

Abstract
The α-(1→2) branching sucrase ΔN123-GBD-CD2 is a transglucosylase belonging to glycoside hydrolase family 70 (GH70) that catalyzes the transfer of d-glucosyl units from sucroseto dextrans or gluco-oligosaccharides via the formation of α-(1→2) glucosidic linkages. The first structures of ΔN123-GBD-CD2 in complex with d-glucose, isomaltosyl, or isomaltotriosyl residues were solved. The glucose complex revealed three glucose-binding sites in the catalytic gorge and six additional binding sites at the surface of domains B, IV, and V. Soaking with isomaltotriose or gluco-oligosaccharides led to structures in which isomaltosyl or isomaltotriosyl residues were found in glucan binding pockets located in domain V. One aromatic residue is systematically identified at the bottom of these pockets in stacking interaction with one glucosyl moiety. The carbohydrate is also maintained by a network of hydrogen bonds and van der Waals interactions. The sequence of these binding pockets is conserved and repeatedly present in domain V of several GH70 glucansucrases known to bind α-glucans. These findings provide the first structural evidence of the molecular interaction occurring between isomalto-oligosaccharides and domain V of the GH70 enzymes.
Keywords: carbohydrate-binding protein; crystal structure; enzyme; glycoside hydrolase; oligosaccharide; alpha-1,2 branching sucrase; α-glucan; family GH70; glucan-binding domain; glucansucrase
Introduction
The glucosyltransferases of glycoside hydrolase family 70 (GH70),4 namely the glucansucrases, the 4,6-α-glucanotransferases, and the branching sucrases, are α-retaining transglucosylases produced by various lactic acid bacteria from Leuconostoc, Streptococcus, Weissella, and Lactobacillus genera (1–3). Glucansucrases from Leuconostoc spp. have been mainly studied for their industrial applications in the production of dextrans or prebiotic gluco-oligosaccharides (4–7), whereas investigations on streptococcal glucansucrases were mostly motivated by their involvement in the accumulation of streptococci on tooth enamel and subsequent human dental caries formation (8). Glucansucrases catalyze the transfer of d-glucopyranosyl moieties from sucrose to acceptor molecules through the formation of a β-d-glucosyl-enzyme intermediate. They synthesize α-glucans, which vary in terms of size, osidic linkages as well as degree and arrangement of branching. Devoid of polymerase activity, the α-(1→2) branching sucrases are specialized in dextran branching through the formation of α-(1→2) glucosidic linkages from sucrose to dextran acceptor (Fig. 1) (9, 10). In the absence of acceptor, these enzymes essentially catalyze sucrose hydrolysis.
FIGURE 1.

The three types of reactions catalyzed by the α-(1→2) branching sucrase ΔN123-GBD-CD2.
Glucansucrases are also known to bind dextrans via a carbohydrate-binding domain remote from the active site (2, 11). This domain is referred to as the glucan-binding domain (GBD) and is not classified in the CAZy database. Carbohydrate-protein interactions often play critical roles in the catalytic activities of carbohydrate active enzymes involved in polysaccharide synthesis, degradation, or decoration. Carbohydrate-binding sites may be intrinsic components of catalytic sites, but may also exert essential functions when located in non-catalytic modules (12). Evidence of the presence of non-catalytic domains showing α-glucan binding ability in GH70 enzymes were first provided by analyzing dextran interactions with non-catalytic polypeptides isolated from tryptic digestion of various streptococcal glucansucrases (13). Based on sequence and biochemical analyses, the streptococcal glucan binding regions were shown to consist of a series of repeats that were classified into four different groups (A, B, C, and D) (14–19). These repeats were proposed to have evolved from a common motif, the so called YG repeat, characterized by conserved aromatic amino acids and glycine ((N/X)X(D/X)X(G/X)φYφφXXXXGXXΨx(G/N/X)X(Ψ/X)Ψ(Ψ/X), where X represents a non-conserved residue, Ψ a hydrophobic residue, and φ an aromatic residue) (for a full description, see Ref. 20). They were also identified at the N-terminal or C-terminal domains of glucansucrases from Leuconostoc spp. and shown to share homology with the cell wall binding repeats of choline-binding proteins or toxins (Pfam CW_binding 1 (PF01473)) (21–23). In particular, the five repeats found at the C-terminal extremity of the GBD of DsrP glucansucrase are capable of binding the cell surface of Leuconostoc cells (24). Mutagenesis of conserved aromatic or basic residues of the repeats found in Streptococcus downei GtfI was performed and led to a reduction of the glucan binding capability (25). The elimination of some repeats by truncation was also shown to influence transglucosylation, modify glucan binding properties, and affect the polymer size (26–29). In particular, the GBD was suggested to provide a glucan anchoring platform, which may affect the single-chain elongation in a semi-processive manner (29). A few studies focused on isolated GBDs disconnected from their catalytic domain, and further demonstrated that truncated forms can autonomously fold and bind α-glucans with very low Kd values estimated in the nanomolar range (30–34).
