

Abstract
The ClpP protease complex and its regulatory ATPases, ClpC1 and ClpX, in Mycobacterium tuberculosis (Mtb) are essential and, therefore, promising drug targets. The Mtb ClpP protease consists of two heptameric rings, one composed of ClpP1 and the other of ClpP2 subunits. Formation of the enzymatically active ClpP1P2 complex requires binding of N-blocked dipeptide activators. We have found a new potent activator, benzoyl-leucine-leucine (Bz-LL), that binds with higher affinity and promotes 3–4-fold higher peptidase activity than previous activators. Bz-LL-activated ClpP1P2 specifically stimulates the ATPase activity of Mtb ClpC1 and ClpX. The ClpC1P1P2 and ClpXP1P2 complexes exhibit 2–3-fold enhanced ATPase activity, peptide cleavage, and ATP-dependent protein degradation. The crystal structure of ClpP1P2 with bound Bz-LL was determined at a resolution of 3.07 Å and with benzyloxycarbonyl-Leu-Leu (Z-LL) bound at 2.9 Å. Bz-LL was present in all 14 active sites, whereas Z-LL density was not resolved. Surprisingly, Bz-LL adopts opposite orientations in ClpP1 and ClpP2. In ClpP1, Bz-LL binds with the C-terminal leucine side chain in the S1 pocket. One C-terminal oxygen is close to the catalytic serine, whereas the other contacts backbone amides in the oxyanion hole. In ClpP2, Bz-LL binds with the benzoyl group in the S1 pocket, and the peptide hydrogen bonded between parallel β-strands. The ClpP2 axial loops are extended, forming an open axial channel as has been observed with bound ADEP antibiotics. Thus occupancy of the active sites of ClpP allosterically alters sites on the surfaces thereby affecting the association of ClpP1 and ClpP2 rings, interactions with regulatory ATPases, and entry of protein substrates.
Keywords: enzyme mechanism, protease, protein complex, protein degradation, x-ray crystallography
Introduction
Members of the Clp family of ATP-dependent protease complexes are found in all phylogenetic kingdoms and constitute a primary protein degradation system in bacteria as well as within the mitochondria and chloroplasts of eukaryotes (1). The ClpP protease and its regulatory ATPases have been extensively studied in Escherichia coli where they were first discovered (2, 3). ClpP is the proteolytic core of the complex and is composed of 14 subunits arranged in layered heptameric rings that enclose a chamber in which proteins are degraded (4, 5). In vivo, ClpP functions in protein degradation in association with either of two hexameric AAA+ ATPase complexes, ClpA or ClpX (6), which activates the protease and catalyzes ATP-dependent unfolding and translocation of substrates into the ClpP chamber (7–9). In Gram-positive bacteria and in Actinobacter the ClpA homologs belong to the ClpC class of Hsp100 proteins (10). Most bacteria have a single clpP gene, and their ClpPs are homomeric tetradecamers. However, some bacteria, including Mycobacterium tuberculosis (Mtb),4 have two clpP genes that encode structurally similar but divergent forms of ClpP (11, 12). The addition of N-terminally blocked dipeptides or tripeptides can induce allosteric changes in Mtb ClpP1 and/or ClpP2 and allow them to associate with each other to form the active heteromeric tetradecamer. This complex, composed of one ClpP1 and one ClpP2 ring, also interacts with Mtb ClpX or ClpC1, allowing the complex to degrade proteins in an ATP-dependent manner (11, 13).
ClpA and ClpX, together with cognate adaptor proteins, recognize specific sets of cellular proteins that are targeted to ClpP for degradation (14). In many bacteria degradation by ClpAP or ClpXP serves important or even essential roles, such as ClpXP-dependent degradation of the master cell cycle control protein, CtrA, in Caulobacter (15, 16) or ClpCP-dependent degradation of ComK, a transcriptional regulator involved in competence development, DNA repair, DNA transformation, and recombination in Bacillus (17). ClpXP and ClpCP affect expression of virulence factors in several human pathogens, including enterohemorrhagic E. coli, Enterococcus faecalis, Staphylococcus aureus, Salmonella enterica serovar Typhimurium, and Yersinia enterocolitica (18–20). ClpP1 and ClpP2 are essential for viability of M. tuberculosis, as are the regulatory ATPases, ClpX and ClpC1 (21), although the proteins that are targeted by the Clp protease complexes have not been identified (22). Novel antibiotics that prevent ATP-dependent proteolysis by ClpC1P1P2 (23, 24) and compounds that block ClpP1P2 (13) have been shown to selectively kill M. tuberculosis. Consequently, the components of the Clp protease complexes are of special interest as potential targets for new classes of antimicrobial compounds.
One exceptional feature of ClpP1P2 from Mtb is that in vitro it can be assembled from inactive ClpP1 or ClpP2 complexes only in the presence of low millimolar concentrations of certain N-blocked dipeptide activators or dipeptide derivatives (11). In our earlier studies, benzyloxycarbonyl-Leu-Leu (Z-LL) was the most effective activator from among a group of related blocked dipeptides and dipeptide derivatives. We showed that these molecules activated formation of ClpP1P2 complexes by promoting the dissociation of the inactive homotetradecameric ClpP1 and ClpP2 complexes into heptameric rings and that association into the heterotetradecameric ClpP1P2 complex increased peptidase activity >1000-fold (11). The plots of the concentration dependence of activation of ClpP1P2 by dipeptide activators are sigmoidal and reach saturation over a very narrow concentration range (Hill coefficient of about six), suggesting that six or more molecules must bind to ClpP1 and/or ClpP2 to promote co-assembly and catalytic competence. However, the addition of dipeptides at concentrations in excess of those needed for activation led to competitive inhibition of peptidase activity, which suggested that activators might bind at the catalytic sites in addition to possible allosteric sites in ClpP. Binding of activators to the active site was confirmed recently by x-ray crystallography (25); however, a mechanism that could reconcile the binding of activators to the active sites with their ability to enhance peptidase activity remained to be defined. Previously, binding of acyldepsipeptide antibiotics (ADEPs) were shown to stabilize assembly of homomeric ClpP tetradecamers (26). ADEPs bind to the AAA+ protein-docking site on the apical surface of the ClpP heptamer and induce the opening of the axial channel, which activates peptidase activity and allows non-processive degradation of unfolded domains in proteins (26–29). However, no previous reports describe an assembly mechanism solely involving ligand binding to the ClpP active site. Studies to date with Mtb ClpP1P2 have involved recombinant proteins expressed in heterologous systems, and it is unclear whether dipeptide or analogous activator mechanisms are required for active enzyme formation in vivo. In any event, this mechanism has facilitated analysis of allosteric communication and enzyme activation in Mtb ClpP1P2.
Here we report the crystal structure of Mtb ClpP1P2 obtained in the presence of the activator peptide, benzoyl-Leu-Leu (Bz-LL). Bz-LL and certain other activators, such as benzoyl-norvaline-isoleucine (Bz-Nva-Ile), were found to support severalfold higher peptidase activity than the activators that were investigated earlier (11). Also unlike those dipeptides, Bz-LL showed maximal activation at the high concentrations used for crystallization. While this work was in progress Schmitz et al. (25) reported the structure of Mtb ClpP1P2 with the less potent peptide activator, benzyloxycarbonyl-Ile-Leu (Z-IL), bound at active sites. In addition, that structure included ADEP molecules bound to the docking site on ClpP used by the cognate ATPases, which complicated interpretation of the allosteric effects of the dipeptide activator alone. Our structure with Bz-LL bound was obtained in the absence of an ADEP and also differs from the previously published one in several features, including the orientation of the dipeptide in the active sites of ClpP2. Importantly, ClpP1P2 with Bz-LL bound specifically enhanced ATP hydrolysis by Mtb ClpX and ClpC1 and ATP-dependent degradation of model protein substrates. These findings make it very likely that the structure described here is the physiologically active form of the ClpP1P2 complex.
Experimental Procedures
Chemicals and Other Reagents
Dipeptide activator Z-LL was obtained from Bachem, and Bz-LL was synthesized in the laboratory of Dr. William Bachovchin (Tufts University). The fluorogenic peptide, acetyl-PKM-7-amido-4-methylcoumarin (Ac-PKM-amc), was obtained from AnaSpec Inc. Pyruvate kinase, lactate dehydrogenase, ATP, NADH, phosphoenol pyruvate, and FITC-casein were purchased from Sigma. All reagents for crystallization were obtained from Hampton Research (Aliso Viejo, CA).
