

Abstract
Toll-like receptor 4 (TLR4) promotes vascular inflammatory disorders such as neointimal hyperplasia and atherosclerosis. TLR4 triggers NFκB signaling through the ubiquitin ligase TRAF6 (tumor necrosis factor receptor-associated factor 6). TRAF6 activity can be impeded by deubiquitinating enzymes like ubiquitin-specific protease 20 (USP20), which can reverse TRAF6 autoubiquitination, and by association with the multifunctional adaptor protein β-arrestin2. Although β-arrestin2 effects on TRAF6 suggest an anti-inflammatory role, physiologic β-arrestin2 promotes inflammation in atherosclerosis and neointimal hyperplasia. We hypothesized that anti- and proinflammatory dimensions of β-arrestin2 activity could be dictated by β-arrestin2's ubiquitination status, which has been linked with its ability to scaffold and localize activated ERK1/2 to signalosomes. With purified proteins and in intact cells, our protein interaction studies showed that TRAF6/USP20 association and subsequent USP20-mediated TRAF6 deubiquitination were β-arrestin2-dependent. Generation of transgenic mice with smooth muscle cell-specific expression of either USP20 or its catalytically inactive mutant revealed anti-inflammatory effects of USP20 in vivo and in vitro. Carotid endothelial denudation showed that antagonizing smooth muscle cell USP20 activity increased NFκB activation and neointimal hyperplasia. We found that β-arrestin2 ubiquitination was promoted by TLR4 and reversed by USP20. The association of USP20 with β-arrestin2 was augmented when β-arrestin2 ubiquitination was prevented and reduced when β-arrestin2 ubiquitination was rendered constitutive. Constitutive β-arrestin2 ubiquitination also augmented NFκB activation. We infer that pro- and anti-inflammatory activities of β-arrestin2 are determined by β-arrestin2 ubiquitination and that changes in USP20 expression and/or activity can therefore regulate inflammatory responses, at least in part, by defining the ubiquitination status of β-arrestin2.
Keywords: deubiquitylation (deubiquitination), inflammation, TNF receptor-associated factor (TRAF), Toll-like receptor 4 (TLR4), ubiquitin, NFκB, USP20, β-arrestin, neointimal hyperplasia, scaffold
Introduction
β-Arrestin2 (βarr2) is an ∼46-kDa multifunctional scaffolding protein that was discovered originally for its ability to desensitize G protein-mediated signaling evoked by seven-transmembrane receptors (7TMRs)4 (1, 2). However, βarr2 modulates the signaling and/or endocytosis of not only most 7TMRs but also of several receptor protein tyrosine kinases, cytokine receptors, ion channel receptors, and the LDL receptor (3, 4). Both the endocytic and signaling functions of βarr2 are intertwined with its ubiquitination, which in turn is stimulus-driven and regulated by specific E3 ubiquitin ligases or deubiquitinases (DUBs) (4). βarr2 not only undergoes dynamic ubiquitination/deubiquitination but also recruits E3 ubiquitin ligases to other substrates. Indeed, βarr2 is integral to the ubiquitination of cell surface receptors, channels, and non-receptor proteins (4, 5). However, thus far there is no clear demonstration that βarr2 can scaffold a DUB to specific substrates and affect signal transduction by mediating deubiquitination.
A role in deubiquitination could help explain the ability of βarr2 to inhibit proinflammatory signaling that culminates in the activation of NFκB (6–8). Canonical activation of NFκB involves agonist-mediated TLR4 or interleukin-1 receptor dimerization, which engenders MyD88-dependent activation of the E3 ubiquitin ligase TRAF6—a process that involves TRAF6 oligomerization, autoubiquitination, and subsequent synthesis of Lys-63-linked polyubiquitin chains that are either covalently or noncovalently attached to other proteins (9). Such Lys-63-linked polyubiquitin chains activate TAK1 and co-localize TAK1 with IκB kinase (IKK) through noncovalent interactions (9). Consequently, TAK1 phosphorylates and thereby activates IKKβ. IKKβ-mediated phosphorylation of IκBα triggers Lys-48-linked polyubiquitination and proteasomal degradation of IκBα, with subsequent deinhibition of NFκB p65/p50 heterodimers (9, 10). In this schema, βarr2 can inhibit NFκB signaling at two levels: by binding to and thereby impeding oligomerization and autoubiquitination of TRAF6 (6) and by binding to and preventing the degradation of IκBα (7, 8). Whether the association of βarr2 with TRAF6 and/or IκBα facilitates deubiquitination of these proteins remains enigmatic.
Although βarr2 appears to inhibit NFκB activation and consequent inflammation in certain systems (6–8, 11–13), it also appears to augment inflammatory signaling in distinct systems (14–16). Physiologic βarr2 expression in SMCs of endothelium-denuded arteries promotes neointimal hyperplasia, a pathology involving inflammation-induced proliferation and migration of SMCs from the tunica media into the subendothelial tunica intima (14, 17). In Ldlr−/− mice, βarr2 promotes atherosclerosis (14), a chronic vasculitis that fundamentally involves canonical NFκB signaling (18–20). Similar proinflammatory roles of βarr2 have been reported in allergic asthma and in lysophosphatidic acid-induced NFκB activation (16, 21). Thus, current data paint a paradoxical picture for βarr2 with respect to NFκB signaling and inflammation.
Apparent paradoxes in βarr2-regulated inflammatory signaling may be reconciled by extrapolating from studies demonstrating reciprocal functions of ubiquitinated βarr2 and non-ubiquitinated βarr2 in 7TMR signaling (22, 23). Does reversible ubiquitination of βarr2 explain the proinflammatory versus anti-inflammatory dimensions of βarr2 activity? Deubiquitination of βarr2 itself is mediated by USP33 (23), and the USP33 homolog USP20 has been shown to deubiquitinate TRAF6 in heterologous systems (24). By scaffolding TRAF6 and USP20, could βarr2 block canonical NFκB activation? Conversely, by sequestering USP20, could βarr2 inhibit TRAF6 deubiquitination and thereby promote canonical NFκB activation? And could these reciprocal roles of βarr2 be regulated by reversible ubiquitination of βarr2? This study uses a variety of in vivo and in vitro approaches to address these questions and to determine whether USP20 and βarr2 function in concert to regulate TRAF6 ubiquitination and canonical NFκB activation.
Experimental Procedures
Generation of Transgenic Mice
All animal experiments were performed in accordance with protocols approved by the Duke University Institutional Animal Care and Use Committee. Transgenic mice were generated to overexpress mouse USP20 or its catalytically inactive mutant, dominant negative USP20 (DN-USP20), which possesses two mutations (C154S and H645Q) in the catalytic domain of the protein. The QuikChangeTM site-directed mutagenesis kit (Stratagene) was used to insert the mutations on the basis of protocols described previously (25). N-terminal HA-tagged USP20 or DN-USP20 coding sequences were inserted into a cloning vector, pBluescript II, so that they were flanked upstream by a 481-base pair portion of the SM22α promoter (−440 to +41 relative to transcription start) for smooth muscle cell-specific expression and downstream by a bovine growth hormone poly(A) signal (26–28). The plasmid constructs were linearized, purified, and microinjected into the pronuclei of B6SJLF1/J zygotes and subsequently implanted into surrogate mice by the Duke Transgenic Core Facility. Positive animals were identified by PCR amplification using a 5′ primer in the SM22α promoter region and a 3′ primer in the USP20 transgene.
Reagents
Protein G Plus/protein A-agarose was purchased from Calbiochem. LPS from Escherichia coli, M2 anti-FLAG affinity agarose beads, and N-ethylmaleimide were obtained from Sigma-Aldrich. The following IgGs were from the sources listed: mouse monoclonal anti-FLAG M2 (200471) and mouse monoclonal anti-β-actin (A5441), Sigma-Aldrich; anti-ubiquitin FK1 (BML-PW8805), Enzo Life Sciences; rabbit polyclonal anti-USP20 (A301-189A) and rabbit polyclonal anti-USP33 (A300-925A), Bethyl Laboratories, Inc.; rabbit monoclonal anti-phospho-p65(Ser-536) (3033) and rabbit polyclonal anti-IκBα (9242), Cell Signaling; rabbit polyclonal anti-NFκB p65 (sc-372), anti-TRAF6 (sc-7221), anti-hemagglutinin (sc-805), anti-MD-2 (sc-20668), anti-CD14 (sc-1182), anti-TLR4 (sc-293072) and anti-VCAM-1 (sc-8304), Santa Cruz Biotechnology; polyclonal anti-β-arrestin1/2 antibodies were generously provided by Dr. Robert J. Lefkowitz (Duke University): A1CT, which recognizes β-arrestin1 and β-arrestin2, and A2CT, which is selective for β-arrestin2 (29). Horseradish peroxidase-conjugated secondary antibodies were from GE/Amersham Biosciences or Rockland Immunochemicals. For USP20 detection, however, we used conjugated secondary antibodies from Bethyl Laboratories, Inc. Lipofectamine 2000TM and GeneSilencer were purchased from Invitrogen and Genlantis, respectively.
Plasmids
Plasmids encoding rat β-arrestin2, βarr2-Ub, and βarr2–0K were described in earlier studies from our laboratory (22), as were HA-tagged USP20 and DN-USP20 plasmids (25). The FLAG-tagged TRAF6 plasmid (with a pcDNA3 backbone) was provided by Dr. Zhijian Chen (University of Texas Southwestern Medical Center).