The crystal structures of several GH70 enzymes truncated at their N-terminal ends shed light on the GBD-fold, although no structures with entire GBDs have been solved yet (9, 35–37). These enzymes follow a U-path course and are organized in five domains, A, B, C, IV, and V. Domains A, B, and C are related to their counterparts found in GH13 α-amylases. In the GH70 family, domains A, B, IV, and V are formed by non-contiguous sequences distributed at the N terminus or C terminus. Only domain C at the basis of the U-fold is built up by a contiguous sequence. Domain V, representing the truncated GBD, is the most distant from the active site. This domain adopts variable positions in GTF180-ΔN glucansucrase due to motions around a hinge located between domains IV and V (38). The branching sucrase ΔN123-GBD-CD2 derived from the bi-functional glucansucrase DSR-E from Leuconostoc citreum NRRL B-1299 (formerly Leuconostoc mesenteroides NRRL B-1299) has its domain V interacting with the domain IV and shows a more compact structure than GTF180-ΔN (9). Of note, the repeats that have been proposed to interact with α-glucans are found in domain V of the GH70 enzymes, either at the N or C terminus depending on the enzyme. They are present in several copies that contain three consecutive β2/β3 elements of about 20 residues that form either three-stranded antiparallel β-sheets or β-hairpins that are rotated ∼60° relative to one to another in a fold related to the β-solenoid fold, first described for C-LytA (2, 21, 35). Unfortunately, no structures displaying carbohydrates bound to domain V have yet been solved and structural data are still lacking to support the role of this domain in glucan binding and the relevance of this interaction with regard to dextran elongation or branching.
The crystal structures of ΔN123-GBD-CD2, a branching sucrase that catalyzes α-(1→2) branches formation onto dextran, were solved in complex with d-glucose, isomaltosyl, and isomaltotriosyl groups at 2.3-, 2.2-, and 1.85-Å resolution, respectively. These structures all revealed the presence of carbohydrates bound in several domains of ΔN123-GBD-CD2. In particular, several carbohydrate-binding sites never before revealed were found near subsite +1 of the catalytic site. Complex analyses also allowed for the first time mapping of the amino acid network constituting the glucan binding pockets of domain V of ΔN123-GBD-CD2 and a description of the carbohydrate-protein interactions. Structural alignment with domain V of other glucansucrases capable of binding α-glucans further allowed us to identify putative glucan binding pockets in domain V of these enzymes. The role of these pockets relative to the enzyme function is discussed.
Experimental Procedures
Cloning, Expression, Purification, and Crystallization of ΔN123-GBD-CD2
Gene cloning, expression, and purification of the α-(1→2) branching sucrase ΔN123-GBD-CD2 was previously described (9). Crystals of ΔN123-GBD-CD2 were obtained by mixing 1 μl of protein in 20 mm sodium acetate, 150 mm NaCl, 2.5% (v/v) glycerol, and 1 mm CaCl2, pH 5.75, with 1 μl of reservoir solution containing 15% PEG 3,350 and 0.1 m NH4NO3. For soaking experiments, all ligands were solubilized using the reservoir solution. Four types of carbohydrate ligands were tested: d-glucose, isomaltotriose, a mixture of α-gluco-oligosaccharides mainly composed of isomaltotriosyl-maltose and isomaltosyl-maltose ([6)-α-d-Glcp-(1→]n-α-d-Glcp-(1→4)-d-Glcp with an average n of 3), and dextran T1 (molecular mass 1 kDa, Pharmacosmos, Holbæk, Denmark) consisting in a mixture of isomalto-oligosaccharides of average degree of polymerization of 6. The glucooligosaccharides produced and purified as previously described were dissolved at 40 mm (39). Soaking of crystals with isomaltotriose (Sigma-Aldrich Chemie S.a.r.l., Saint Quentin-Fallavier, France) was performed at 50 mm ligand. The concentration of PEG 3,350 was increased to 28.5% (w/v) for cryoprotection. d-Glucose (Sigma) was dissolved at 100 mm. Soaking solutions of dextran T1 were prepared at concentrations varying from 50 mm to saturation. No cryo-protectant was necessary for those two last ligands. Crystals were then cryo-cooled in a flux of nitrogen gas at 100 K.