Protein Purification and Preparation of the Heterotetradecameric ClpP1P2 Complex
M. tuberculosis ClpP1 and ClpP2 with His6 C-terminal extensions were individually expressed and purified from E. coli cells as described previously (11). Proteins were dialyzed into 20 mm potassium phosphate, pH 7.5, containing 0.1 m KCl and 5% (v/v) glycerol. The purified proteins were mixed in equimolar amounts at a final concentration of 2–3 mg/ml and Bz-LL or Z-LL was added to a final concentration of 5 mm. After 2 h at room temperature the proteins were concentrated by centrifugation through a Microcon 100 membrane to a final concentration of 15–20 mg/ml. Proteins were frozen in a dry ice/ethanol bath and stored at −80 °C until thawed on ice for crystallization. Mtb ClpX and ClpC1 were purified as described previously (11) and stored in 50 mm Tris HCl buffer, pH 7.5, with 100 mm KCl and 10% glycerol.
Assays of Peptidase Activity
Peptidase assays were performed at 37 °C in black 96-well plates using a Plate Reader SpectraMax M5 (Molecular Devices). Each well contained 10 μm fluorogenic peptide, 20–50 nm ClpP1P2 in 80 μl of buffer A (20 mm phosphate buffer, pH 7.6, with 100 mm KCl, 5% glycerol, and 2 mm Bz-LL). Peptidase activity was followed in the linear range by monitoring the rate of production of fluorescent 7-amino-4-methylcoumarin from peptide-amc substrates at 460 nm (Ex at 380 nm). The deviation of fluorescence value in three independent measurements was not >5%.
Proteinase Assay
ClpP1P2 was assayed continuously in 96-well plates using the fluorescent protein substrates, GFP-SsrA and FITC-casein. To measure GFP-SsrA degradation by the ClpXP1P2 complex, each well contained 500 nm GFP-SsrA, 75–100 nm ClpP1P2 tetradecamer and 300–400 nm ClpX hexamer, and 2 mm Mg-ATP in 100 μl of buffer A and GFP-SsrA fluorescence was measured at 510 nm (excitation at 470 nm). To measure FITC-casein degradation by ClpC1P1P2, each well contained 150–200 nm ClpP1P2, 500–700 nm ClpC1 hexamer, 1–1.2 μm FITC-casein, and 2 mm Mg-ATP in 100 μl of buffer A. FITC-casein fluorescence was monitored at 518 nm (excitation at 492 nm), and the deviation of fluorescence value in three independent measurements was ≤10%.
ATPase Assay
ATP hydrolysis was measured with the enzyme-linked assay using pyruvate kinase (PK) and lactic dehydrogenase. 2 μg of pure ClpC1 or ClpX were mixed with 100 μl of the assay buffer B containing 1 mm phosphoenolpyruvate, 1 mm NADH, 2 units of pyruvate kinase/lactic dehydrogenase, 4 mm MgCl2, and 1 mm ATP, and the ATPase activity was followed by measuring the oxidization of NADH to NAD spectrometrically at 340 nm. Measurements were performed in triplicate and agreed within 5%.
Protein Crystallization and Structure Determination
Crystals of the complexes of ClpP1 and ClpP2 with activator peptides were grown in hanging drops. The well solution consisted of 25% (w/v) PEG3350 in 0.1 m Bis-Tris buffer, pH 6.5. A drop contained a mixture of 2 μl of ClpP1P2 in phosphate buffer as described above and 2 μl of well solution. Crystals were grown with either Bz-LL or Z-LL under the same conditions, and the choice of the activator peptide did not appear to affect crystallization. Two data sets were collected at beamline ID-22 (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory. The Bz-LL data set was processed with XDS (30), and the structure was solved by molecular replacement using Phaser (31). The data set with the PDB code 2CBY was used as a search model for ClpP1, whereas PDB code 4JCT was used for ClpP2. The asymmetric unit contained four heptameric rings, two consisting of only ClpP1 and two consisting of only ClpP2. The four heptameric rings were divided between two tetradecamers made up of a ClpP1 heptamer bound to a ClpP2 heptamer in a manner analogous to functional ClpP tetradecamers from other species. The structure of the Bz-LL complex was refined with REFMAC5 (32). Data collected from Z-LL complex crystal were processed with HKL2000 (33), and the structure of the final model of the Bz-LL complex was used directly to initiate refinement using REFMAC5. Data collection and refinement statistics are shown in Table 1.
TABLE 1.
Data collection and structure refinement
r.m.s.d., root mean square deviation.
| ClpP1P2 with Bz-Leu-Leu | ClpP1P2 with Z-Leu-Leu | |
|---|---|---|
| Data collection | ||
| Space group | C2 | C2 |
| Molecules/asymmetric unit | 2 (28 chains) | 2 (28 chains) |
| Unit cell a, b, c (Å); β | 205.94, 183.35, 188.45; 94.44° | 205.18, 183.54, 188.37; 94.53° |
| Resolution (Å) | 100.0-3.07 (3.15-3.07)a | 50.0-2.9 (2.95-2.9) |
| Rmergeb (%) | 10.5 (66.6) | 9.1 (80.4) |
| No. of reflections (measured/unique) | 545,500/129,368 | 560,142/147,557 |
| 〈I /σI〉 | 11.33 (2.29) | 14.1 (1.2) |
| Completeness (%) | 99.5 (100) | 94.6 (84.9) |
| Redundancy | 4.22 (4.29) | 3.8 (2.9) |
| Refinement | ||
| Resolution (Å) | 72.2-3.07 | 49.27-2.9 |
| No. of reflections (refinement/Rfree) | 115,022/6,092 | 143,061/4,469 |
| R / Rfreec | 0.197/0.231 | 0.202/0.234 |
| No. atoms | ||
| Protein | 40,432 | 40,563 |
| Ligands | 490 | 0 |
| Water | 53 | 105 |
| r.m.s.d. from ideal targets | ||
| Bond lengths (Å) | 0.016 | 0.018 |
| Bond angles (°) | 1.94 | 1.99 |
| PDB accession code | 5DZK | 5E0S |
a The highest resolution shell is shown in parentheses.
b Rmerge = ΣhΣi|Ii − 〈I〉|/ΣhΣiIi, where Ii is the observed intensity of the ith measurement of reflection h, and 〈I〉 is the average intensity of that reflection obtained from multiple observations.
c R = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively, calculated for all data. Rfree was defined in Ref. 56.
Results
Bz-Leu-Leu and Bz-Nva-Ile Are Potent Activators of Mtb ClpP1P2
In our earlier studies (11) we found that Z-LL stimulated formation of the active the Mtb ClpP1P2 complex but that maximal activation required 5 mm activator and no activity was evident below 2 mm (Fig. 1). In searching for more potent activators, we identified two new N-terminally blocked dipeptides, Bz-LL and Bz-Nva-Ile, which produced severalfold greater stimulation of peptidase activity. When peptidase activity was measured with the preferred fluorogenic substrate, Ac-PKM-amc (11, 13), a half-maximal rate of hydrolysis was obtained at ∼1.5 mm Bz-LL, and maximum stimulation occurred at 2.5 mm (Fig. 1). The Vmax for peptidase activity stimulated by Bz-LL was 3–4 times greater than that obtained with 4 mm Z-LL (Fig. 1). Similar effects were obtained with Bz-Nva-Ile (data not shown). In contrast, Z-Ile-Leu (Z-IL), the activator used by Schmitz et al. (25) in their crystallographic and kinetic studies, produced weaker activation than both Bz-LL and Z-LL (Fig. 1). Activation by Bz-LL was sigmoidal with a Hill coefficient of about four, suggesting that binding of multiple molecules of Bz-LL per tetradecamer might be needed for full activation. At higher concentrations (severalfold over that required for maximal activation) Bz-LL, Z-LL, and similar molecules become inhibitory (Fig. 1 and Ref. 11), suggesting that the activators might interfere with substrate binding at the active sites (see below). ClpP1P2 activity was lost immediately upon dilution into buffer without Bz-LL (data not shown). Thus, the remarkable activation is readily reversible and requires the persistent presence of bound activator to maintain the activated conformation.