Purified Proteins
C-terminal MYC/DDK-tagged recombinant human TRAF6 (TP319528) and C-terminal MYC/DDK-tagged human USP20 (TP308051) were purchased from OriGene Technologies, Inc. Both proteins were supplied at >80% purity. (DDK is an alternative appellation for the FLAG epitope DYKDDDDK and is referred to as FLAG in this article.) Purified rat β-arrestin2 was provided by Dr. Robert J. Lefkowitz (Duke University) (30). Using protocols we have reported previously (23, 25), we purified HA-USP20 from COS-7 cells transfected with a pcDNA3-HA construct that contained the human USP20 cDNA insert (23, 25).
Cell Lines
Smooth muscle cells (SMCs) were isolated by enzymatic digestion of aortas stripped of adventitia and endothelial cells, according to published protocols (14, 31, 32). They were split 1:4 for each passage and not used after passage 7 (freshly isolated cells = passage 1). β-Arrestin1/2 double knockout mouse embryo fibroblasts (MEFs) were obtained from Dr. Robert J. Lefkowitz (Duke University) and were characterized previously (29, 33, 34). SMCs and double knockout MEFs were kept in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 1% penicillin/streptomycin. HEK-293 cells were obtained from the American Type Culture Collection. These cells were grown in minimal essential medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For generation of stable cell lines expressing TRAF6, HEK-293 cells were transfected with a plasmid encoding the mouse TRAF6 with a FLAG epitope at its N terminus. Transfectant clones were selected by cultivation in growth medium supplemented with G418: initially at 1 mg/ml and in later passages at 400 μg/ml, as described previously (35).
Transient expression of YFP-tagged βarr2 constructs in double knockout MEFs was achieved with Lipofectamine 2000TM transfection using a modified protocol (36). The use of YFP-tagged βarr2 constructs allowed assessment of transfection efficiency: 30–50% for the βarr2-WT and βarr2–0K constructs and 20–30% for the βarr2-Ub construct. Relative to endogenous βarr2 in HEK-293 cells, these βarr2 construct-transfected double knockout MEFs expressed 50% as much βarr2-WT and βarr2–0K protein and 25–30% as much βarr2-Ub protein (assessed by βarr2 immunoblotting).
Immunoprecipitation and Immunoblotting
Plasmid transfections were performed in HEK-293 cells and MEFs at 50% confluency with Lipofectamine 2000TM 48 h before experiments. For signaling experiments, SMCs and MEFs were starved overnight in serum-free medium. HEK-293 cells were starved for 4 h prior to stimulation with LPS or vehicle. Cells were washed with ice-cold phosphate-buffered saline (pH 7.4) and solubilized in an ice-cold lysis buffer (50 mm HEPES (pH 7.5), 2 mm EDTA, 250 mm NaCl, 10% (v/v) glycerol, and 0.5% (v/v) IGEPAL CA-630) that was supplemented with phosphatase and protease inhibitors (1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, and 100 μm benzaminidine; all from Sigma-Aldrich). The lysis buffer used in the ubiquitination assays for immunoprecipitating TRAF6 and βarr2 was supplemented with 10 mm N-ethylmaleimide and 20 μm MG132 to inhibit cellular DUB and 26S proteasome activities, respectively. The cell lysates were centrifuged at 13,000 rpm for 20 min at 4 °C to remove cell debris, and protein concentrations were determined on the resulting supernatant whole cell extracts by Bradford protein assay. Cell lysate proteins (∼800 μg) were immunoprecipitated using either anti-FLAG M2 resin or A1CT antibody with protein G Plus/protein A-agarose beads. Samples were incubated overnight (4 °C) with end-over-end rotation for immunoprecipitation. Immune complexes were washed three times with lysis buffer, and bound proteins were eluted in 2× SDS-PAGE sample buffer. Samples were resolved on 4–20% gradient or 10% Tris-glycine gels along with 20 μg of corresponding lysates (which corresponded to ∼2.5% of that used for the IP) and then transferred onto nitrocellulose membranes. Membranes were blocked and probed in 5% (w/v) dried skim milk powder dissolved in TTBS (2% (v/v) Tween 20, 10 mm Tris-Cl, (pH 8.0), and 150 mm NaCl), and washes were performed in TTBS. Enhanced chemiluminescence (SuperSignal West Pico reagent, Pierce) was used for protein detection. Blot imaging was performed with a charge-coupled device camera system (Bio-Rad Chemidoc-XRS), and band densities were quantified with Image-Lab software (Bio-Rad).
In Vitro Binding of USP20
For the analysis of USP20/βarr2 binary interaction, 125 ng of purified FLAG-USP20 was incubated for 30 min with increasing doses of βarr2 in a total volume of 50 μl of KOAc buffer (34) containing 100 mm K+ acetate, 50 mm HEPES, 0.5 mm MgSO4, 0.2 mm DTT, 0.2% bovine serum albumin, and protease inhibitors (pH 7.4). The protein complex was subsequently diluted to 500 μl with lysis buffer supplemented with 10 mm N-ethylmaleimide (as in immunoprecipitation assays) and rotated with M2 anti-FLAG affinity agarose beads for 2 h at 4 °C. Beads were pelleted, washed three times in lysis buffer, eluted in SDS sample buffer, run on 4–12% gradient Tris-glycine gels, and immunoblotted. Reactions containing either only USP20 or only βarr2 served as negative controls. To determine USP20-TRAF6-βarr2 complex formation, 80 ng of purified FLAG-TRAF6 was mixed with 125 ng of HA-tagged USP20 and incubated for 30 min in KOAc buffer with varied doses of purified βarr2. FLAG pull-down and subsequent steps were similar to those used for the binary complex above.
RNA Interference
Non-targeting control siRNA and siRNA targeting βarr2, USP20, or USP33 were purchased from Dharmacon GE Healthcare Life Sciences and described previously (25, 36, 37). For rescue experiments, siRNA specifically targeting human βarr2 or human USP20 were co-transfected along with siRNA-resistant, YFP-tagged rat βarr2 (2 μg in a 100-mm dish) or HA-tagged mouse USP20 (2 μg in a 100-mm dish). GeneSilencer was used for βarr2 silencing, and Lipofectamine 2000TM was used for USP20/USP33 silencing, following the protocol of the manufacturer. Early-passage cells that were 40–50% confluent were transfected with 20 μg of siRNA with or without plasmid DNA and incubated for 4 h (HEK-293) or 12–14 h (SMCs) at 37 °C in serum-free medium and then for 48 h in serum-containing medium prior to assays. Cells with >85% reduction in target protein expression were used for experimental analyses.
In SMC experiments, each well of a 6-well dish was independently transfected with siRNA because trypsinizing siRNA-transfected SMCs engendered excessive cell toxicity. Consequently, each well of SMCs constituted a single experimental replicate. For this reason, interassay variability was greater in these assays than in assays of transgenic SMC lines. To compensate for this variability, SMC RNAi experiments included a greater number of experimental replicates (Fig. 9).
FIGURE 9.

NFκB signaling is reduced in βarr2−/− SMCs. A, SMCs of the indicated genotype were transfected with siRNA targeting no mRNA (Control) or USP20 mRNA. Cells were stimulated with 100 ng/ml LPS at 37 °C for the indicated times. Lysates were resolved by SDS-PAGE on BisTris gels (10% polyacrylamide) and immunoblotted (IB) for the indicated proteins. Arrowheads indicate the electrophoretic mobility for USP20 (second panel) and βarr2 (fourth panel). B, band intensities for IκBα were normalized to corresponding β-actin band intensities. These ratios were normalized to those obtained from unstimulated WT SMCs transfected with control siRNA to obtain percent of control, plotted as mean ± S.E. from a total of nine independent siRNA transfections in three independent pairs of WT and βarr2−/− SMC lines. From two-way analysis of variance with Tukey's multiple comparison test, we found p < 0.05 for the following comparisons, designated by SMC genotype/(siRNA transfected): *, compared with WT/(control); ‡, compared with βarr2−/−/(control); and †, compared with βarr2−/−/(USP20).
Carotid Endothelial Denudation
Carotid endothelial denudation was performed on mice anesthetized with pentobarbital (50 mg/kg) using a 0.36-mm-diameter coronary guide wire (Cordis), as we described previously (14, 31). Four weeks after endothelial denudation, injured common carotids were harvested from anesthetized mice after 20 min of perfusion-fixation (80 mm Hg) with 10% formalin in PBS. Subsequently, carotids were fixed in formalin for 20 h and then embedded in paraffin.
Histology
For carotid artery morphometry, paraffin-embedded specimens were sliced at 5 μm and stained with a modified Masson's trichrome and Verhoeff's elastic tissue stain as we described previously (14, 31). Computerized planimetry with ImageJTM was performed as described previously (14, 31) by observers blinded to sample identity. Immunofluorescence staining was performed on aortas embedded in OCT compound or paraffin-embedded carotids after samples were sliced at 5 μm (14, 31). Tissue sections were probed with rabbit IgGs specified above, followed by anti-rabbit IgG conjugated to Alexa-488, Alexa-548, or Alexa-594, as described previously (14, 31), or with Cy3-conjugated 1A4 anti-SMC α-actin (Sigma-Aldrich), as described previously (14, 31). Nuclei were counterstained with Hoechst 33342 (10 μg/ml) during the secondary antibody incubation. Nonspecific fluorescence (determined on serial sections probed with equivalent concentrations of nonimmune rabbit IgG) was subtracted from the total signal to obtain antigen-specific fluorescence. Imaging and analyses were performed by observers blinded to specimen identity. Specimens from all three groups (non-Tg, USP20-Tg, and DN-USP20-Tg) were stained and imaged batchwise to minimize variation in staining and imaging among groups.
Statistical Analyses
All experiments were reproduced at least three independent times. Data averaged from three or more independent experiments are presented as means ± S.E. Statistical significance was determined by analysis of variance followed by post hoc test for multiple comparisons (GraphPad Prism 6 from GraphPad, Inc.), and p < 0.05 was considered significant.