Data Collection, Phasing, and Refinement
Diffraction data were collected at the ESRF (Grenoble, France). Intensities were indexed and integrated using iMOSFLM or XDS and scaled using SCALA or XSCALE (40–42). Crystals belong to space group P212121 with one molecule per asymmetric unit, which corresponds to a solvent content of 52.5% and Matthews coefficient of 2.59 Å3 Da−1. Structures were solved using molecular replacement with Phaser (43) and the atomic coordinates of the apo-form of ΔN123-GBD-CD2 (PDB entry 3TTQ (9)) as a search model. Models were manually constructed in σA weighted 2Fo − Fc electron density maps using Coot and refined using Refmac5 (44, 45). Carbohydrate fitting in electron density maps was also guided by simulated annealing (Fo − Fc) omit maps were calculated with PHENIX (46). Data collection and refinement statistics are shown in Table 1.
TABLE 1.
Data collection and refinement statistics for structures of ΔN123-GBD-CD2 in complex with different sugars
| Complex A | Complex B | Complex C | |
|---|---|---|---|
| Soaking | d-Glucose | Isomaltotriose | Gluco-oligosaccharides |
| Observed ligand | d-Glucose | Isomaltosyl residue | Isomaltotriosyl residue |
| Data collection | |||
| Beamline | ID29 | ID23–1 | ID23–1 |
| Wavelength (Å) | 0.9763 | 0.8726 | 0.9763 |
| Unit cell parameters (Å) | a = 67.77 b = 98.98 | a = 68.29 b = 100.10 | a = 68.31 b = 100.14 |
| c = 181.91 | c = 184.43 | c = 186.24 | |
| Space group | P212121 | P212121 | P212121 |
| Molecules per AUa | 1 | 1 | 1 |
| Resolution (Å) | 47.8–2.3 (2.36–2.30) | 44.0–2.2 (2.30–2.18) | 27.2–1.9 (1.95–1.85) |
| No. of unique reflections | 54,661 (3,770) | 66,920 (9,657) | 109,405 (15,715) |
| Completeness (%) | 99.5 (94.2) | 100.0 (100.0) | 99.7 (99.1) |
| Multiplicity | 6.5 (4.6) | 5.4 (5.3) | 3.5 (3.2) |
| Rmeasb/Rmergeb | 0.108 (0.735)b | 0.111 (0.439)c | 0.090 (0.497)c |
| Mean I/σ(I) | 13.7 (2.1)b | 11.0 (3.6)c | 8.7 (2.0)c |
| Overall B factor–Wilson plot (Å2) | 34.2 | 23.8 | 24.7 |
| Refinement | |||
| No. of reflections | 51,888 (2,772)d | 63,475 (3,388)d | 103,857 (5,458)d |
| Rwork (%) | 16.9 | 16.8 | 17.0 |
| Rfree (%) | 22.6 | 22.0 | 21.0 |
| Mean B values (Å2) | 40.0 | 28.0 | 30.0 |
| No of non-H atoms | |||
| Protein | 8,113 | 8,196 | 8,189 |
| Ligands | 127 | 42 | 64 |
| Water molecules | 395 | 715 | 922 |
| R.m.s. deviation from ideal | |||
| Bond lengths (Å) | 0.013 | 0.015 | 0.019 |
| Bond angles (°) | 1.531 | 1.591 | 1.840 |
| Ramachandran plot (%) | |||
| Favored | 95.0 | 96.6 | 96.9 |
| Allowed | 99.8 | 99.9 | 100.0 |
| Outliers | 0.2 | 0.1 | 0.0 |
Structural Analysis
Electrostatic potential of the ΔN123-GBD-CD2 surface, without any ligand, was calculated via the PDB2PQR server using the program APBS, specifying the pH at 5.75, which is the optimum pH for activity (47). From complex B, the groove search from subsite −1 was done using CAVER 3.01 as a PyMOL plugin (48).