FIGURE 1.

Bz-LL is a more potent activator of ClpP1P2 than dipeptides described previously. Hydrolysis of fluorogenic peptide Ac-PKM-amc was assayed at 37 °C as described under “Experimental Procedures.” Dipeptide activators (Bz-Leu-Leu, Z-Leu-Leu, or Z-Ile-Leu) were added at the concentrations shown. The production of fluorescent 7-amino-4-methylcoumarin from Ac-PKM-amc was monitored by the increase in fluorescence at 460 nm (excitation at 380 nm). Each point is the average of 3 measurements, which agreed within 5%. Similar results were obtained in at least three independent experiments. RFU, relative fluorescence.
Bz-Leu-Leu and Bz-Nva-Ile Synergize with ClpX and ClpC1 to Enhance Peptidase Activity of ClpP1P2
In our earlier studies we had found that the peptide activator Z-LL was required to obtain ClpX- and ClpC1-promoted protein degradation by ClpP1P2 (11). We wanted to know whether peptide activators would also synergize with ClpX or ClpC1 to enhance ClpP1P2 activity against peptide substrates. For these experiments we used the more potent activators, Bz-LL and Bz-Nva-Ile. With only ClpX or ClpC1 hexamers present in equimolar amounts with ClpP1P2 tetradecamers no peptidase activity could be detected (Fig. 2). However, inclusion of Bz-LL or Bz-Nva-Ile in addition to ClpX or ClpC1 produced a dramatic stimulation of ClpP1P2 peptidase activity (Fig. 2). With saturating concentrations of the dipeptide activator, the maximal peptidase activity was the same with or without the ClpX or ClpC1; however, half-maximal activation occurred at an order of magnitude lower concentration of activator when either ATPase was present (Fig. 2). These data indicate that ClpC1 and ClpX induce a conformation of ClpP1P2 that has higher affinity for Bz-LL and Bz-Nva-Ile. Interestingly, the apparent Hill coefficient for activator binding was significantly lower (∼2) when either ClpX or ClpC1was present, suggesting that fewer molecules of bound activator are needed to maintain an active conformation of ClpP1P2 when it is complexed with ClpX or ClpC1.
FIGURE 2.

The presence of ClpC1 or ClpX lowers the concentration of dipeptide activator needed to stimulate peptidase activity of ClpP1P2. Peptidase activity was measured with Ac-PKM-amc as described under “Experimental Procedures” but with varying concentrations of the activator Bz-Nva-Ile. Similar results were obtained with Bz-Leu-Leu (not shown). Where indicated, assay solutions also contained either ClpX or ClpC1, which was added in equimolar concentrations with the ClpP1P2 complex. Each point is the average of three measurements, which agreed within 5%. Similar results were obtained in at least three independent experiments. RFU, relative fluorescence.
ClpP1P2 with Bz-Leu-Leu Bound Stimulates ATP Hydrolysis by ClpX and ClpC1 and Enhances ATP-dependent Proteolysis
Because Bz-LL and Bz-Nva-Ile were more potent than Z-LL in stimulating peptidase activity of ClpP1P2 in the presence of ClpX or ClpC1, we asked whether these peptide activators affected the interaction of ClpP1P2 with either ATPase and whether they were also more potent in promoting protein degradation by ClpXP1P2 or ClpC1P1P2. Previous studies had shown that binding of ClpP to ClpX, ClpA, or ClpC1 affected its ATPase activity, although both inhibitory (34) and activating (35) effects had been reported. We measured the rates of ATP hydrolysis by Mtb ClpX and ClpC1 in the presence of ClpP1P2. ClpP1P2 complex was pre-assembled at high protein concentrations (5 mg/ml) in the presence of either Z-LL or Bz-LL and then diluted together with one of the ATPases into reaction buffer containing (or not containing) peptide activators (Fig. 3A). As shown in Fig. 3A, neither ClpP1 nor ClpP2 alone affected the ATPase activity of ClpX or ClpC1 with or without the added peptide activator. Also, in the absence of a peptide activator, the mixture of ClpP1 and ClpP2 did not affect ATPase activity. However, the Bz-LL-activated ClpP1P2 complex stimulated the ATPase activity of both ClpX and of ClpC1 >2-fold (Fig. 3A). It is noteworthy that binding of the activator was essential to sustain the interaction between ClpP1P2 and the ATPase because the enhanced activity was lost almost immediately upon dilution into reaction buffer lacking the activator (Fig. 3A).
FIGURE 3.

Peptide-activated ClpP1P2 stimulates ATPase activities of ClpX and ClpC1. A, ClpP1P2 activates ATP hydrolysis by ClpC1 and ClpX only in the presence of Bz-LL. ATP hydrolysis was measured in a coupled enzymatic assay with pyruvate kinase and lactate dehydrogenase as described under “Experimental Procedures.” To obtain an active ClpP1P2 complex, ClpP1 (5 mg/ml) and ClpP2 (5 mg/ml) were mixed together in the presence of Bz-LL or Z-LL and incubated for 30 min at room temperature. ClpP1P2 was then diluted 50-fold into 100 μl of reaction mixture that contained 10 μg of ClpC1 (top graph) or 5 μg of ClpX (bottom graph) and the dipeptide activators as indicated. ATPase activity was monitored by the decrease in absorbance at 340 nm. Activities obtained when Z-LL or Bz-LL was present are expressed relative to the activity measured in the absence of activator, which was taken as 100% (Control). Specific activities of ClpC1 and ClpX alone were 1.25 and 1.87 μmol/mg/min, respectively. Each point is the average of 3 measurements, which agreed within 5–10%. Similar results were obtained in at least three independent experiments. B, ratio of ClpP1P2 and ClpC1 or ClpX resulting in maximal activation of ATP hydrolysis. ATP hydrolysis by 10 μg of ClpC1 or 5 μg ClpX was measured in 100 μl of reaction buffer as described in panel A. ClpP1P2 was added in varying concentrations, and Bz-LL (2 mm) was present in all assays. ATPase activity in the absence of ClpP1P2 was taken as 100% (control). Each point is the average of 3 measurements, which agreed within 5–10%. Similar results were obtained in at least three independent experiments.
To determine the stoichiometry of ClpP1P2 and the cognate ATPases in the active complexes, we measured ATPase activity of ClpC1 or ClpX in the presence of varying amounts of ClpP1P2. Maximal activation was reached when the ClpP1P2 tetradecamers were present in equimolar concentrations with ClpC1 or ClpX hexamers (Fig. 3B). These data support the conclusion that the stimulation of ATPase activity reflects a specific interaction and further suggest that only one heptameric ring in ClpP1P2 interacts with ClpC1 or ClpX, as was shown by Leodolter et al. (36) and suggested by the binding of ADEP to only the ClpP2 ring in the ClpP1P2 structure reported by Schmitz et al. (25). The addition of protein substrates for ClpX (GFP-SsrA) or ClpC1 (FITC-casein) to assays with ClpP1P2 and the peptide activator present did not produce any further stimulation of ATPase activity (data not shown). Thus, binding of the peptide induces a conformation of ClpP1P2 that interacts effectively and productively with ClpX and ClpC1. This effect of ClpP1P2 on the ATPase is quite specific, as no activation of the Mtb ATPases was observed with E. coli ClpP (eClpP) (with or without the dipeptide activators) (Fig. 4A). Similarly, Mtb ClpP1P2 was unable to activate ATP hydrolysis by E. coli ClpA (eClpA), although E. coli ClpP (eClpP) binding produced a 3–4-fold activation of eClpA ATPase activity (Fig. 4A).
FIGURE 4.

Specificity of interaction between peptide-activated ClpP1P2 and ClpX or ClpC1. A, E. coli or Mtb ClpP protease complexes stimulate ATPases from the same species only. ATPase activity was measured with 5 μg of Mtb ClpC1, Mtb ClpX, or eClpA in 100 μl of reaction mixture as in Fig. 3A. Assay mixtures had either no ClpP, 10 μg of E. coli ClpP, or 10 μg of Mtb ClpP1P2 plus 2 mm Bz-LL. Activity in the absence of ClpP1P2 was taken as 100% (control). Each point is the average of three measurements, which agreed within 5%. Similar results were obtained in at least three independent experiments. B, ADEP blocks the activation of ClpC1 and ClpX ATPases by ClpP1P2. ATPase assays were conducted as in Fig. 3A. Assay mixtures contained 10 μg of ClpC1 or ClpX and either no addition, 10 μg of ClpP1P2 plus 2 mm Bz-LL, or 10 μg of ClpP1P2 plus 2 mm Bz-LL plus 100 μg ADEP2. Activity in the absence of ClpP1P2 was taken as 100% (control). Each point is the average of 3 measurements, which agreed within 5–10%. Similar results were obtained in at least three independent experiments.