Results
βarr2, USP20, and TRAF6 Associate with Each Other
In the course of regulating 7TMR trafficking and endocytosis, βarr2 associates with and is deubiquitinated by ubiquitin-specific protease 33 (USP33) (23). To determine whether βarr2 could be regulated by other USP family members, we asked whether βarr2 associates with USP20, which shares 59% identity with USP33 (25). Co-immunoprecipitation of βarr2-FLAG in HEK-293 cells showed that endogenous USP20 interacted with βarr2 (Fig. 1A). βarr2 also associates with TRAF6 and thereby inhibits not only TRAF6 oligomerization and autoubiquitination but also NFκB signaling (6). As reported before (6), βarr2-FLAG co-immunoprecipitated with endogenous TRAF6 in our experiments (Fig. 1B). Furthermore, FLAG-TRAF6 co-immunoprecipitated with endogenous USP20 (Fig. 1C). These protein-protein interaction studies suggest that USP20, βarr2 and TRAF6 bind each other and might function together in NFκB signaling.
FIGURE 1.

Protein-protein interaction of βarr2, USP20, and TRAF6. HEK-293 cells were transfected with FLAG-tagged βarr2 (A and B), FLAG-TRAF6 (C) or empty vector (−), and immunoprecipitation was performed with ANTI-FLAG M2 affinity gel (Sigma-Aldrich). Immunoprecipitates and cell lysates were immunoblotted (IB) for endogenous USP20 (A and C) and endogenous TRAF6 (B). Subsequently the blots were stripped and reprobed for their respective bait proteins: βarr2 (A and B) or TRAF6 (C).
To determine whether βarr2 binds to USP20 directly, we tested the interaction of purified βarr2 with purified USP20 (Fig. 2, A–D). As shown in Fig. 2A, the association of βarr2 with USP20 increased roughly linearly with [βarr2] and then saturated over the range of βarr2 concentrations used. We then used this purified protein approach to determine whether βarr2 is required for the interaction of USP20 and TRAF6. In the absence of βarr2, minimal amounts of USP20 were detected in TRAF6 pull-downs. At low concentrations, βarr2 augmented the association of USP20 with TRAF6 by ∼5-fold (Fig. 2B). However, at higher concentrations, βarr2 failed to augment the association of USP20 with TRAF6 at all (Fig. 2B). Thus, under conditions wherein the concentration of βarr2 is limiting, βarr2 can function as a scaffold that conjoins USP20 and TRAF6 in a ternary complex (with βarr2). However, at high concentrations, βarr2 forms only binary complexes with USP20 or perhaps with TRAF6 (Fig. 2, A–C).
FIGURE 2.

The relative abundance of βarr2 determines whether βarr2 forms binary or ternary complexes with TRAF6 and USP20 in purified preparations. A, the indicated concentration of purified βarr2 was incubated in 50 μl (final volume) with or without 20 nm purified FLAG-USP20, as described under “Experimental Procedures.” FLAG pull-downs were immunoblotted (IB) sequentially for βarr2 and USP20. Nonspecific βarr2 pull-down was determined from lanes lacking FLAG-USP20. Shown is an experiment representative of four performed. B, the indicated concentrations of purified βarr2 were incubated in 50 μl (final volume) with or without purified HA-USP20 (20 nm) and/or FLAG-TRAF6 (20 nm), as described under “Experimental Procedures.” FLAG pull-downs were successively immunoblotted for HA, βarr2, and TRAF6. Nonspecific HA-USP20 pull-down was determined from lanes lacking FLAG-TRAF6. Shown are results of a single experiment representative of four performed. C, for the USP20 pull-downs in A, specific (total minus nonspecific) βarr2 band intensities were quantified and normalized to USP20 band intensities. These ratios were normalized to those obtained with 4 nm βarr2 to obtain percent of maximum (% max), plotted (filled squares) as mean ± S.E. from four independent experiments. Compared with 0.4 nm: *, p < 0.05 (analysis of variance). For the TRAF6 pull-downs in B, specific (total minus nonspecific) USP20 band intensities were normalized to cognate TRAF6 band intensities. These ratios were normalized to those obtained with 2 nm βarr2 to obtain the percentage of maximum (% max), plotted (empty circles) as means ± S.E. from four independent experiments. Compared with control: *, p < 0.01. D, Coomassie-stained gels show 1 μg of purified proteins separated on 4–20% gradient Tris-Glycine polyacrylamide gels. Arrows indicate the mobility of purified proteins. The asterisks indicate light chain IgG bands that co-elute during the purification of HA-tagged proteins.
βarr2 Functions as a Scaffold for USP20-mediated Deubiquitination of TRAF6
βarr2 serves as a multifunctional scaffold and adaptor in 7TMR signaling. For example, βarr2 recruits to the receptor different components of the ERK pathway or elements of the endocytosis machinery (4, 38, 39). We hypothesized that the scaffolding abilities of βarr2 were critical for TLR4-dependent NFκB signaling and asked whether βarr2 affects the interaction of USP20 and TRAF6. We first silenced βarr2 in HEK-293 cells and assayed USP20/TRAF6 association. In control cells, immunoprecipitation of TRAF6 pulled down both βarr2 and USP20. This observation suggested the possibility that these proteins form a ternary complex in intact cells (Fig. 3A). Remarkably, silencing βarr2 reduced the amount of endogenous USP20 that co-immunoprecipitated with TRAF6 (by 30 ± 2 [unstimulated] to 60 ± 3% [+LPS]) even though cellular levels of TRAF6 and USP20 did not change with βarr2 knockdown (Fig. 3, A and B). Our siRNA knockdown was specific for βarr2 because the levels of its homolog βarr1 were unchanged. Thus, βarr2 appears to promote the binding of USP20 with TRAF6. On the other hand, USP20 knockdown did not significantly alter the amount of endogenous TRAF6 co-immunoprecipitating with βarr2 (Fig. 3, C and D).
FIGURE 3.

βarr2 functions as a scaffold for USP20-TRAF6 interaction in intact cells. A, HEK-293 cells stably expressing FLAG-TRAF6 were transfected with control (CTL) or βarr2 siRNA, stimulated with LPS (37 °C, 10 min), and solubilized. TRAF6 (FLAG) immunoprecipitates and whole cell lysates were immunoblotted (IB) for the indicated proteins. Shown are results of a single experiment representative of three performed. B, USP20 in each IP was normalized to the cognate amount of TRAF6 immunoprecipitated. These ratios were normalized to those obtained in unstimulated cells transfected with control siRNA to obtain fold over control non-stimulated (NS), plotted as mean ± S.E. from three independent experiments. Compared with control: *, p < 0.05. C, HEK-293 cells transiently overexpressing βarr2-FLAG were transfected with control or USP20 siRNA, stimulated with LPS, and solubilized as in A. βarr2 (FLAG) immunoprecipitates and whole cell lysates were immunoblotted for the indicated proteins. Shown are results of a single experiment representative of five performed. D, TRAF6 in each IP was normalized to the cognate amount of βarr2 immunoprecipitated. These ratios were normalized and plotted from five experiments as in B.
If βarr2 affects the association of USP20 with TRAF6, then one should expect βarr2 to affect the ubiquitination of TRAF6. Indeed, TRAF6 ubiquitination increased after siRNA-mediated knockdown of either βarr2 or USP20 (Fig. 4A). In these assays, there was no additive augmentation of TRAF6 ubiquitination when βarr2 and USP20 were knocked down simultaneously (Fig. 4, A and B). Thus, βarr2 and USP20 appear to use a shared mechanism to prevent TRAF6 ubiquitination (Fig. 4B). In these experiments, silencing USP20 or βarr2 had no effect on the expression level of USP33 (lysate blots in Fig. 4A). Hence, USP20 appears to have a distinct role from its homolog USP33 in deubiquitinating TRAF6.
FIGURE 4.

βarr2 and USP20 cooperatively inhibit TRAF6 autoubiquitination. A, HEK-293 cells stably expressing FLAG-TRAF6 were transfected with control (CTL), βarr2, and/or USP20 siRNA, stimulated with LPS (37 °C, 10 min), and then solubilized for immunoprecipitation of FLAG-TRAF6. TRAF6 (FLAG) immunoprecipitates and whole cell lysates were immunoblotted (IB) sequentially for the indicated proteins. Shown are results of a single experiment representative of six performed. B, the intensity of the ubiquitin smear in each IP was normalized to the cognate TRAF6 (FLAG) band in each lane. These ratios were normalized to that obtained with non-stimulated (NS) cells transfected with control siRNA, and plotted as the mean ± S.E. from six experiments. Compared with control: *, p < 0.05. C, HEK-293 cells stably expressing FLAG-TRAF6 were transfected with siRNA targeting no protein or human βarr2. Simultaneously, these cells were transfected with pcDNA3 containing only YFP or βarr2-YFP (rat βarr2 cDNA). D, the ubiquitin smear in each IP was normalized to the cognate TRAF6 (FLAG) band in each lane. These ratios were normalized to those obtained in stimulated cells co-transfected with control siRNA and the YFP vector to obtain fold over control, plotted as mean ± S.E. from five independent experiments. Compared with control: *, p < 0.05. E, HEK-293 cells stably expressing FLAG-TRAF6 were transfected with siRNA targeting no protein or human USP20. Simultaneously, these cells were transfected with pcDNA3 containing only HA tag or HA-USP20 (mouse USP20 cDNA). After LPS stimulation (37 °C, 10 min), cells were solubilized for IP. TRAF6 (FLAG) immunoprecipitates and whole cell lysates were immunoblotted for the indicated proteins. Shown are results of a single experiment representative of three performed. F, the ubiquitin smear in each IP was normalized to the cognate TRAF6 (FLAG) band in each lane. These ratios were normalized to those obtained in stimulated cells co-transfected with control siRNA and the HA vector to obtain fold over control, plotted as mean ± S.E. from three independent experiments. Compared with control: *, p < 0.05.