Molecular Dynamics
The starting point for the molecular dynamics (MD) simulation was the crystal structure of complex A, composed of the protein and 10 d-glucose molecules. The calculations were performed with the Amber 14 software (49–51) using the FF03 force field for the protein, the GAFF force field for the ligands, and the TIP3P water model for the solvent (52). The complex was placed at the center of a periodic box. To hydrate the system, the volume occupied by the complex was extended by 10 Å on each side, and 40,251 water molecules were added to the box. To neutralize the charge of the system, 37 Na+ ions were added by random replacement of the equivalent number of solvent molecules. The final system had the dimensions of 101 × 141 × 113 Å and contained 136,985 atoms. The equilibration of the entire system was achieved in several steps. Initially, the energy of the system was minimized by 1,000 cycles of the steepest descent algorithm, with the solute held fixed, by constraining its Cartesian coordinates using a harmonic potential with the force constant k equal to 100 kcal/mol/Å2. In the second step, the energy was minimized by 500 cycles of steepest descent and 1,500 cycles of the conjugate gradient algorithm, with weakly restrained solute (k = 10 kcal/mol/Å2). Next, a short 20-ps run of NVT-MD (constant volume and temperature) was performed on weakly restrained solute with temperature varying linearly from 0 to 300 K. The temperature control was achieved using the Langevin dynamics with the collision frequency parameter γ set to 1.0 ps−1. The integration step used in this run was 1 fs. Throughout the calculations, a cut-off of 12 Å was used for electrostatic interactions, the particle mesh Ewald method (53) was used to treat long-range electrostatic interactions and the SHAKE algorithm (54) was used to constrain hydrogen bond lengths. The MD simulation continued for 100 ps with constant pressure of 1 bar (NPT-MD) at 300 K with no restraints, with the integration step of 2 fs. Finally, a 50-ns run with constant pressure of 1 bar was launched, with atomic coordinates saved every 10 ps. The Langevin dynamics was used to control the temperature, with γ = 1.0 ps−1, whereas the pressure was controlled by the Berendsen barostat with the pressure relaxation time τp = 2 ps. Calculations were performed using a GeForce GTX TITAN Black GPU card, which worked at the speed of about 8.22 ns/day. To obtain a detailed view of the studied system, nine simulations of this type have been performed, for the total time length of 450 ns. The resulting MD trajectories were analyzed with the AmberTools 14 programs as well as with in-house software. The last 20 ns of each equilibrated trajectory were used to calculate intermolecular interaction energy for all protein-ligand pairs with the MM-GBSA approach (55).
Identification of Glucan Binding Pockets by Multiple Sequence Alignments
Sequence alignment of the three repeats forming the sugar-binding pockets, V-J, V-K, and V-L in domain V of ΔN123-GBD-CD2, was first performed using Clustal Omega (56). This alignment was inspected and corrected using the structural superimposition of each of the three binding pockets. Then, the primary structure of the full-length GBD of DSR-E (57) was analyzed using the online repeat detection tool TRUST (58), and this allowed detecting 12 sequence repeats in this domain. Similarly, repeats corresponding to glucan binding pockets were identified by sequence analyses of domain V of several glucansucrases that were experimentally shown to bind α-glucans (namely GBD4RS of GTF-I from Streptococcus sobrinus, GBD1A of GTF-I from S. downei, and GBD5 of DSR-S from L. mesenteroides B-512F) (25, 31, 32). These repeats were then aligned using the web service Expresso (from T-coffee suite), providing structure-based multiple sequence alignment. In our case, the structural information of the binding pockets V-J, V-K, and V-L was used as templates (59, 60). From this alignment, we deduced the LOGO sequence of one repeat using WebLogo 3 (61).
Affinity Gel Electrophoresis
For affinity gel electrophoresis, ΔN123-GBD-CD2 fused with a His6 tag at the N terminus and a Strep-tag® at the C terminus was expressed in a soluble form and purified as previously described (62). The dissociation constant of the purified α-(1→2) branching sucrase ΔN123-GBD-CD2 with dextrans was determined by affinity gel electrophoresis in 6.5% polyacrylamide native gels containing increasing amounts of dextran (63, 64). Two types of dextran were tested, a high molar mass dextran with an estimated molar mass of 22.5 MDa and a medium molar mass dextran with an estimated molar mass of 68.4 kDa (Sigma). Their concentrations ranged from 0 to 0.9% (w/v). Bovine serum albumin (BSA, Sigma) was used as a standard. Electrophoresis migration was performed for 2 h at 20 °C, pH 8.3, in a mini PROTEAN system (Bio-Rad) at constant intensity (10 mA per gel). The gels were stained with Coomassie Blue.