ADEP antibiotics have been shown to bind to the same sites on ClpP used for docking ClpX and ClpC1 (26, 29). As a further confirmation that activation of ClpX or ClpC1 ATPase activity seen in the presence of ClpP1P2 was dependent on the same functional interactions that support ATP-dependent proteolysis, we tested whether the stimulated ATPase activity could be blocked by an ADEP antibiotic. The addition of saturating levels of ADEP2 completely inhibited the ability of ClpP1P2 to stimulate ATP hydrolysis by ClpX and ClpC1, whereas ADEP2 had no effect on the basal (non-stimulated) rate of ATP hydrolysis by either ATPase (Fig. 4B).
As shown above (Fig. 3A), although both Z-LL and Bz-LL promoted ClpP1P2 formation and ATP-dependent protein degradation, only Bz-LL gave measureable stimulation of ATPase activity. To determine if the increase in ATP hydrolysis was associated with enhanced proteolysis, we compared protein degradation rates by ClpXP1P2 and ClpC1P1P2 in the presence of Bz-LL and Z-LL. The rates of degradation of GFP-SsrA by ClpXP1P2 and FITC-casein by ClpC1P1P2 were 2-fold higher in the presence of Bz-LL than with Z-LL (Fig. 5A). This enhanced protein degradation in the presence of Bz-LL correlated with the 2–3-fold stimulation of ATP hydrolysis. To confirm that the protein degradation was dependent on ATP hydrolysis, we assayed degradation with Bz-LL-activated ClpP1P2 in the presence of the ATPases but added the non-hydrolyzed analog ATPγS instead of ATP. No degradation of the tightly folded protein, GFP, or the very loosely folded substrate, casein, was observed with ATPγS present (Fig. 5A). Thus, the increased ATP consumption is apparently linked to increased capability of protein degradation. The tight coupling of ATPase and proteolytic activities also supports the conclusion that the conformation of ClpP1P2 promoted by N-benzoyl dipeptide activators is physiologically relevant.
FIGURE 5.

Bz-LL stimulates proteolytic activity of ClpP1P2 more than Z-LL in the presence or absence of the regulatory ATPases. A, Bz-LL enhanced the ability of ClpC1 or ClpX to promote protein degradation by ClpP1P2. Degradation of FITC-casein by ClpP1P2 in the presence of ClpC1 and degradation of GFP-SsrA by ClpP1P2 in the presence of ClpX were measured continuously as described under “Experimental Procedures” in buffer A containing either 2 mm Mg-ATP or 100 μm ATPγS and the dipeptide activators as indicated. For ease of comparison, data were normalized to the rate of degradation in the presence of Z-LL, which was taken as 100% (control). Each point is the average of 3 measurements, which agreed within 5–10%. Similar results were obtained in at least three independent experiments. B, Bz-LL promotes degradation of peptides and unfolded proteins by ClpP1P2 in the absence of ATPases. FITC-casein was incubated with a mixture of ClpP1 and ClpP2 in the presence of 2 mm Bz-LL or 5 mm of Z-LL. Degradation was monitored by the decrease in fluorescence of FITC-casein as described under “Experimental Procedures.” Peptidase activity was measured as described in Fig. 1. Protein degradation or peptidase activity in the presence of Z-LL was designated as 100% (control). Each point is the average of 3 measurements, which agreed within 5–10%. Similar results were obtained in at least three independent experiments.
Crystallization of ClpP1P2 in the Presence of Bz-Leu-Leu and Z-Leu-Leu
The ClpP1P2 complexes were prepared in the presence of either Z-LL or Bz-LL; formation of active complexes was confirmed by enzymatic assay using fluorogenic peptide, Ac-GGL-amc (11). Complexes assembled with either Bz-LL or Z-LL produced isomorphous monoclinic crystals, in a form not previously reported for either ClpP1 or ClpP2 (Table 1). The structures were solved by molecular replacement at a resolution of 3.07 Å for the Bz-LL complex and 2.90 Å for the Z-LL complex (Table 1). In each case, the asymmetric unit contained two tetradecameric assemblies. The two tetradecamers in the asymmetric unit were nearly identical, differing primarily in their crystal contacts. The following description will be limited to just a single tetradecamer and will refer primarily to the molecule with Bz-LL bound, as the peptide was visible in that structure. Also, as the secondary and tertiary folds of ClpP are highly conserved, the numbering of helices and β-strands used for E. coli ClpP (4) is adopted here.
Each tetradecamer consist of two heptameric rings (Fig. 6A), one made up of seven ClpP1 molecules (chains H-N and h-n), and the other made up of seven ClpP2 molecules (chains A-G and a-g). With the exception of two N-terminal residues, the entire coding region of ClpP2 is seen, whereas the ClpP1 molecule is missing seven N-terminal residues and eight C-terminal residues. No density is seen for the C-terminal linker or the His6 and FLAG tags, which are presumably mobile. The -fold of the core of the ClpP1 and ClpP2 monomers is very similar, with an root mean square deviation of 0.69 Å for 170 Cα pairs in a superposition of molecules A and H of the Bz-LL complex (Fig. 6B). The two molecules differ primarily in the N-terminal region, where residues 15–32 of ClpP2 form a very stable β-hairpin that extends away from the body of the compact tetradecamer. As observed in a recent crystal structure of ClpP1P2 (25), the extended conformation is stabilized by interaction with the loop extensions from another ClpP2 molecule related by crystallographic symmetry. The first seven residues of ClpP1 are not visible and presumably form a flexible coil. The visible portion of the N-terminal region contributes to helix A, which is slightly longer in ClpP1 than in ClpP2 and extends into the axial channel, which is consequently narrower in ClpP1 than in ClpP2 (Fig. 6C). The missing residues in ClpP1 are not sufficient to form the type of β hairpin seen in ClpP2 and in other ClpP structures (26) but would at least to some degree further occupy the axial channel and be positioned to interact with translocating substrates.
FIGURE 6.

Structural features of Mtb ClpP1P2. A, the tetradecamer is a complex of ClpP1 and ClpP2 heptamers. The mixed tetradecamer formed by joining the heptamers of ClpP1 (cyan helices and magenta β-strands) and ClpP2 (red helices and yellow β-strands) is shown in side view in schematic representation. In ClpP2 the N-terminal β-hairpins are in an extended conformation (yellow and green). B, ribbon representation showing the overlap of ClpP1 and ClpP2 subunits. Backbone atoms between residues 20–185 align with root mean square deviation of 1.2 Å. ClpP2 (green ribbon) has an extended N-terminal β-hairpin that is shorter and disordered in ClpP1 (cyan). ClpP2 also has a bulge in the coiled portion of the alpha-β handle that forms the interface between the heptamers in the tetradecameric complex. The activators are depicted as narrow sticks colored according to the subunits to which they are bound. C, overlap of the ClpP1 and ClpP2 heptamers. Stick representations of ClpP1 and ClpP2 are overlaid. Residues 13–19 of ClpP1 and 15–32 of ClpP2 are shown in surface rendering to illustrate the difference in the axial channel diameter in the two heptamers. D, 2Fo − Fc electron density of the activator bound in ClpP1 and ClpP2. The molecules lie in opposite orientations relative to the catalytic serine shown as sticks.