To ascertain the specificity of our knockdown experiments, we performed rescue experiments for both βarr2 (Fig. 4, C and D) and USP20 (Fig. 4, E and F). In these assays, βarr2 knockdown induced an increase in TRAF6 ubiquitination that was reversed when a plasmid encoding rat βarr2 cDNA was co-transfected with the siRNA (Fig. 4, C and D). Similarly, co-transfection of a mouse HA-USP20 construct reversed the increase of TRAF6 ubiquitination observed with USP20 knockdown (Fig. 4, E and F). Together our results strongly suggest that USP20 is a deubiquitinase for TRAF6 and that βarr2 functions as a critical adaptor for TRAF6 deubiquitination by USP20.
SMC-specific Expression of USP20 and DN-USP20 in Transgenic Mice
To determine the vascular effects of USP20, we generated transgenic mice overexpressing either USP20 or a catalytically inactive, dominant-negative mutant USP20 (DN-USP20-Tg) (Fig. 5A) (25) under the control of the SMC-specific SM22α promoter (28, 40, 41). Immunoblots of aortic extracts demonstrated that the total level of USP20 in the USP20-Tg was 2 ± 1-fold more than endogenous USP20 levels in non-Tg and that DN-USP20-Tg expression was about 3 ± 1-fold greater than endogenous USP20 levels (Fig. 5B). By immunostaining for the HA tag of the transgenes in aortic cross-sections, we found that actin and DNA staining were comparable in littermate control and Tg aortas, but only Tg aortas stained for HA-USP20. Furthermore, ∼90% of HA-USP20 (WT or DN) co-localized with SMC-actin (colocalization plugin, ImageJ software), indicating SMC-specific expression (Fig. 5C).
FIGURE 5.

Generation of transgenic mice with SMC-targeted USP20 expression. A, the USP20 transgenes comprised (from 5′ to 3′) a 481-base pair portion of the SM22α promoter (−440 to +41 relative to transcription start), an N-terminal HA epitope, the 2751-base pair murine USP20 coding sequence, and the bovine growth hormone poly(A) signal. The two catalytic domain mutations contained in the DN-USP20 mutant are depicted as Xs: C154S and H645Q. B, 30 μg of aorta extracts from congenic C57BL/6 mice with SM22α-driven expression of USP20, DN-USP20, or no transgene were immunoblotted (IB) with IgGs specific for the indicated proteins. Shown is an immunoblot from a single experiment representative of three such experiments. C, littermate control and Tg aorta sections were stained with anti-HA (green), Cy3-conjugated anti-SMC actin (red), and DRAQ5 for DNA (blue). Sections were imaged with a ×40 oil objective of a LSM 510 Meta confocal microscope. Scale bars = 50 μm.
Antagonizing USP20 Activity Augments Neointimal Hyperplasia and Vascular NFκB Activation
In model cell lines, USP20 can inhibit TRAF6-dependent NFκB activation (24), which regulates a multitude of inflammatory signaling pathways (20, 42). To determine whether SMC USP20 activity suppresses vascular inflammation, we subjected the transgenic animals and their non-Tg littermates to carotid endothelial denudation. This wire-mediated procedure triggers adhesion to the subendothelial extracellular matrix by neutrophils and platelets, which secrete cytokines and growth factors that provoke proliferation of medial SMCs that migrate across the internal elastic lamina and into the subendothelial, “neointimal” space to create “neointimal hyperplasia,” an inflammatory lesion that compromises the efficacy of arterial stenting (14, 20, 31, 42, 43). Although the carotid arteries of each Tg mouse were morphologically equivalent before intervention, neointimal and medial areas 4 weeks after endothelial denudation were 2.6- and 1.4-fold greater, respectively, in SMC-DN-USP20-Tg than in either Non-Tg or SMC-USP20-Tg mice (Fig. 6). Correspondingly, the luminal area was 2-fold less in SMC-DN-USP20-Tg mice (Fig. 6). Thus, antagonizing USP20 activity in SMCs augments neointimal hyperplasia, which is triggered by vascular inflammation (14, 20, 42, 43).
FIGURE 6.
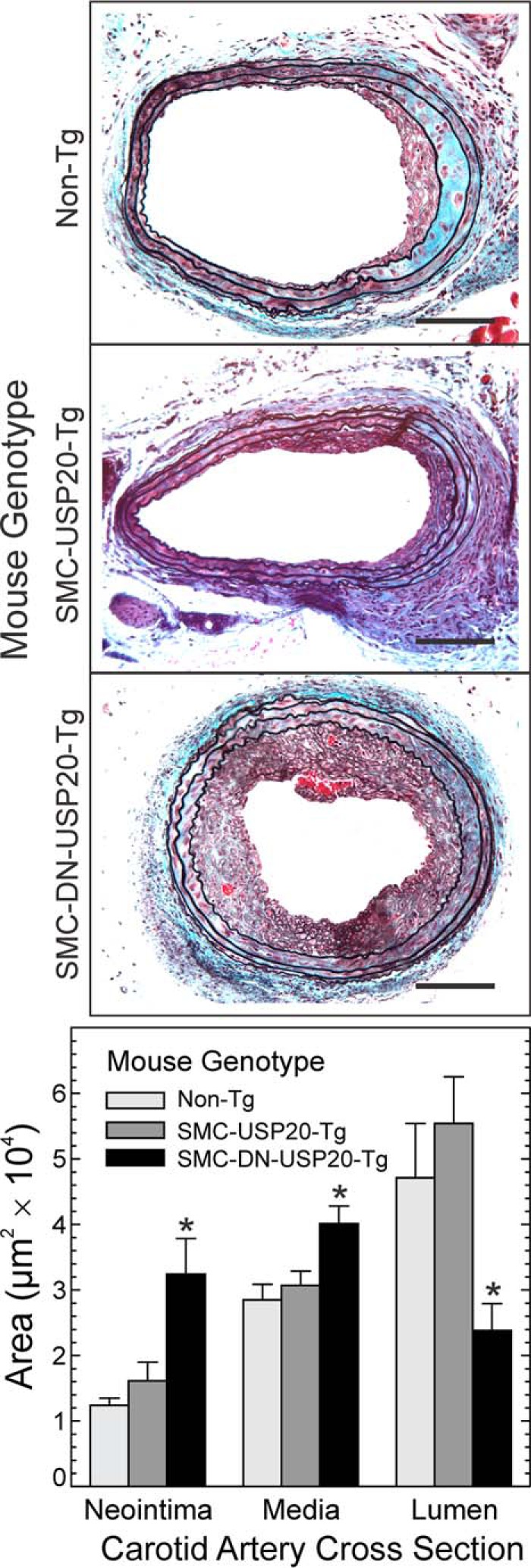
Antagonizing USP20 activity augments neointimal hyperplasia. A, congenic C57BL/6 mice with SM22α-driven expression of USP20, DN-USP20, or no transgene (non-Tg) were subjected to wire-mediated carotid endothelial denudation. Carotids were harvested 4 weeks later after perfusion-fixation, and sections were stained with a modified connective tissue stain. Scale bars = 50 μm. B, the areas of the indicated arterial layers were measured by observers blinded to specimen identity. Areas are plotted as means ± S.E. of ≥6 carotids per group. Compared with non-Tg: *, p < 0.01.
To determine the extent to which NFκB was activated in our endothelium-denuded carotid arteries (42), we employed two readouts: NFκB p65 Ser-536 phosphorylation, which is effected by IκB kinase-β and which augments NFκB transcriptional activity (44–46), and expression of VCAM-1 (CD106), an integrin-binding protein that facilitates adhesion of monocytes and lymphocytes and is encoded by an NFκB-dependent gene (20, 47). We found equivalent NFκB activation (phospho-p65(Ser-536)) in Non-Tg and SMC-USP20-Tg carotid arteries but greater NFκB activation in SMC-DN-USP20-Tg carotid arteries (Fig. 7). Phosphorylation of NFκB p65 (on Ser-536) was 70% ± 20% greater in SMC-DN-USP20-Tg than in SMC-USP20-Tg and non-Tg carotid arteries (Fig. 7) even though total p65 levels were equivalent in all carotid arteries. Congruently, VCAM-1 levels were 50% greater in SMC-DN-USP20-Tg arteries than in either SMC-USP20-Tg or non-Tg arteries (Fig. 7). Thus, antagonizing USP20 in SMCs augmented NFκB activation in the context of arterial injury in mice.
FIGURE 7.