Determination of Binding Constants
Migration distances of BSA (dBSA) or ΔN123-GBD-CD2 (dENZ) were measured relative to the top and bottom of gels using the software associated with the GelDocTM EZ System (Bio-Rad). Relative mobilities (R) were calculated as the migration distance of ΔN123-GBD-CD2 divided by the migration distance of BSA (R = dENZ/dBSA). In the absence of dextran, the relative mobility is defined as RMAX. This relative mobility is normalized to 1.0 (R′MAX). The normalized relative mobilities are R′ (R′ = R/RMAX). In the presence of a large excess of dextran, the relative normalized mobility is defined as R′MIN. Assuming that the total concentration of dextran in gels greatly exceeds the total polypeptide concentration, the affinity constant (Ka) of ΔN123-GBD-CD2 for dextrans was calculated by plotting the normalized relative mobility, expressed as 1/(R′MAX − R′) of ΔN123-GBD-CD2 versus the molar polysaccharide concentration expressed as 1/[Dex]. As described by Abbot and Boraston (65), the relationship between normalized relative mobilities and polysaccharide concentrations is,
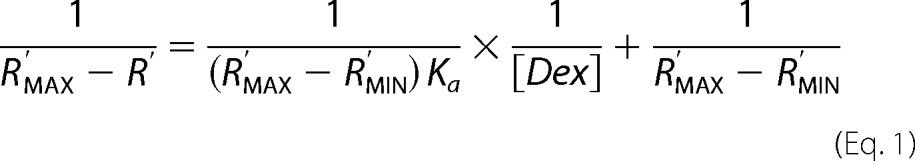 |
where R′MIN, R′MAX, and Ka are constants,
Accession Numbers
The atomic coordinates were deposited in the Protein Data Bank with accession codes 4TVD, 4TTU and 4TVC for structures in complex with d-glucose, isomaltosyl, or isomaltotriosyl residues, respectively.
Results
Evidence of the Dextran Binding Ability of ΔN123-GBD-CD2
The α-(1→2) branching sucrase ΔN123-GBD-CD2 is a truncated enzyme that was designed from DSR-E, a GH70 glucansucrase with two catalytic domains (CD1 and CD2) separated by a putative GBD rich in repeats proposed to be involved in the glucan binding ability of several enzymes from the GH70 family (27, 57). The ΔN123-GBD-CD2 enzyme catalyzes transglucosylation reaction from sucrose to dextran or isomalto-oligosaccharide acceptors, through the formation of α-(1→2) branches. In the absence of an acceptor, the enzyme mainly catalyzes sucrose hydrolysis (9). As dextran is the preferred acceptor of ΔN123-GBD-CD2, we evaluated its dissociation constant by affinity gel electrophoresis. High molar mass and low molar mass dextrans were immobilized in native polyacrylamide gels at various concentrations. For both cases, the presence of dextran decreased the enzyme electrophoretic mobility, providing evidence of carbohydrate-protein interactions (Fig. 2). The dissociation constants were calculated from the double-reciprocal plots of the inverse of electrophoretic mobility versus the inverse of dextran concentration and estimated at 0.3 and 104 μm polysaccharide for 22.5 MDa and 68.4 kDa dextran, respectively. To investigate the molecular determinants at the origin of this binding, crystals of the branching enzyme were soaked with various ligand including d-glucose, isomaltotriose, gluco-oligosaccharides (mainly isomaltotriosyl- and isomaltosyl-maltose), and a mixture of isomalto-oligosaccharides of average degree of polymerization 6–7 (1 kDa dextran). Despite reasonable resolution, i.e. from 3.35 to 2.60 Å, the three-dimensional structures obtained from crystals soaked with 1-kDa dextran did not reveal any clear electron density attributable to a ligand.
FIGURE 2.

Affinity gel electrophoresis of ΔN123-GBD-CD2 in gels containing variable amounts of dextrans. Left panel, affinity gels for which the content in dextran is expressed in % (w/v). Lanes 1 correspond to BSA, lanes 2 correspond to ΔN123-GBD-CD2. Right panel, double-reciprocal plots of the inverse of the relative electrophoretic mobility versus the inverse of dextran concentrations. The relative mobility was determined from the migration distances observed on the gels. * estimated average molecular weight.
Structure of the Complex of ΔN123-GBD-CD2 with d-Glucose
The crystal structure of ΔN123-GBD-CD2 in complex with d-glucose has been solved at 2.3-Å resolution and superimposed with the apo structure (PDB entry 3TTQ) with a r.m.s. deviation on Cα atoms of 0.6 Å. Nine d-glucose molecules are observed at the enzyme surface (Figs. 3A and 4). Four of them are found in domain A in sites A-1, A2, A3, and A4. Additionally, two glucose molecules were bound in domain IV (sites IV1 and IV2). Two others are at the interface of domains IV and V (site IV-V) or domains B, IV, and V (site B-IV-V) and one glucose unit is bound to the domain V (site V-L). Finally, one additional glucose molecule is located at the interface of two symmetry-related protein molecules. This binding site will not be described further due to its biological inconsistency.
FIGURE 3.

