H., Beckham G. T., and Sandgren M. (2014) Structural and electronic snapshots during the transition from a Cu(II) to Cu(I) metal center of a lytic polysaccharide monooxygenase by x-ray photoreduction. J. Biol. Chem. 289, 18782–18792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chiu E., Hijnen M., Bunker R. D., Boudes M., Rajendran C., Aizel K., Oliéric V., Schulze-Briese C., Mitsuhashi W., Young V., Ward V. K., Bergoin M., Metcalf P., and Coulibaly F. (2015) Structural basis for the enhancement of virulence by viral spindles and their in vivo crystallization. Proc. Natl. Acad. Sci. U.S.A. 112, 3973–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Peisach J., and Blumberg W. E. (1974) Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins. Arch. Biochem. Biophys. 165, 691–708 [DOI] [PubMed] [Google Scholar]
- 70. Simpson P. J., Xie H., Bolam D. N., Gilbert H. J., and Williamson M. P. (2000) The structural basis for the ligand specificity of family 2 carbohydrate-binding modules. J. Biol. Chem. 275, 41137–41142 [DOI] [PubMed] [Google Scholar]
- 71. Hildén L., Daniel G., and Johansson G. (2003) Use of a fluorescence labelled, carbohydrate-binding module from Phanerochaete chrysosporium Cel7D for studying wood cell wall ultrastructure. Biotechnol. Lett. 25, 553–558 [DOI] [PubMed] [Google Scholar]
- 72. DeBoy R. T., Mongodin E. F., Fouts D. E., Tailford L. E., Khouri H., Emerson J. B., Mohamoud Y., Watkins K., Henrissat B., Gilbert H. J., and Nelson K. E. (2008) Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J. Bacteriol. 190, 5455–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hazlewood G. P., and Gilbert H. J. (1998) Structure and function analysis of Pseudomonas plant cell wall hydrolases. Prog. Nucleic Acids Res. Mol. Biol. 61, 211–241 [DOI] [PubMed] [Google Scholar]