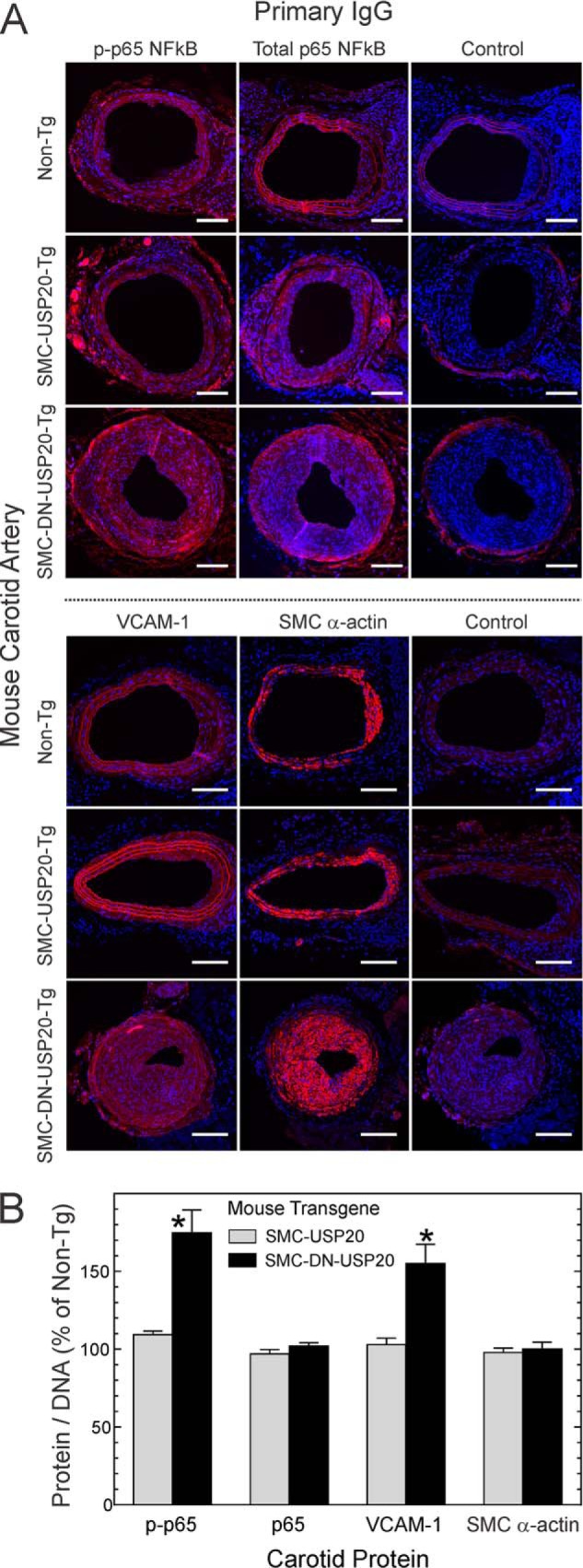
Antagonizing USP20 activity promotes neointimal NFκB activation in vivo. A, serial sections from the injured carotid arteries used for Fig. 6 were fluorescently stained for DNA (blue) and immunostained with rabbit IgG specific for phospho-p65(Ser-536) (p-p65), total p65, VCAM-1, or no particular protein (Control). In addition, serial sections were stained for SMC α-actin. Scale bars = 50 μm. B, the ratios of protein immunofluorescence to DNA fluorescence intensity within the neointima were normalized to those obtained from WT samples from the same immunostaining batch to yield percent of non-Tg. Plotted are the means ± S.E. from four or more carotid arteries of each genotype. Compared with non-Tg: *, p < 0.05.
USP20 Inhibits TLR4-induced NFκB Activation in SMCs
In the pathogenesis of arterial injury, one of the important triggers for neointimal hyperplasia and SMC NFκB activity is TLR4 signaling in SMCs (48). Therefore, we studied the effects of USP20 on NFκB activation in SMCs in vitro by assaying NFκB p65 Ser-536 phosphorylation (as in our in vivo studies above) as well as the rate of IκBα degradation (31) triggered upon TLR4 stimulation with LPS. In SMCs from SMC-DN-USP20-Tg mice, LPS induced NFκB p65 phosphorylation on Ser-536 to an extent that was 3- and 10-fold greater, respectively, than that in non-Tg and SMC-USP20-Tg SMCs (Fig. 8, A and B). Congruently, the LPS-induced rate of IκBα degradation followed this rank order: SMC-DN-USP20-Tg > Non-Tg > SMC-USP20-Tg. Indeed, after 30 min of LPS stimulation, the levels of IκBα in non-Tg and SMC-DN-USP20-Tg SMCs were 5-fold lower than those in SMC-USP20-Tg SMCs (Fig. 8, C and D). These short-term signaling data were corroborated by long-term NFκB activity data, assessed as the expression level of the NFκB-dependent VCAM-1. In response to 24 h of LPS stimulation, SMCs from non-Tg and SMC-DN-USP20-Tg mice evinced VCAM-1 protein levels that were 2.7-fold higher than those in SMCs from SMC-USP20-Tg mice (Fig. 8, E and F). Thus, USP20 activity in SMCs inhibits TLR4-induced NFκB activation.
FIGURE 8.

Modulation of USP20 activity alters LPS-induced NFκB signaling in SMCs. SMCs from non-Tg, SMC-USP20-Tg, and SMC-DN-USP20-Tg mice were serum-starved overnight and then exposed at 37 °C to serum-free medium lacking (−, control) or containing 1 μg/ml LPS for 10 min (A and B), 10 and 30 min (C and D), and 24 h (E and F). Cells were solubilized in 2× sample buffer and immunoblotted for the indicated proteins. A, SMC lysates were serially immunoblotted (IB) for phospho-p65(Ser-536), total p65, and β-actin. B, band intensities for phospho-p65 were normalized to those of cognate β-actin. These ratios were normalized to those obtained in unstimulated non-Tg SMCs to obtain fold over control basal, plotted as mean ± S.E. from six independent experiments. Compared with control (*) or with USP20-Tg (#): p < 0.0001. C, SMC lysates were serially blotted for IκBα and β-actin. D, band intensities for IκBα were normalized to those of corresponding β-actin bands. These ratios were normalized to those obtained from unstimulated SMCs of the cognate genotype to obtain fold over basal, plotted as mean ± S.E. from seven independent experiments. Compared with non-Tg and USP20-Tg: #, p < 0.05. Compared with Non-Tg and DN-USP20-Tg: *, p < 0.05. E, SMCs stimulated with LPS for 24 h were immunoblotted for VCAM-1. F, band intensities for VCAM-1 were normalized to those of corresponding β-actin bands. These ratios were normalized to those obtained from unstimulated non-Tg SMCs to obtain fold over basal, plotted as mean ± S.E. from six independent experiments. Compared with non-Tg (*) or DN-USP20-Tg (#): p < 0.0001. G, equivalent protein samples of the indicated SMC lysates were immunoblotted for TLR4, MD-2, CD14, and β-actin.
To evaluate whether USP20 activity affects LPS-induced signaling upstream of TRAF6, we quantitated in our three SMC lines the protein levels of three cell-surface proteins required for LPS-induced signaling: TLR4, CD14, and MD-2 (49). As shown in Fig. 8G, all three SMC lines expressed equivalent levels of TLR4, CD14, and MD-2. Consequently, differences among SMCs with regard to LPS-evoked signaling were not attributable to differences in LPS-binding proteins; rather, these differences accorded with their relative levels of USP20 activity in the SMCs.
NFκB Signaling Is Reduced in βarr2−/− SMCs
To determine whether the effects of USP20 on NFκB signaling are regulated by βarr2, we performed USP20 RNAi in SMCs from congenic WT and βarr2−/− mice. In WT and βarr2−/− SMCs, silencing USP20 reduced levels of IκBα in unstimulated SMCs as well as in LPS-stimulated SMCs (Fig. 9). Thus, USP20 appears to regulate NFκB activity in SMCs in the absence or presence of βarr2. However, with or without USP20 silencing, βarr2−/− SMCs demonstrated less IκBα degradation than WT SMCs at each time point (Fig. 9). Nevertheless, βarr2−/− and WT SMCs expressed equivalent levels of TLR4 (Fig. 9). These findings suggest that βarr2 contributes to NFκB activation and, thereby, to proinflammatory phenotypic changes in SMCs.
LPS-induced βarr2 Ubiquitination Is Reversed by USP20
βarr2 seems to play paradoxically reciprocal roles in TLR4-dependent NFκB signaling in SMCs. The first apparent role is anti-inflammatory: βarr2 scaffolds USP20 and TRAF6 and thereby facilitates TRAF6 deubiquitination and, consequently, diminishes NFκB activation. The second βarr2 role is proinflammatory: βarr2 appears to promote IκBα degradation. To elucidate how βarr2 could be either anti- or proinflammatory, we investigated whether the reciprocal effects of βarr2 in TLR4-dependent NFκB signaling could be influenced by ubiquitination of βarr2 itself. We pursued this strategy because ubiquitination regulates the function of βarr2 in GPCR trafficking and endocytosis (4, 5). To examine βarr2 ubiquitination in NFκB signaling, we immunoprecipitated endogenous βarr isoforms from non-Tg and SMC-DN-USP20-Tg SMCs challenged with LPS. Both basal and LPS-induced βarr ubiquitination were greater in SMC-DN-USP20-Tg than in non-Tg SMCs (Fig. 10, A and B). Thus, βarr isoforms are ubiquitinated downstream of TLR4 activation, and βarr appears to be deubiquitinated by USP20. With prolonged stimulation, βarr ubiquitination was undetectable in both non-Tg and SMC-DN-USP20-Tg SMCs, suggesting that deubiquitinases distinct from USP20 may also deubiquitinate βarr isoforms.
FIGURE 10.