Structure of ΔN123-GBD-CD2 in complex with carbohydrates. A, structure of ΔN123-GBD-CD2 in complex with d-glucose (complex A). Nine glucose-binding sites have been identified at the surface of the enzyme. Four sites in domain A (A-1, A2, A3, and A4), two sites in domain IV (IV1 and IV2), one site in domain V (named binding pocket V-L), and two sites spanning over domains B, IV, and V (site B-IV-V and site IV-V). Domains A, B, C, IV, and V are colored in blue, green, magenta, yellow, and fire-brick, respectively. Glucose molecules are shown as spheres (with carbon and oxygen atoms in gray and red, respectively). B, grooves at the surface of the enzyme in complex B. Grooves were searched using CAVER starting in the active site from subsite −1. Glucose molecules from the complex A have been superimposed. C, electrostatic potential surface color-coded from red (−5 kBT/e) to blue (+5 kBT/e) for complex B computed at pH 5.75; white is neutral. The nine glucose molecules were superimposed and figured as yellow and red spheres.
FIGURE 4.

Glucose-binding sites found over the ΔN123-GBD-CD2 enzyme. The simulated annealing omit (Fo − Fc) electron density maps (in green) around the nine glucose molecules bound to ΔN123-GBD-CD2 were contoured at 2σ. ΔN123-GBD-CD2 residues in the neighborhood of each glucose molecule are shown as lines and colored by atom types. Stereo view of subsites A-1, A2, A3, A4, B-IV-V, IV1, IV2, IV-V, and VL are shown on panels A–I, respectively.
Among the nine glucose binding sites, site A-1 corresponds to subsite −1 of GH70 transglucosylases, which was shown to accommodate the glucopyranosyl ring of sucrose (66). The β-d-glucopyranose in A-1 adopts a 4C1 conformation, similar to that observed for the d-glucopyranosyl unit of sucrose described for the GTF180-ΔN·sucrose complex (35). The oxygen atom O1 of glucose is hydrogen bound to the side chain atoms of the catalytic residues Glu-2248 and Asp-2210 at a distance of 2.35 and 2.67 Å, respectively. The atom O2 can establish interactions with the side chains of Arg-2208, Glu-2248, His-2321, and Asp-2322. The O3 and O4 atoms interact with His-2321 and Asp-2643, respectively, and with the same water molecule. Finally, the O6 atom makes hydrogen bonds with the side chains of Asp-2210 and Gln-2694 and with a water molecule. Interestingly, two other glucose molecules were clearly visible in the sites A2 and A3, also close to the catalytic site (Figs. 3A and 5). In site A2, the β-d-glucopyranose molecule adopts a 4C1 conformation and is firmly maintained by hydrogen bonds between the O2 atom and Glu-2265, the O3 atom and Glu-2270 and the main chain carbonyl group of Ser-2266, the O4 atom and Asn-2267, and the O6 atom and Asp-2294 (Fig. 5). In site A3, the β-d-glucopyranose is likely in 4C1 conformation and in a stacking interaction with Tyr-2764. The O1 atom interacts with Tyr-2764, O3 with Lys-2765, and O4 with Asn-2161 and Asn-2217. The O2 atom is less well defined in simulated annealing omit map and may interact with the oxygen atom of the carbonyl function of Lys-2765. Two surrounding water molecules interact with the O2, O3, and O4 (Fig. 5).
FIGURE 5.

Glucose-binding sites A2 (left panel) and A3 (right panel). The simulated annealing omit (Fo − Fc) electron density maps around glucose were contoured at 3σ or 2.3σ for A2 and A3, respectively. Interacting water molecules are depicted as red spheres. Residues of domains A or B are represented as blue or green sticks, respectively. For residues that are involved in sugar binding through their backbone atoms, side chains are omitted. In the left panel, residues forming the catalytic triad (Asp-2210, Glu-2248, and Asp-2322) are shown as turquoise sticks.
Molecular dynamics simulation of ΔN123-GBD-CD2 in complex with d-glucose revealed that the glucose molecules at subsites A-1 and A2 have respective average r.m.s. deviation values of 2.9 ± 0.1 and 3.4 ± 0.2 Å, respectively, indicating that they are stable in their respective binding sites. They form on the average 3 intermolecular contacts with the protein residues. MM-GBSA calculations were performed to estimate the interaction energy between ΔN123-GBD-CD2 and d-glucose molecules. Interaction energy values for glucose molecules at subsites A-1 and A2 were comparable (about −20 kcal/mol). Altogether, these observations reveal that the binding sites A-1 and A2 of the catalytic gorge have strong affinities for glucose molecules. The A3 site was found to bind the ligand for a limited time (about 90 ns), after which the ligand left for the solvent.