Opposite Orientations of the Activating Dipeptide in ClpP1 and ClpP2 Rings
The Bz-LL peptide is visible in all 14 molecules of the ClpP1P2 complex bound to the active site in the vicinity of the catalytic serine (Fig. 6D). Surprisingly, Bz-LL is oriented differently in ClpP1 and ClpP2. The peptide is aligned in antiparallel manner to flanking β-strands 6 and 8 of ClpP1. The side chain of the C-terminal leucine extends into the S1 binding pocket at the catalytic site, and the carboxyl oxygen is positioned with one carboxyl oxygen making a polar contact with the active site serine and the other forming polar contacts with the backbone amide nitrogens of Gly-69 and Met-99 (Fig. 7A), which have been shown to comprise the oxyanion hole in other ClpPs. Superposition of a ClpP1 subunit with Bz-LL bound onto the structure of E. coli ClpP with a peptide methyl ketone (chloromethyl ketone) covalently bound in the active site (37) showed an excellent overlap of the two peptide ligands, which engage in identical backbone hydrogen bonding interactions with the flanking β-strands (Fig. 7B).
FIGURE 7.
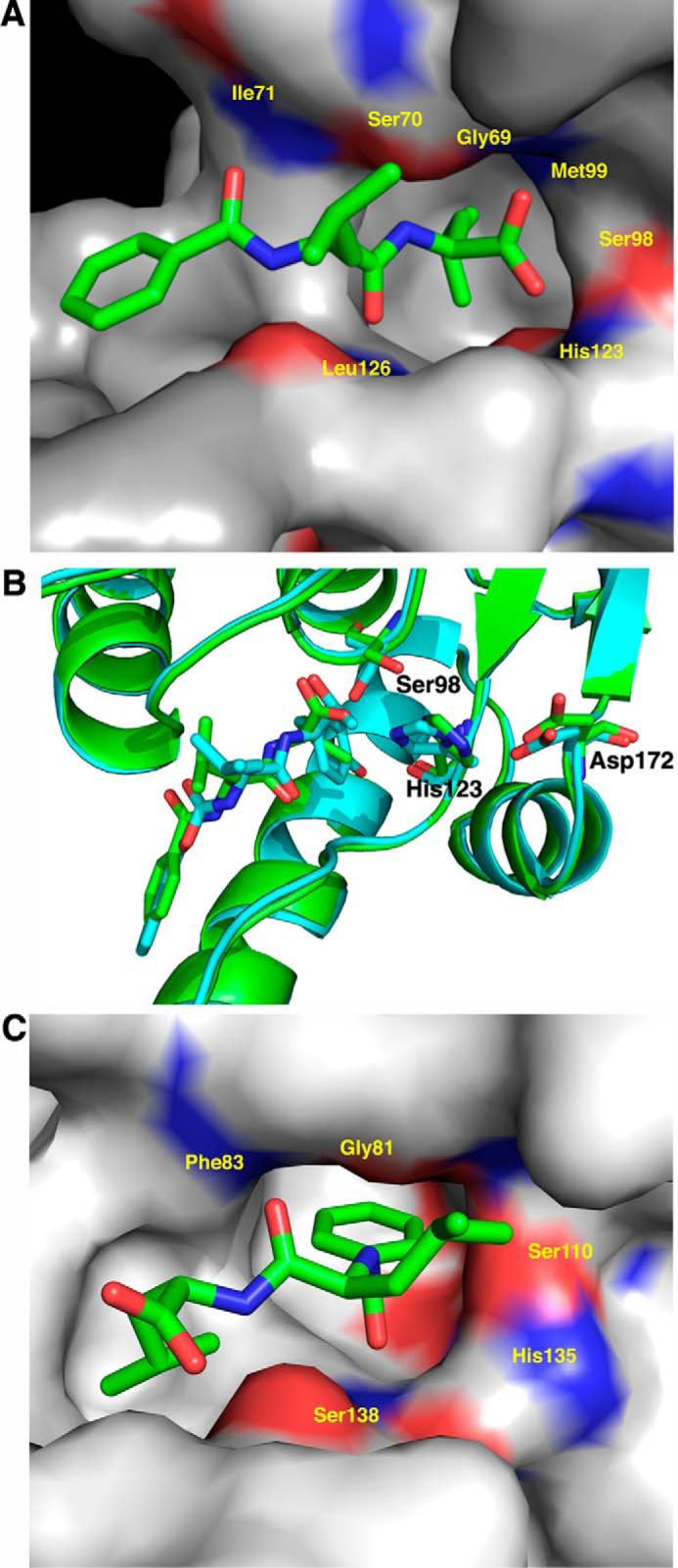
The activator is bound in opposite orientations in ClpP1 and ClpP2. Subunits are shown in surface rendering and colored red (negative) and blue (positive) to illustrate the electrostatic surfaces in the vicinity of the catalytic serine and histidine residues (shown as sticks). The S1 pocket, which accepts the P1 side chain of bound substrates, is identifiable as a deep gray cavern. A, Bz-LL in ClpP1. Bz-LL (cyan stick) is also held in place by hydrogen bonding with backbone atoms but lies in the opposite orientation with the side chain of leucine in the S1 pocket. One of the C-terminal oxygens of Bz-LL makes weak hydrogen-bonding interactions with the catalytic serine and histidine residues. The other C-terminal oxygen interacts with the oxyanion hole created by the backbone amides from Gly-69 and Met-99. B, Bz-LL in ClpP1 is aligned in the same manner as the covalently bound active site inhibitor, Z-LY-chloromethyl ketone. Overlap of E. coli ClpP with Z-LY-chloromethyl ketone covalently bound at the active site (PDB code 2FZS) (cyan) with Mtb ClpP1 with Bz-LL bound (green). Both ligands make identical backbone contacts with ClpP and engage the catalytic residues. C, Bz-LL in ClpP2. Bz-LL (cyan stick) is held in place by backbone hydrogen bonding and the bonding of the benzoyl moiety in the S1 pocket. The catalytic serine and histidine residues are rotated toward each other, but there are no interactions between Bz-LL and the catalytic residues.
Remarkably, Bz-LL is bound to ClpP2 subunits in the opposite orientation relative to its position in ClpP1 and is aligned parallel to β-strands 6 and 8 of ClpP2 (Fig. 7C). The benzoyl group is inserted into the S1 pocket, and the C terminus extends toward the lumen of the ClpP chamber. Because the latter mode of binding was unexpected and the resolution of the map was limited, we tested the model by assuming the “standard” mode of binding and refining both models independently. The unbiased electron density map, which was calculated after an additional round of refinement that did not include the ligand at all (Fig. 6D), indicated unambiguously that the non-standard mode of binding to all seven molecules was correct. This conclusion was made even stronger by the fact that each ligand was seen in 14 crystallographically independent molecules, with good agreement among the independent electron densities. This reverse orientation of a slightly different activator peptide was also seen in a recent crystal structure of ClpP1P2 (25), although in that structure the ligands were bound in the same orientation in both ClpP1 and ClpP2.
The structure of ClpP1P2 tetradecamers formed in the presence of Z-LL was virtually identical to that observed in the presence of Bz-LL, with the exception that no electron density attributable to Z-LL was visible. As crystals of ClpP1P2 were only obtained when the activator was included in the crystallization solution, we conclude that Z-LL promoted formation of the heterologous ClpP1P2 complex but that binding is highly dynamic in the crystal. A rapid off-rate for Z-LL would be consistent with the weaker binding and lower amplitude of activation observed with this activator.
The monoclinic crystals of both the Bz-LL and Z-LL complexes obtained in this study were different from the orthorhombic crystals of the ClpP1P2 complex grown in the presence of ADEP and Z-IL (PDB code 4U0G; Ref. 25). Nevertheless, the conformations of the tetradecamers were virtually identical. Superposition of one tetradecamer from each of these crystals with the program ALIGN (38) gave a root mean square deviation 0.84 Å for 2480 Cα. Therefore, it is not immediately clear why binding of Bz-LL leads to greater enzymatic activity. The activator was bound with the C terminus directed toward the catalytic residues in ClpP1. This orientation is similar to that expected for a degradation product immediately after cleavage of the acyl enzyme intermediate, which would allow the activator to be easily displaced by incoming tripeptide substrates in ClpP1, which mutagenesis has shown has almost all of the peptidase activity (13). If the Bz-LL were not easily displaced by incoming tripeptides in ClpP2, which lacks peptidase activity, the ClpP1P2 complex would persist and remain in the active conformation. Presumably, Bz-LL can be at least transiently displaced by protein substrates during ATP-driven degradation by ClpC1P1P2 or ClpXP1P2, because both ClpP1 and ClpP2 have been shown to participate in protein degradation. One other possible implication of these observations is that binding to ClpP2 is probably primarily responsible for inducing the conformational changes required to stabilize the active heterologous ClpP1P2 complex, because peptide substrates that should bind in the manner seen with Bz-LL bound to ClpP1 do not promote formation of the activate complex.