USP20 reverses TLR4-induced ubiquitination of βarr2. A, SMCs from congenic non-transgenic or SMC-DN-USP20-Tg mice were exposed to serum-free medium lacking (−) or containing 1 μg/ml LPS at 37 °C for the indicated times and then solubilized. SMC lysates were immunoprecipitated with anti-βarr1/2 IgG, and IPs were immunoblotted (IB) serially for Ub with FK1 IgG and then for βarr1/2. Tris-glycine 4–20% gradient gels were used to facilitate resolution of ubiquitinated proteins. However, these gels do not optimally resolve the two βarr isoforms. (Nonetheless, the A1CT IgG used for IP pulls down βarr2 as well as or even better than it does βarr1 (69)). B, ubiquitin smears (Mr ≥64) in each lane were normalized to their corresponding βarr band intensities. These ratios were normalized to those obtained from unstimulated non-Tg SMCs to obtain “ubiquitinated βarr,” plotted as mean ± S.E. from three independent experiments. Compared with unstimulated Non-Tg: *, p < 0.05. C, HEK-293 cells were transfected with FLAG-tagged βarr2 and either empty vector plasmid (control) or untagged DN-USP20 plasmid, exposed to serum-free medium lacking or containing LPS (1 μg/ml) for 10 min at 37 °C, and then solubilized. βarr2 was immunoprecipitated with anti-FLAG IgG, and immunoprecipitates were immunoblotted serially for total ubiquitin and then βarr2. Lysates were immunoblotted for USP20. D, for each βarr2 IP, the density of the entire lane of bands staining for ubiquitin was normalized to the cognate βarr2 band. Data were plotted as mean ± S.E. from five experiments. Compared with unstimulated control cells: *, p < 0.05. E, HEK-293 cells were transfected with FLAG-tagged βarr2 and siRNA targeting no known protein (control, CTL), USP20, USP33, or USP20 + USP33. Cells were challenged ± LPS and processed as in C. F, βarr2 ubiquitination was calculated as in D and plotted as mean ± S.E. from five experiments. Compared with unstimulated control cells: *, p < 0.05, **, p < 0.01.
To corroborate findings obtained with DN-USP20 in SMCs, we tested βarr2 ubiquitination in HEK-293 cells. First, just as we found in SMCs, DN-USP20 in HEK-293 cells increased the ubiquitination of βarr2 (Fig. 10, C and D). We then silenced USP20 with siRNA transfection and observed similar effects: βarr2 ubiquitination increased when USP20 expression was reduced (Fig. 10, E and F). To determine whether USP33 can deubiquitinate βarr2 and whether USP20 and USP33 jointly effect βarr2 deubiquitination, we silenced USP33 alone or in combination with USP20 (Fig 10, E and F). Interestingly, βarr2 ubiquitination levels increased with silencing of either USP20 or USP33 even though USP20 RNAi did not decrease USP33 levels and USP33 RNAi did not decrease USP20 levels. Although these findings suggested the possibility that USP20 and USP33 deubiquitinate distinct sites in βarr2, silencing USP20 and USP33 simultaneously failed to augment βarr2 ubiquitination above levels observed with individual USP silencing (Fig 10, E and F). (This latter observation may be attributable, in part, to incomplete efficacy of the double knockdown). Together, these data strongly suggest that, upon TLR4 stimulation, USP20 deubiquitinates βarr2 as well as TRAF6.
βarr2 Ubiquitination Promotes NFκB Activation
Temporally, βarr2 ubiquitination coincided with NFκB activation (Figs. 8 and 10), and both processes were inhibited by USP20. To test whether ubiquitination of βarr2 itself affects NFκB signaling, we used βarr2 mutant constructs: βarr2-Ub, a chimeric fusion protein that is resistant to deubiquitination (50), and βarr2–0K, in which all Lys residues were replaced with Arg to remove all sites of ubiquitination while preserving charge density, to model βarr2 that cannot be ubiquitinated (22). βarr2-Ub, βarr2–0K, and WT βarr2 were each expressed in βarr1−/−/βarr2−/− MEFs (29): at equivalent βarr2 levels for the WT and 0K constructs and ∼50% of WT βarr2 levels for the βarr2-Ub construct (Fig. 11A, βarr2 blot). In response to LPS, a modest increase in NFκB activation (phospho-p65(Ser-536)) was observed in cells that lack both βarr isoforms. Similar augmentation of 30% above basal activity was obtained in cells transfected with WT βarr2. On the other hand, βarr2-Ub transfection evoked a 2-fold increase in NFκB activity, which was significantly higher than all the other conditions, whereas βarr2–0K-expressing cells showed weak p65 activation of 10–15% above basal signals (Fig. 11, A and B). Thus, whereas ubiquitinated βarr2 appears to promote NFκB activation, non-ubiquitinated βarr2 does not.
FIGURE 11.

Reciprocal roles of βarr2 in NFκB signaling are defined by the scaffolding efficiency of βarr2 for USP20. A, βarr1−/−/βarr2−/− MEFs were transfected with plasmids encoding YFP (control) or the indicated YFP-tagged construct: WT-βarr2; a βarr2-ubiquitin chimera that is resistant to deubiquitination (βarr2-Ub); or βarr2 in which all Lys residues are mutated to Arg (βarr2–0K). MEFs were stimulated ± LPS (1 μg/ml) for 10 min (37 °C), and extracts were immunoblotted (IB) serially for phospho-p65(Ser-536), βarr2, and β-actin. B, band intensities for p-p65(Ser-536) were normalized to corresponding β-actin band intensities. These ratios were normalized to those obtained from unstimulated, control-transfected βarr1−/−/βarr2−/− MEFs to obtain fold over control, plotted as mean ± S.E. from five independent experiments. Compared with the cognate basal signal (*) or compared with all LPS-stimulated groups (#): p < 0.05. C, HEK-293 cells were transiently transfected with plasmids encoding no protein (−) or the indicated FLAG-tagged construct: βarr2 WT, βarr2-Ub (Ub), or βarr2–0K (0K). βarr2 immunoprecipitates and cognate lysates were immunoblotted serially for (endogenous) TRAF6 and βarr2. D, band intensities for co-immunoprecipitated TRAF6 were normalized to corresponding βarr2 band intensities. These ratios were normalized to those obtained in WT βarr2 IPs to obtain fold over control, plotted as mean ± S.E. from three independent experiments performed in triplicate. *, p < 0.05 compared with βarr2 WT or with βarr2-Ub. E, HEK-293 cells were transfected and immunoprecipitated as in C, but βarr2 immunoprecipitates and cognate whole cell lysates were immunoblotted serially for (endogenous) USP20 and βarr2. F, band intensities for co-immunoprecipitated USP20 were normalized to corresponding βarr2 band intensities. These ratios were normalized to those obtained in WT βarr2 IPs to obtain fold over control, plotted as mean ± S.E. from three independent experiments performed in triplicate. *, p < 0.05 compared with βarr2 WT; #, p < 0.01 compared with βarr2-Ub.
Non-ubiquitinated βarr2 Is an Efficient Scaffold for USP20
Because βarr2 ubiquitination promoted TLR4-induced activation of NFκB, we asked whether βarr2 ubiquitination affects the ternary complex of USP20, βarr2, and TRAF6. To this end, we first immunoprecipitated FLAG-tagged βarr2-WT, βarr2-Ub, and βarr2–0K from HEK-293 cells and immunoblotted for endogenous TRAF6. This approach showed that the association of TRAF6 with βarr2 was greatest when βarr2 was not ubiquitinated (Fig. 11, C and D). The same approach showed that the association of βarr2 with endogenous USP20 is 2-fold greater when βarr2 is not ubiquitinated (Fig. 11, E and F). In contrast, the association of βarr2 with endogenous USP20 was equivalent whether the βarr2 construct was constitutively ubiquitinated (βarr2-Ub) or the WT (presumably because a substantial fraction of the WT βarr2 was ubiquitinated, as seen in Fig. 10, C and E). These results indicate that the non-ubiquitinated form of βarr2 is a better scaffold for simultaneously engaging TRAF6 and USP20 than the ubiquitinated form of βarr2. Thus, perhaps by scaffolding TRAF6 and USP20, deubiquitinated (or non-ubiquitinated) βarr2 blocks NFκB activity and reduces inflammation (Fig. 12). In contrast, ubiquitinated βarr2 (or the βarr2-Ub chimera) promotes NFκB signaling because it cannot scaffold USP20 to activated TRAF6.
FIGURE 12.
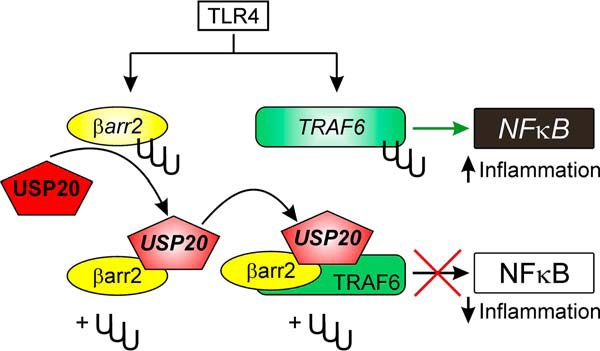
Proposed model for βarr2-mediated regulation of ubiquitin-dependent NFκB signaling. Activation of TLR4 triggers ubiquitination of βarr2 and TRAF6. Ubiquitinated βarr2 scaffolds USP20 to TRAF6 with low avidity. Therefore, when βarr2 is ubiquitinated, there is less deubiquitination of TRAF6 and greater NFκB signaling. However, after it is deubiquitinated by USP20, βarr2 scaffolds USP20 to TRAF6 with high avidity. Consequently, USP20-mediated TRAF6 deubiquitination and inhibition of NFκB signaling transpires.
Discussion
Our data demonstrate that USP20 inhibits inflammatory signaling in SMCs, and that USP20 may do so, in part, by deubiquitinating TRAF6 in a manner that requires scaffolding by non-ubiquitinated βarr2. Non-ubiquitinated βarr2 thereby serves to diminish inflammatory signaling. However, ubiquitinated βarr2 augments inflammatory signaling in a manner that can be triggered by TLR4 signaling. TLR4 signaling promotes βarr2 ubiquitination, reduces USP20/βarr2 association, and thereby potentiates TRAF6 ubiquitination and downstream NFκB signaling (Fig. 12). Consequently, our study helps to elucidate apparently paradoxical findings showing that βarr2 can be anti-inflammatory in some systems (6–8, 11–13) and proinflammatory in models of vascular disease (14–16, 21).