The d-glucose molecule at site A4 is bound in a pocket at the rear of the (β/α)8 barrel of domain A (Fig. 3A) and establishes hydrogen bonds with Asn-2241, Asn-2588, Asn-2683, and Gln-2685. The oxygen atoms of the hydroxyl groups O6 and O1 are oriented toward the enzyme. Molecular dynamics simulations suggest that the molecule is loosely bound to the protein. In domain IV, two glucose molecules that are located 4.2 Å from each other occupy the glucose-binding sites IV1 and IV2. d-Glucose at site IV1 is stacked over Trp-2014 and is stabilized by a hydrogen bond network involving side chains of Asp-2002, His-2003, Asp-2098, and Gln-2102. In site IV2, d-glucose is buried in a cleft with the O1 atom making two hydrogen bonds with Asp-2034 and Arg-2036 and the O6 atom interacting with Thr-2098. As shown by molecular dynamics simulations, both molecules are relatively stable and have respective average r.m.s. deviation values of 4.3 ± 0.3 and 5.3 ± 0.1 Å. These values indicate their reorientations within the binding site, confirmed by the constant distance of their center of mass to the neighboring protein residues and by the formation of only 1 intermolecular contact. Their interaction energies are similar to those found for the A-1 and A2 sites (about −20 kcal/mol), probably because of abundance of OH groups capable of forming electrostatic interactions with neighboring residues, nearly independently of the orientation of the ligand within the binding site, revealing for the first time a possible carbohydrate binding function of GH70 glucansucrase domain IV. The glucose bound in site IV-V is labile in half of the simulated trajectories. In the site B-IV-V, d-glucose is orientated with O6 and O1 exposed to the solvent and was shown to be labile by molecular dynamics.
Finally, one glucose molecule was bound to the surface of domain V. This is the first carbohydrate molecule ever identified in domain V of a GH70 enzyme. The corresponding binding site expanding from Ala-1906 to Asp-1981 was named the V-L pocket (Fig. 3A). The d-glucose molecule in the 4C1 conformation is stacked over Tyr-1914, with its oxygen atoms O2, O3, and O4 engaged in hydrogen bonds with the side chains of Gln-1942, Gln-1951, Lys-1953 and with the main chain carbonyl group of Lys-1969 (not shown). Glucose binding in the V-L site is labile because the r.m.s. deviation value was about 46.6 ± 6.6 Å over the last 10 ns of molecular dynamics simulation. A non-interpretable electron density was also observed in domain V over residue Tyr-1834, suggesting the presence of another binding site, named V-K, expanding from Met-1828 to Val-1905.
Structures of ΔN123-GBD-CD2 in Complex with Isomaltosyl or Isomaltotriosyl Residues
ΔN123-GBD-CD2 was soaked with pure isomaltotriose or with a mixture of gluco-oligosaccharides (mainly composed of isomaltosyl-maltose and isomaltotriosyl-maltose). The crystal structures of the corresponding complexes B and C were solved at 2.2 and 1.85 Å, respectively. These structures did not reveal any interpretable electron density in the catalytic gorge or in the previously described glucose-binding sites (A-1, A2, A3, A4, IV1, IV2, B-IV-V, or IV-V). In contrast, electron density corresponding to glucose, isomaltosyl, or isomaltotriosyl groups were visible in pockets V-K and V-L of domain V. Both binding pockets are made up of three consecutive super-secondary elements, either β-hairpins or three-stranded β-sheets. In site V-K, a clear electron density was observed, which corresponds to an isomaltosyl group or an isomaltotriosyl group in complexes B and C, respectively (Fig. 6). The electron density maps indicate that the oligosaccharides may possess an additional glucosyl residue oriented toward the solvent. In the V-L binding site, one glucosyl residue was visible in the electron density maps of both complexes (Fig. 7).
FIGURE 6.

Binding pocket V-K found in the GBD of ΔN123-GBD-CD2 in complexes B (upper panels) and C (lower panels). For this pocket, in complexes B or C, an isomaltosyl or isomaltotriosyl residue was observed and shown as yellow sticks. The network of hydrogen bonding is shown on the right panels, whereas the electron density map around carbohydrates is displayed on left panels. The residues and water molecules involved in binding are represented as pale red sticks and red spheres, respectively (right panels). The simulated annealing omit (Fo − Fc) electron density maps (in green) around carbohydrates were contoured at 3σ or 2σ for isomaltosyl and isomaltotriosyl, respectively. The σA weighted 2Fo − Fc electron density maps in blue were contoured at 0.9 σ.
FIGURE 7.

View of the V-L binding pocket in the glucan-binding domain of ΔN123-GBD-CD2 with gluco-oligosaccharide. For gluco-oligosaccharide or isomaltotriose ligands, a glucosyl residue, shown as yellow stick, was visible in the electron density map. Residues and water molecules involved in binding are represented as pale red sticks and red spheres, respectively. The σA weighted 2Fo − Fc electron density map was contoured at 0.9 σ.