Activating Dipeptides at the Active Sites Influences the Substrate Entry Channel
The N-terminal β-hairpin in ClpP2 was observed in an extended conformation in our structure (Fig. 6, A and C) and in the published PDB 4U0G structure (25). The latter crystals (unlike the crystallographic studies) were grown in the presence of an ADEP antibiotic, which was bound to the ClpC1/X docking site on the apical surface of Mtb ClpP heptamers. Binding of different ADEPs has been shown to activate degradation of unfolded proteins by ClpP (28). ADEPs allosterically induce a change in binding interactions between the N-terminal β-hairpin and residues on the surface of ClpP, causing a reorientation of the β-hairpins and widening of the axial channel, which presumably allows unfolded polypeptides to pass into the degradation chamber (26, 39). In the crystal structures Bz-LL and Z-LL both promoted an open axial channel configuration in ClpP2. To determine if Bz-LL also promotes opening of the ClpP2 channel in the activated ClpP1P2 complex, we assayed the degradation of an intrinsically unfolded protein, FITC-casein, by ClpP1P2. Although ClpP1 and ClpP2 alone or a mixture of the two proteins in the absence of activator could not degrade FITC-casein (11), ClpP1P2 in the presence of Bz-LL actively degraded FITC-casein (Fig. 5B), although the rate of degradation was several times slower than ATP-dependent degradation observed with Bz-LL and ClpC1. Z-LL also stimulated degradation of FITC-casein by ClpP1P2 (11) but at ∼25% of the rate seen with Bz-LL (Fig. 5B). Because these activators associate with the active sites, they must alter the substrate entry channel and the interaction with the ATPase by an allosteric mechanism. Presumably allosteric effects of peptides within the active site also are important during degradation of proteins.
Discussion
Compartmentalized proteases like ClpP1P2, ClpP, and the proteasome face two potential barriers to efficient protein degradation. Access to the proteolytic chamber is normally restricted to the relatively narrow axial channels, which prevent the excessive degradation of cell proteins but in the presence of the complementary ATPase allow passage of one or at most two closely packed polypeptide chains. This gating mechanism ensures smooth entry of unfolded polypeptides through the channel, whereas the products of digestion (peptides of 3–20 residues) (40, 41) are released from the chamber through the same axial channels or through equatorial channels without disassembly of the tetradecamer. Studies over the past few years have revealed that ClpPs, like the proteasome (42), possess conformational flexibility that ensures both efficient substrate uptake and product release (43–46). The N-terminal loops that form the entrance to the axial channel are mobile and undergo transitions between closed and open states in response to binding of ClpX, ClpA/C, or ADEPs to surface docking sites (26, 47). In addition, the axial loops interact with the proximal surface of the bound ClpX heptamer (and presumably ClpA/C) to prevent motions that might interfere with polypeptide translocation (48). Conformational dynamics at the tetradecamer interface have also been revealed by crystallization of S. aureus and other ClpPs in two different states, one with all seven of the “handle domains” (a long αβ-extension consisting of strand-9 and helix-E) extended and making multiple contacts with the apposing heptamers (closed state) and one with the handle domains contracted and folded away from the apposing heptamer leaving gaps at the interface large enough for peptides to pass (open state) (46, 49). Heptamers in the contracted state are not active because the orientation of the catalytic residues depends on a hydrogen-bonding network affected by interactions with the handles from an adjacent subunit and from an apposing subunit across the tetradecamer (44).
The Role of the Dipeptide Activators
By themselves, Mtb ClpP1 and Mtb ClpP2 assemble into homomeric heptamers that then form tetradecamers, but neither has enzymatic activity by itself (11). Crystals of ClpP1 show that it adopts a collapsed form of the tetradecamer with the handle region contracted (50). The addition of peptide activators is needed to weaken the homomeric tetradecamer interactions and allow the ClpP1 and ClpP2 rings to assemble with each other and assume active conformations. As shown here, the nature of the activating peptide and its concentration are critical for these interactions and the amount of activity. Both ClpP1 and ClpP2 have the conserved catalytic triad found in other ClpPs, and both proteins are essential for ATP-dependent proteolysis with ClpC1 or ClpX (11, 13).
Prior studies of Mtb ClpP1P2 used ClpP1 and ClpP2 expressed separately in heterologous systems and combined under various conditions after purification. Unexpectedly, we found that the formation of functional tetradecamers in vitro required the binding of either a dipeptide or dipeptide derivative (11). Furthermore, the activity and integrity of the complex are lost very rapidly upon removal of the activator by dilution. The presence of an activator is also needed to assemble active complexes of ClpP1P2 with ClpC1 or ClpX (11, 13), and the enhanced ATP hydrolysis and ATP-dependent protein degradation were also lost immediately upon dilution into the buffer lacking the activator. This unusual requirement for activators appears to be unique to ClpPs from Actinobacteria, because in other species that contain two ClpP subunits, such as Listeria monocytogenes, formation of the active enzyme does not show a similar requirement (51).
The effects of the peptide activators on Mtb ClpP1P2 highlight the important role of allostery in the function of ClpP proteases. For various ClpP proteins, binding of the ClpX, ClpA/C ATPases, or ADEP antibiotics induces conformational changes that highly stimulate peptidase and protease activities. The conformational changes involve several regions in ClpP and include rearrangement of the axial loops that control access to the degradation chamber, collapse and extension of the α-β arms that form the interface between heptameric rings, and orientation of the active site residues into catalytically competent configurations. Several observations suggest that these three conformational changes are linked. For example, binding of ADEP induces a change in the N-terminal axial loops and also stabilizes the interactions between the heptameric rings, leading to stimulation of peptidase activity. Also, active site-directed reagents (e.g. fluorophosphate inhibitors) stabilize the tetradecamer and promote tighter binding between ClpP and ClpA or ClpX (35, 52). Direct evidence that changes in one structural region influence distant binding sites was obtained with S. aureus ClpP, in which the compressed conformation in the equatorial tetradecamer interface is accompanied by closing of the docking sites for ClpX and ClpC on the apical surface (46).
The ability of dipeptides bound at the active site to potentiate catalytic activity must depend on allosteric communication among the active sites. In our initial studies (11) it was unclear if the activators were bound to some of the active sites or to a separate regulatory site, such as the docking site where ADEP antibiotics bind. Ligands bound at the active sites would be expected to be inhibitory, and as expected, all the peptide activators we have identified (>15), including Bz-LL, competitively inhibit peptidase activity when present at concentrations >3-fold over those required for half-maximal activation. Because of the clustering of 14 active sites within the confined volume of the degradation chamber in ClpPs, the capacity to degrade peptide bonds far exceeds that required for maximum rates of proteolysis. With E. coli ClpP each subunit can hydrolyze in excess of 800 peptide bonds per minute (>14,000 per tetradecamer) (53). Such rates are much greater than that required for protein degradation, whose rate is limited by the ATP-dependent unfolding and translocation by ClpX or ClpA/C of polypeptides through narrow axial channels in ClpP. In fact, nearly half the active sites of E. coli ClpP (53) or the functionally similar HslUV (54) can be inactivated by a covalent inhibitor with almost no effect on the maximal rate of peptide or protein degradation. Thus, a high percentage of the active sites would need to be bound by a competitive inhibitor to result in a decreased rate of cleavage. At low concentrations of dipeptide activators, when only a few actives sites would be occupied, allosterically induced changes in the remaining active sites would be sufficient to enhance proteolytic activity. Activity of ClpP is also dependent on rapid transit of substrates through the axial channel, as has been observed when the channel is widened either by deletion of the axial loops (55) or by induced conformational changes upon binding of ADEP antibiotics (26). Allosteric communication between the active sites and the axial loops would also provide a mechanism of enhancing proteolytic activity.