In earlier studies, βarr2 failed to deubiquitinate TRAF6 in cell-free assays (6). Consequently, the inhibitory effect of βarr2 on TLR4-dependent NFκB signaling was previously attributed to βarr2-mediated inhibition of TRAF6 oligomerization and subsequent TRAF6 autoubiquitination (6). In this study, however, we report that βarr2 facilitates TRAF6 deubiquitination by serving as a scaffold for the deubiquitinase USP20. Thus, the ternary complex of TRAF6-βarr2-USP20 conforms to a common theme: that deubiquitinases associate with scaffolding proteins to facilitate association with their substrate and, consequently, to enhance their substrate affinity and specificity (51). Although the absence of USP20 did not affect the association of βarr2 with TRAF6, the absence of βarr2 abrogated the association of USP20 with TRAF6 in cells and reduced by 5-fold the association of purified USP20 with purified TRAF6. Indeed, the βarr2 dependence of TRAF6/USP20 association may, along with possible βarr2-mediated inhibition of TRAF6 oligomerization, account for the increase in TRAF6 ubiquitination observed in βarr2-deficient cells (6).
βarr2 appears to regulate NFκB activation through cell- and signaling context-specific mechanisms. In response to LPS, bone marrow-derived macrophages from βarr2−/− mice show more IKK activity than WT macrophages (6), but they show equivalent LPS-induced secretion of the NFκB-dependent gene products TNF and IL-6 (11). Although βarr2 appears to reduce secretion of TNF and IL-6 from fibroblast-like synoviocytes (13), it has no effect on the secretion of the NFκB-dependent gene products (52–54) hyaluronan and plasminogen activator inhibitor-1 from lung fibroblasts (15). However, in SMCs, βarr2 augments TLR4-dependent IκB degradation and inflammation-associated SMC proliferation (Fig. 9 and Ref. 14). By performing βarr2 reconstitution experiments in βarr1/2-double knockout MEFs, distinct groups have shown both (a) that βarr2 decreases LPS-induced TRAF6 ubiquitination and IκBα phosphorylation (6), and (b) that βarr2 increases lysophosphatidic acid-induced activation of nuclear NFκB (21). In the intact mouse, βarr2 exerts similarly diverse effects on a variety of endpoints regulated substantially by canonical NFκB activation (20, 55, 56). βarr2 attenuates the effects of LPS-induced or septic shock, which transpires over hours (6, 11). However, βarr2 has no effect on LPS-induced asthma (16), and βarr2 augments allergic asthma (16), arterial neointimal hyperplasia, and atherosclerosis (14), which develop over many weeks. To reconcile these diverse (and in some cases divergent) findings in vitro and in vivo, one could in some cases invoke the diversity of signaling mechanisms in play. However, the current work with dynamic βarr2 ubiquitination enables us to invoke more specific and novel mechanisms, too. We speculate that (a) βarr2 augments NFκB activity under conditions where deubiquitination of βarr2 is relatively slow, or impaired, so that βarr2 cannot serve to tether USP20 to TRAF6 and (b) βarr2 attenuates NFκB activity under conditions where βarr2-mediated scaffolding of USP20 is important for negatively regulating NFκB activation. Such conditions may be found in systems wherein the ratios of βarr2:USP20 and βarr2:TRAF6 are sufficiently low to favor the ternary complex of βarr2-USP20-TRAF6 rather than the binary complexes of βarr2-USP20 and βarr2-TRAF6, as demonstrated by our studies with purified proteins (Fig. 2). Our transgenic mice with SMC-specific expression of USP20 or DN-USP20 provide the first in vivo evidence that USP20 serves an anti-inflammatory role. This finding is remarkable because USP20 is only one of ∼85 DUBs in the mammalian proteome (57, 58), and very few of these have been implicated in the regulation of NFκB signaling. For example, the USP-family DUB known as CYLD can bind to p62/TRAF6 complexes, inhibit TRAF6 ubiquitination, and regulate RANK signaling in osteoclast precursor cells (59). Furthermore, CYLD also inhibits TNF receptor-triggered NFκB signaling by deubiquitinating TRAF2 (60, 61). A somewhat contrary example is provided by the ovarian tumor protease DUB subfamily member A20, which can also deubiquitinate TRAF6, regulate NFκB signaling (62–64), and reduce NFκB-dependent gene product expression and atherosclerosis in Apoe−/− mice (65). Knockin studies with a deubiquitinase-defective A20 demonstrate that domains distinct from the DUB domain appear to achieve A20-mediated NFκB regulation (66). Whether βarr2 functions as an adaptor for additional DUBs that may regulate the NFκB pathway remains to be determined.
This study reveals the importance of dynamic ubiquitination as a major modulator of the reciprocal roles of βarr2 in NFκB signaling. Although ubiquitinated βarr2 scaffolds proteins in 7TMR pathways (22), non-ubiquitinated βarr2 appears to scaffold USP20 and its substrate TRAF6 in canonical NFκB pathways. Our data also suggest the possibility that activation and inactivation of USP20 may regulate the signaling properties of βarr2 in the canonical NFκB pathways. In the context of 7TMR trafficking, USP20 activity is regulated by cAMP-dependent kinase (PKA)-mediated phosphorylation of USP20 (67). Whether seryl phosphorylation of USP20 by TLR4-activated kinases such as IRAK1 (68) can modulate USP20 activity and thereby regulate βarr2 functions remains an interesting possibility that warrants further scrutiny.
Author Contributions
P. Y. J. C., L. Z., J. H. W., S. H., L. B., and S. K. S. performed the experiments and analyzed the data. P. Y. J. C., S. K. S., and N. J. F. wrote the manuscript. All authors approved the contents of this manuscript.
This work was supported by National Institutes of Health Grants HL080525 (to S. K. S.) and HL118369 (to S. K. S. and N. J. F.) and Grant 25550051 from the American Heart Association (to S. K. S.). We also acknowledge funding support from the Edna and Fred L. Mandel Jr. Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- 7TMR
- seven-transmembrane receptor
- DUB
- deubiquitinase
- IκB
- inhibitor of NFκB
- IKK
- IκB kinase
- DN
- dominant negative
- SMC
- smooth muscle cell
- IP
- immunoprecipitation
- Tg
- transgenic
- USP
- ubiquitin-specific protease
- Ub
- ubiquitin
- MEF
- mouse embryo fibroblast.
References
- 1. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 2. Lefkowitz R. J. (2013) Arrestins come of age: a personal historical perspective. Prog. Mol. Biol. Transl. Sci. 118, 3–18 [DOI] [PubMed] [Google Scholar]
- 3. Lefkowitz R. J., Rajagopal K., and Whalen E. J. (2006) New roles for β-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol. Cell 24, 643–652 [DOI] [PubMed] [Google Scholar]
- 4. Shenoy S. K., and Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kommaddi R. P., and Shenoy S. K. (2013) Arrestins and protein ubiquitination. Prog. Mol. Biol. Transl. Sci. 118, 175–204 [DOI] [PubMed] [Google Scholar]
- 6. Wang Y., Tang Y., Teng L., Wu Y., Zhao X., and Pei G. (2006) Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 7, 139–147 [DOI] [PubMed] [Google Scholar]
- 7. Gao H., Sun Y., Wu Y., Luan B., Wang Y., Qu B., and Pei G. (2004) Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 14, 303–317 [DOI] [PubMed] [Google Scholar]
- 8. Witherow D. S., Garrison T. R., Miller W. E., and Lefkowitz R. J. (2004) β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc. Natl. Acad. Sci. U.S.A. 101, 8603–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z. J. (2012) Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 246, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skaug B., Jiang X., and Chen Z. J. (2009) The role of ubiquitin in NF-κB regulatory pathways. Annu. Rev. Biochem. 78, 769–796 [DOI] [PubMed] [Google Scholar]
- 11. Fan H., Bitto A., Zingarelli B., Luttrell L. M., Borg K., Halushka P. V., and Cook J. A. (2010) β-Arrestin 2 negatively regulates sepsis-induced inflammation. Immunology 130, 344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan H., Luttrell L. M., Tempel G. E., Senn J. J., Halushka P. V., and Cook J. A. (2007) β-Arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol. Immunol. 44, 3092–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li P., Cook J. A., Gilkeson G. S., Luttrell L. M., Wang L., Borg K. T., Halushka P. V., and Fan H. (2011) Increased expression of β-arrestin 1 and 2 in murine models of rheumatoid arthritis: isoform specific regulation of inflammation. Mol. Immunol. 49, 64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J., Zhang L., Peppel K., Wu J. H., Zidar D. A., Brian L., DeWire S. M., Exum S. T., Lefkowitz R. J., and Freedman N. J. (2008) β-Arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ. Res. 103, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lovgren A. K., Kovacs J. J., Xie T., Potts E. N., Li Y., Foster W. M., Liang J., Meltzer E. B., Jiang D., Lefkowitz R. J., and Noble P. W. (2011) β-Arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci. Transl. Med. 3, 74ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker J. K., Fong A. M., Lawson B. L., Savov J. D., Patel D. D., Schwartz D. A., and Lefkowitz R. J. (2003) β-Arrestin-2 regulates the development of allergic asthma. J. Clin. Invest. 112, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zernecke A., and Weber C. (2005) Inflammatory mediators in atherosclerotic vascular disease. Basic Res. Cardiol. 100, 93–101 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Peppel K., Sivashanmugam P, Orman ES, Brian L, Exum ST, Freedman NJ (2007) Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27, 1087–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michelsen K. S., Wong M. H., Shah P. K., Zhang W., Yano J., Doherty T. M., Akira S., Rajavashisth T. B., and Arditi M. (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A. 101, 10679–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Libby P. (2012) Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun J., and Lin X. (2008) β-Arrestin 2 is required for lysophosphatidic acid-induced NF-κB activation. Proc. Natl. Acad. Sci. U.S.A. 105, 17085–17090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shenoy S. K., Barak L. S., Xiao K., Ahn S., Berthouze M., Shukla A. K., Luttrell L. M., and Lefkowitz R. J. (2007) Ubiquitination of β-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J. Biol. Chem. 282, 29549–29562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shenoy S. K., Modi A. S., Shukla A. K., Xiao K., Berthouze M., Ahn S., Wilkinson K. D., Miller W. E., and Lefkowitz R. J. (2009) β-Arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. U.S.A. 106, 6650–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yasunaga J., Lin F. C., Lu X., and Jeang K. T. (2011) Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-κB signaling. J. Virol. 85, 6212–6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berthouze M., Venkataramanan V., Li Y., and Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 28, 1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kemp P. R., Osbourn J. K., Grainger D. J., and Metcalfe J. C. (1995) Cloning and analysis of the promoter region of the rat SM22 α gene. Biochem. J. 310, 1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osbourn J. K., Weissberg P. L., and Shanahan C. M. (1995) A regulatory element downstream of the rat SM22 α gene transcription start point enhances reporter gene expression in vascular smooth muscle cells. Gene 154, 249–253 [DOI] [PubMed] [Google Scholar]