Short peptide substrates bind weakly to the active sites (Km ∼ 5–10 mm), and thus ClpP cannot be fully saturated with peptide substrates under the conditions used here. Because most active sites are underutilized at typical peptide concentrations or during protein breakdown, the activator binding to unoccupied sites can promote the active conformation of ClpP while causing only little or no interference with substrate hydrolysis. Although the crystal structure indicated the presence of an activator molecule in each of the 14 active sites of ClpP1P2, the precise number of sites that must be occupied to activate ClpP1P2 is not known. The strong concentration dependence of activation (Hill coefficient of four-five) suggests that several peptides must be bound to obtain full activation. Even if six peptides must bind for maximal activation, peptidase activity of ClpP1P2 will only be partially inhibited by the activators. Our crystal structure shows that Bz-LL binds in different modes in the two subunits. Bz-LL binding to ClpP2 does not mimic substrate binding, but it induces a change in the access channel to enhance peptide entry. This unexpected orientation of the activator in ClpP2 seems to explain the finding that ClpP1 is responsible for the peptidase activity, whereas ClpP2 exhibits little or none (13, 25). We propose that Bz-LL binding to ClpP2 opens the axial channel of the complex so that substrates can be cleaved at the active site in ClpP1. Although Bz-LL was seen in all the active sites of ClpP1 under crystallization conditions, it causes little or no inhibition of peptide hydrolysis at comparable concentrations under assay conditions. Bz-LL was positioned in the ClpP1 active site in the manner of a peptide cleavage product, which would not be expected to bind tightly. Product inhibition is rare in serine proteases, and the cleaved peptides usually bind much more weakly than peptide substrates and are easily displaced by incoming substrate molecules.
Because both ClpP1 and ClpP2 are active in ATP-dependent protein degradation, the activators in at least some of the active sites in ClpP2 would need to be displaced by incoming proteins without disrupting the active complex. One possibility is that protein substrates are bound to fewer than seven active sites, leaving a sufficient number of sites with the activators still bound to preserve the ClpP1P2 complex. Another possibility is that, once the ClpP1P2 complex is assembled, it is stabilized by interaction with ClpX or ClpC1 (as is indicated by the enhanced binding seen in Fig. 2) or that the presence of partially digested proteins within the ClpP chamber stabilizes the complex in a manner similar to peptide activators, as was also suggested by Schmitz et al. (25).
Recently, Leodolter et al. (36) reported that after incubation for several hours at room temperature, ClpP1 and ClpP2 formed a tetradecameric complex in the absence of a dipeptide activator. That complex apparently lacked peptidase activity, because the N-terminal propeptides remained unprocessed after extended incubation unless a dipeptide activator was added. Leodolter et al. (36) also showed that protein substrates could stimulate processing of the propeptides when they were translocated into ClpP1P2 complex with either ClpX or ClpC1 in the presence of ATP. The protein substrates were subsequently degraded over a course of 5–7 h by the activated complex. We estimate that the proteolysis rate observed by Leodolter et al. (36) was <2% that shown in Fig. 4 for ClpXP1P2 or ClpC1P1P2 in the presence of the Bz-LL. Moreover, as noted above and previously (11), removal of the activator causes a dramatic loss of activity. Schmitz et al. (25) also reported very low proteolytic activity in the absence of dipeptide activators, but that activity was very much less than shown here in the presence of Bz-LL. Thus, whatever ClpP1P2 complex is formed in the absence of an activator, it appears to be functionally quite different from the Bz-LL activated form studied here. Our studies suggest that, because of its greater ability to hydrolyze peptides and unstructured proteins and to support ATP hydrolysis and ATP-dependent proteolysis by ClpC1 or ClpX, the active conformation induced by dipeptides is very likely to be the functional form in in vivo. It is possible that in Mtb cells endogenous peptides or proteins meet the requirement for peptide activators in order to assemble the ClpP1P2 complex. Our findings emphasize the importance of identifying the factors that activate Mtb ClpP1P2 complex and maintain its activity in vivo.
Author Contributions
A. L. G. and M. R. M. conceived and coordinated the study. A. L. G., O. K., T. A., M. R. M., M. R. M., and A. W. wrote the paper. O. K. and T. A. designed, performed, and analyzed the experiments shown in Figs. 1–5. M. L., P. D., M. R. M., and A. W. designed, performed, and analyzed the experiments shown in Figs. 6 and 7. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Diffraction data were collected on the SER-CAT 22ID beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract W-31-109-Eng-38.
This work was supported in part by National Institutes of Health Grant RO-051923-170 (NIGMS; to A. L. G.) and in part with federal funds from the NCI, National Institutes of Health, under Contract HHSN261200800001E and by the Intramural Research Program of the NCI, Center for Cancer Research. This work was also supported by the Harvard Catalyst program. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does the mention of trade names, commercial products, or organizations imply endorsement by the United States government. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5DZK and 5E0S) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Mtb
- M. tuberculosis
- eClpA
- E. coli ClpA
- Z-LL
- benzyloxycarbonyl-Leu-Leu
- Bz-LL
- benzoyl-Leu-Leu
- ADEP
- acyldepsipeptide
- Bz-Nva-Ile
- benzoyl-norvaline-isoleucine
- Z-IL
- benzyloxycarbonyl-Ile-Leu
- Ac-PKM-amc
- acetyl-PKM-7-amido-4-methylcoumarin
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
References
- 1. Porankiewicz J., Wang J., and Clarke A. K. (1999) New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol. 32, 449–458 [DOI] [PubMed] [Google Scholar]
- 2. Katayama-Fujimura Y., Gottesman S., and Maurizi M. R. (1987) A multiple-component, ATP-dependent protease from Escherichia coli. J. Biol. Chem. 262, 4477–4485 [PubMed] [Google Scholar]
- 3. Hwang B. J., Park W. J., Chung C. H., and Goldberg A. L. (1987) Escherichia coli contains a soluble ATP-dependent protease (Ti) distinct from protease La. Proc. Natl. Acad. Sci. U.S.A. 84, 5550–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang J., Hartling J. A., and Flanagan J. M. (1997) The structure of ClpP at 2.3 Å resolution suggests a model for ATP- dependent proteolysis. Cell 91, 447–456 [DOI] [PubMed] [Google Scholar]
- 5. Kessel M., Maurizi M. R., Kim B., Kocsis E., Trus B. L., Singh S. K., and Steven A. C. (1995) Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26 S proteasome. J. Mol. Biol. 250, 587–594 [DOI] [PubMed] [Google Scholar]
- 6. Baker T. A., and Sauer R. T. (2006) ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem. Sci. 31, 647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ortega J., Singh S. K., Ishikawa T., Maurizi M. R., and Steven A. C. (2000) Visualization of substrate binding and translocation by the ATP-dependent protease, ClpXP. Mol. Cell 6, 1515–1521 [DOI] [PubMed] [Google Scholar]
- 8. Aubin-Tam M. E., Olivares A. O., Sauer R. T., Baker T. A., and Lang M. J. (2011) Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maillard R. A., Chistol G., Sen M., Righini M., Tan J., Kaiser C. M., Hodges C., Martin A., and Bustamante C. (2011) ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schirmer E. C., Glover J. R., Singer M. A., and Lindquist S. (1996) HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem. Sci. 21, 289–296 [PubMed] [Google Scholar]
- 11. Akopian T., Kandror O., Raju R. M., Unnikrishnan M., Rubin E. J., and Goldberg A. L. (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 31, 1529–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benaroudj N., Raynal B., Miot M., and Ortiz-Lombardia M. (2011) Assembly and proteolytic processing of mycobacterial ClpP1 and ClpP2. BMC Biochem. 12, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Akopian T., Kandror O., Tsu C., Lai J. H., Wu W., Liu Y., Zhao P., Park A., Wolf L., Dick L. R., Rubin E. J., Bachovchin W., and Goldberg A. L. (2015) Cleavage specificity of Mycobacterium tuberculosis ClpP1P2 protease and identification of novel peptide substrates and boronate inhibitors with anti-bacterial activity. J. Biol. Chem. 290, 11008–11020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sauer R. T., Bolon D. N., Burton B. M., Burton R. E., Flynn J. M., Grant R. A., Hersch G. L., Joshi S. A., Kenniston J. A., Levchenko I., Neher S. B., Oakes E. S., Siddiqui S. M., Wah D. A., and Baker T. A. (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jenal U., and Fuchs T. (1998) An essential protease involved in bacterial cell-cycle control. EMBO J. 17, 5658–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryan K. R., Judd E. M., and Shapiro L. (2002) The CtrA response regulator essential for Caulobacter crescentus cell-cycle progression requires a bipartite degradation signal for temporally controlled proteolysis. J. Mol. Biol. 324, 443–455 [DOI] [PubMed] [Google Scholar]