- 28. Keys J. R., Zhou R. H., Harris D. M., Druckman C. A., and Eckhart A. D. (2005) Vascular smooth muscle overexpression of G protein-coupled receptor kinase 5 elevates blood pressure, which segregates with sex and is dependent on Gi-mediated signaling. Circulation 112, 1145–1153 [DOI] [PubMed] [Google Scholar]
- 29. Kohout T. A., Lin F. S., Perry S. J., Conner D. A., and Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao K., Shenoy S. K., Nobles K., and Lefkowitz R. J. (2004) Activation-dependent conformational changes in β-arrestin 2. J. Biol. Chem. 279, 55744–55753 [DOI] [PubMed] [Google Scholar]
- 31. Wu J. H., Zhang L., Fanaroff A. C., Cai X., Sharma K. C., Brian L., Exum S. T., Shenoy S. K., Peppel K., and Freedman N. J. (2012) G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arterioscler. Thromb. Vasc. Biol. 32, 308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai X., Wu J. H., Exum S. T., Oppermann M., Premont R. T., Shenoy S. K., and Freedman N. J. (2009) Reciprocal regulation of the platelet-derived growth factor receptor-β and G protein-coupled receptor kinase 5 by cross-phosphorylation: effects on catalysis. Mol. Pharmacol. 75, 626–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shenoy S. K., McDonald P. H., Kohout T. A., and Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β 2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 34. Wu J. H., Peppel K., Nelson C. D., Lin F. T., Kohout T. A., Miller W. E., Exum S. T., and Freedman N. J. (2003) The adaptor protein β-arrestin2 enhances endocytosis of the low density lipoprotein receptor. J. Biol. Chem. 278, 44238–44245 [DOI] [PubMed] [Google Scholar]
- 35. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., and Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 36. Han S. O., Xiao K., Kim J., Wu J. H., Wisler J. W., Nakamura N., Freedman N. J., and Shenoy S. K. (2012) MARCH2 promotes endocytosis and lysosomal sorting of carvedilol-bound β(2)-adrenergic receptors. J. Cell Biol. 199, 817–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han S. O., Kommaddi R. P., and Shenoy S. K. (2013) Distinct roles for β-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Rep. 14, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luttrell L. M., and Miller W. E. (2013) Arrestins as regulators of kinases and phosphatases. Prog. Mol. Biol. Transl. Sci. 118, 115–147 [DOI] [PubMed] [Google Scholar]
- 39. Kang D. S., Tian X., and Benovic J. L. (2014) Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L., Miano J. M., Mercer B., and Olson E. N. (1996) Expression of the SM22α promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J. Cell Biol. 132, 849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Solway J., Seltzer J., Samaha F. F., Kim S., Alger L. E., Niu Q., Morrisey E. E., Ip H. S., and Parmacek M. S. (1995) Structure and expression of a smooth muscle cell-specific gene, SM22 α. J. Biol. Chem. 270, 13460–13469 [DOI] [PubMed] [Google Scholar]
- 42. Squadrito F., Deodato B., Bova A., Marini H., Saporito F., Calò M., Giacca M., Minutoli L., Venuti F. S., Caputi A. P., and Altavilla D. (2003) Crucial role of nuclear factor-κB in neointimal hyperplasia of the mouse carotid artery after interruption of blood flow. Atherosclerosis 166, 233–242 [DOI] [PubMed] [Google Scholar]
- 43. Wang X., Chai H., Lin P. H., Lumsden A. B., Yao Q., and Chen C. (2006) Mouse models of neointimal hyperplasia: techniques and applications. Med. Sci. Monit. 12, RA177–185 [PubMed] [Google Scholar]
- 44. Sakurai H., Suzuki S., Kawasaki N., Nakano H., Okazaki T., Chino A., Doi T., and Saiki I. (2003) Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 278, 36916–36923 [DOI] [PubMed] [Google Scholar]
- 45. Yang F., Tang E., Guan K., and Wang C. Y. (2003) IKK β plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 170, 5630–5635 [DOI] [PubMed] [Google Scholar]
- 46. Li Q., and Verma I. M. (2002) NF-κB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734 [DOI] [PubMed] [Google Scholar]
- 47. Neish A. S., Williams A. J., Palmer H. J., Whitley M. Z., and Collins T. (1992) Functional analysis of the human vascular cell adhesion molecule 1 promoter. J. Exp. Med. 176, 1583–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang L. L., Gao C. Y., Fang C. Q., Wang Y. J., Gao D., Yao G. E., Xiang J., Wang J. Z., and Li J. C. (2011) PPARγ attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc. Res. 92, 484–493 [DOI] [PubMed] [Google Scholar]
- 49. Stoll L. L., Denning G. M., and Weintraub N. L. (2006) Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr. Pharm. Des. 12, 4229–4245 [DOI] [PubMed] [Google Scholar]
- 50. Shenoy S. K., and Lefkowitz R. J. (2003) Trafficking patterns of β-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J. Biol. Chem. 278, 14498–14506 [DOI] [PubMed] [Google Scholar]
- 51. Ventii K. H., and Wilkinson K. D. (2008) Protein partners of deubiquitinating enzymes. Biochem. J. 414, 161–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hou B., Eren M., Painter C. A., Covington J. W., Dixon J. D., Schoenhard J. A., and Vaughan D. E. (2004) Tumor necrosis factor α activates the human plasminogen activator inhibitor-1 gene through a distal nuclear factor κB site. J. Biol. Chem. 279, 18127–18136 [DOI] [PubMed] [Google Scholar]
- 53. Lokeshwar V. B., Gomez P., Kramer M., Knapp J., McCornack M. A., Lopez L. E., Fregien N., Dhir N., Scherer S., Klumpp D. J., Manoharan M., Soloway M. S., and Lokeshwar B. L. (2008) Epigenetic regulation of HYAL-1 hyaluronidase expression: identification of HYAL-1 promoter. J. Biol. Chem. 283, 29215–29227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Z. J. (2005) Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 7, 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Poole E., King C. A., Sinclair J. H., and Alcami A. (2006) The UL144 gene product of human cytomegalovirus activates NFκB via a TRAF6-dependent mechanism. EMBO J. 25, 4390–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nijman S. M., Luna-Vargas M.P., Velds A., Brummelkamp T.R., Dirac A.M., Sixma T.K., and Bernards (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 [DOI] [PubMed] [Google Scholar]
- 58. Clague M. J., Coulson J. M., and Urbé S. (2012) Cellular functions of the DUBs. J. Cell Sci. 125, 277–286 [DOI] [PubMed] [Google Scholar]
- 59. Jin W., Chang M., Paul E. M., Babu G., Lee A. J., Reiley W., Wright A., Zhang M., You J., and Sun S. C. (2008) Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J. Clin. Invest. 118, 1858–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harhaj E. W., and Dixit V. M. (2011) Deubiquitinases in the regulation of NF-κB signaling. Cell Res 21, 22–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang J., Stirling B., Temmerman S. T., Ma C. A., Fuss I. J., Derry J. M., and Jain A. (2006) Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Invest. 116, 3042–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beyaert R., Heyninck K., and Van Huffel S. (2000) A20 and A20-binding proteins as cellular inhibitors of nuclear factor-κ B-dependent gene expression and apoptosis. Biochem. Pharmacol. 60, 1143–1151 [DOI] [PubMed] [Google Scholar]
- 63. Boone D. L., Turer E. E., Lee E. G., Ahmad R. C., Wheeler M. T., Tsui C., Hurley P., Chien M., Chai S., Hitotsumatsu O., McNally E., Pickart C., and Ma A. (2004) The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060 [DOI] [PubMed] [Google Scholar]
- 64. Shembade N., and Harhaj E. W. (2012) Regulation of NF-κB signaling by the A20 deubiquitinase. Cell. Mol. Immunol. 9, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wolfrum S., Teupser D., Tan M., Chen K. Y., and Breslow J. L. (2007) The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-κB target genes. Proc. Natl. Acad. Sci. U.S.A. 104, 18601–18606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De A., Dainichi T., Rathinam C. V., and Ghosh S. (2014) The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep. 15, 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kommaddi R. P., Jean-Charles P. Y., and Shenoy S. K. (2015) Phosphorylation of the deubiquitinase USP20 by protein kinase A regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 290, 8888–8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Janssens S., and Beyaert R. (2003) Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell 11, 293–302 [DOI] [PubMed] [Google Scholar]
- 69. McDonald P. H., Chow C. W., Miller W. E., Laporte S. A., Field M. E., Lin F. T., Davis R. J., and Lefkowitz R. J. (2000) β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 [DOI] [PubMed] [Google Scholar]