- 17. Turgay K., Hahn J., Burghoorn J., and Dubnau D. (1998) Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 17, 6730–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frees D., Savijoki K., Varmanen P., and Ingmer H. (2007) Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63, 1285–1295 [DOI] [PubMed] [Google Scholar]
- 19. Kajfasz J. K., Abranches J., and Lemos J. A. (2011) Transcriptome analysis reveals that ClpXP proteolysis controls key virulence properties of Streptococcus mutans. Microbiology 157, 2880–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Butler S. M., Festa R. A., Pearce M. J., and Darwin K. H. (2006) Self-compartmentalized bacterial proteases and pathogenesis. Mol. Microbiol. 60, 553–562 [DOI] [PubMed] [Google Scholar]
- 21. Ollinger J., O'Malley T., Kesicki E. A., Odingo J., and Parish T. (2012) Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J. Bacteriol. 194, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raju R. M., Unnikrishnan M., Rubin D. H., Krishnamoorthy V., Kandror O., Akopian T. N., Goldberg A. L., and Rubin E. J. (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gavrish E., Sit C. S., Cao S., Kandror O., Spoering A., Peoples A., Ling L., Fetterman A., Hughes D., Bissell A., Torrey H., Akopian T., Mueller A., Epstein S., Goldberg A., Clardy J., and Lewis K. (2014) Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 21, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao W., Kim J. Y., Anderson J. R., Akopian T., Hong S., Jin Y. Y., Kandror O., Kim J. W., Lee I. A., Lee S. Y., McAlpine J. B., Mulugeta S., Sunoqrot S., Wang Y., Yang S. H., Yoon T. M., Goldberg A. L., Pauli G. F., Suh J. W., Franzblau S. G., and Cho S. (2015) The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 59, 880–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmitz K. R., Carney D. W., Sello J. K., and Sauer R. T. (2014) Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc. Natl. Acad. Sci. U.S.A. 111, E4587–E4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li D. H., Chung Y. S., Gloyd M., Joseph E., Ghirlando R., Wright G. D., Cheng Y. Q., Maurizi M. R., Guarné A., and Ortega J. (2010) Acyldepsipeptide antibiotics induce the formation of a structured axial channel in CIpP: a model for the CIpX/CIpA-bound state of CIpP. Chem. Biol. 17, 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brötz-Oesterhelt H., Beyer D., Kroll H. P., Endermann R., Ladel C., Schroeder W., Hinzen B., Raddatz S., Paulsen H., Henninger K., Bandow J. E., Sahl H. G., and Labischinski H. (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 28. Kirstein J., Hoffmann A., Lilie H., Schmidt R., Rübsamen-Waigmann H., Brötz-Oesterhelt H., Mogk A., and Turgay K. (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 1, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee B. G., Park E. Y., Lee K. E., Jeon H., Sung K. H., Paulsen H., Rübsamen-Schaeff H., Brötz-Oesterhelt H., and Song H. K. (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 17, 471–478 [DOI] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 34. Hwang B. J., Woo K. M., Goldberg A. L., and Chung C. H. (1988) Protease Ti, a new ATP-dependent protease in Escherichia coli, contains protein-activated ATPase and proteolytic functions in distinct subunits. J. Biol. Chem. 263, 8727–8734 [PubMed] [Google Scholar]
- 35. Joshi S. A., Hersch G. L., Baker T. A., and Sauer R. T. (2004) Communication between ClpX and ClpP during substrate processing and degradation. Nat. Struct. Mol. Biol. 11, 404–411 [DOI] [PubMed] [Google Scholar]
- 36. Leodolter J., Warweg J., and Weber-Ban E. (2015) The Mycobacterium tuberculosis ClpP1P2 Protease Interacts Asymmetrically with Its ATPase Partners ClpX and ClpC1. PLoS ONE 10, e0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Szyk A., and Maurizi M. R. (2006) Crystal structure at 1.9A of E. coli ClpP with a peptide covalently bound at the active site. J. Struct. Biol. 156, 165–174 [DOI] [PubMed] [Google Scholar]
- 38. Cohen G. E. (1997) ALIGN: a program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Crystallogr. 30, 1160–1161 [Google Scholar]
- 39. Alexopoulos J., Ahsan B., Homchaudhuri L., Husain N., Cheng Y. Q., and Ortega J. (2013) Structural determinants stabilizing the axial channel of ClpP for substrate translocation. Mol. Microbiol. 90, 167–180 [DOI] [PubMed] [Google Scholar]
- 40. Kisselev A. F., Akopian T. N., Woo K. M., and Goldberg A. L. (1999) The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes: implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 274, 3363–3371 [DOI] [PubMed] [Google Scholar]
- 41. Thompson M. W., and Maurizi M. R. (1994) Activity and specificity of Escherichia coli ClpAP protease in cleaving model peptide substrates. J. Biol. Chem. 269, 18201–18208 [PubMed] [Google Scholar]
- 42. Smith D. M., Kafri G., Cheng Y., Ng D., Walz T., and Goldberg A. L. (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell 20, 687–698 [DOI] [PubMed] [Google Scholar]
- 43. Geiger S. R., Böttcher T., Sieber S. A., and Cramer P. (2011) A conformational switch underlies ClpP protease function. Angew. Chem. Int. Ed. Engl. 50, 5749–5752 [DOI] [PubMed] [Google Scholar]
- 44. Gersch M., List A., Groll M., and Sieber S. A. (2012) Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease P (ClpP) protein. J. Biol. Chem. 287, 9484–9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee B. G., Kim M. K., and Song H. K. (2011) Structural insights into the conformational diversity of ClpP from Bacillus subtilis. Mol. Cells 32, 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J., Ye F., Lan L., Jiang H., Luo C., and Yang C. G. (2011) Structural switching of Staphylococcus aureus Clp protease: a key to understanding protease dynamics. J. Biol. Chem. 286, 37590–37601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Effantin G., Maurizi M. R., and Steven A. C. (2010) Binding of the ClpA unfoldase opens the axial gate of ClpP peptidase. J. Biol. Chem. 285, 14834–14840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin A., Baker T. A., and Sauer R. T. (2007) Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol. Cell 27, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye F., Zhang J., Liu H., Hilgenfeld R., Zhang R., Kong X., Li L., Lu J., Zhang X., Li D., Jiang H., Yang C. G., and Luo C. (2013) Helix unfolding/refolding characterizes the functional dynamics of Staphylococcus aureus Clp protease. J. Biol. Chem. 288, 17643–17653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ingvarsson H., Maté M. J., Högbom M., Portnoï D., Benaroudj N., Alzari P. M., Ortiz-Lombardía M., and Unge T. (2007) Insights into the inter-ring plasticity of caseinolytic proteases from the x-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr. D Biol. Crystallogr. 63, 249–259 [DOI] [PubMed] [Google Scholar]
- 51. Dahmen M., Vielberg M. T., Groll M., and Sieber S. A. (2015) Structure and mechanism of the caseinolytic protease ClpP1/2 heterocomplex from Listeria monocytogenes. Angew. Chem. Int. Ed. Engl. 54, 3598–3602 [DOI] [PubMed] [Google Scholar]
- 52. Singh S. K., Guo F., and Maurizi M. R. (1999) ClpA and ClpP remain associated during multiple rounds of ATP-dependent protein degradation by ClpAP protease. Biochemistry 38, 14906–14915 [DOI] [PubMed] [Google Scholar]
- 53. Thompson M. W., Singh S. K., and Maurizi M. R. (1994) Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli: requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J. Biol. Chem. 269, 18209–18215 [PubMed] [Google Scholar]
- 54. Lee J. W., Park E., Jeong M. S., Jeon Y. J., Eom S. H., Seol J. H., and Chung C. H. (2009) HslVU ATP-dependent protease utilizes maximally six among twelve threonine active sites during proteolysis. J. Biol. Chem. 284, 33475–33484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bewley M. C., Graziano V., Griffin K., and Flanagan J. M. (2009) Turned on for degradation: ATPase-independent degradation by ClpP. J. Struct. Biol. 165, 118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]