A Rearrangement of the Catalytic Gorge in Complex B
Compared with the previously published apo structure, a rearrangement of loop 7 (from His-2319 to Ser-2369) is observed in complex B (Fig. 8). This loop connects strand 7 and helix 7 of the (β/α)8 barrel. The mean B-factor of the corresponding Cα atoms is 39.7 Å2, which is almost twice as much as the value observed for Cα atoms of domains A, B, and C (21.5 Å2). A geometric exploration of the grooves expanding from subsite −1 of the catalytic gorge was performed with CAVER, and four different grooves were found. Grooves A2 and A3 include glucose binding sites A2 and A3, respectively (Fig. 3B). The groove named “groove loop 7” is delineated by the cleft occurring upon loop 7 rearrangement. Finally, groove 4 expands over loop 7. As shown in Fig. 3C, the movement of loop 7 reveals a positively charged patch in the vicinity of subsite −1 in groove 4, which is absent in the apo structure.
FIGURE 8.
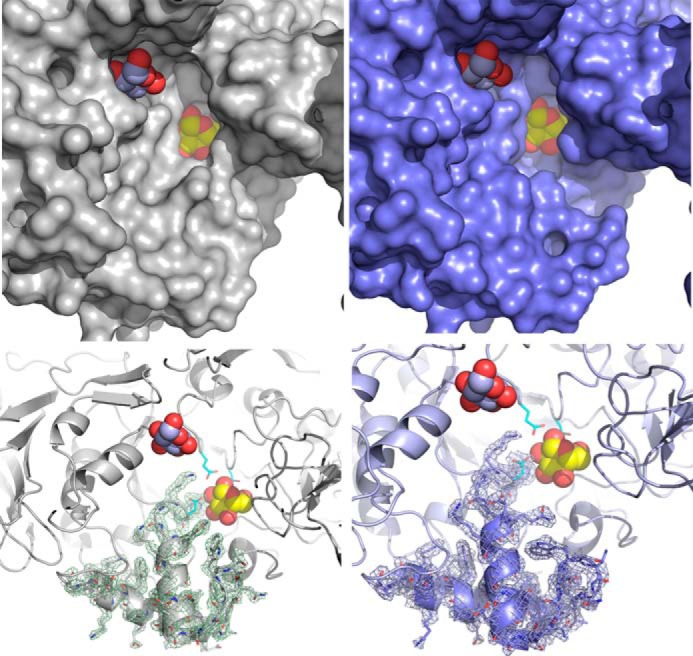
Loop rearrangement in the catalytic gorge of ΔN123-GBD-CD2. The left panels show the catalytic gorge conformation in the apo form. The right panels show the same region for the structure of the complex B. The upper panels represent the protein surface, whereas the schematic representation has been used in the lower panels. The σA weighted 2Fo − Fc electron density maps around loop 7 of domain A were contoured at 1σ. To figure the active site, the d-glucose molecule bound in subsite −1, as found in the structure of the complex with glucose, is superimposed and represented as yellow and red spheres. The d-glucose molecule found in the binding site A2 is also represented (light blue and red spheres). Residues forming the catalytic triad are represented as turquoise sticks.
The V-K Binding Pocket
In complex C, seven residues of the V-K pocket are involved in isomaltotriosyl binding (Fig. 6). The α-d-glucopyranosyl unit 2 (Glc-2) is stacked in the 4C1 conformation over Tyr-1834. The α-d-glucosyl units 1 and 3 (Glc-1 and Glc-3) are also probably stabilized in the 4C1 conformation, although the electron density map is less well defined. The Glc-1 and Glc-2 moieties are essentially stabilized by hydrogen bonding with side chain atoms; the O2 atom of Glc-2 interacts with Gln-1879 and Lys-1881 and the O3 atom with Gln-1870 and Lys-1898 (via the oxygen atom of its main chain carbonyl group). The O5 atom of Glc-1 could make a long-range hydrogen bond with the side chain of Lys-1881, O2 with the oxygen atom of Gly-1897 carbonyl function, and O4 with Lys-1898 and a water molecule. The side chain of Lys-1881 also interacts with the oxygen atom of the osidic bond between Glc-1 and Glc-2. The Glc-3 is weakly stabilized by hydrogen bonding through two water molecules and through van der Waals interactions with Tyr-1834 and Trp-1849.
In complex B, one isomaltosyl residue is found in the V-K pocket. The backbone atoms of the V-K pocket of complexes B and C superimpose well. The glucopyranosyl ring at the non-reducing end is stacked in the 4C1 conformation over the side chain of Tyr-1834, similar to Glc-2 in complex C (Fig. 6). The ring is also maintained by hydrogen bonds between O3 and the main chain carbonyl group of Lys-1898 and the side chain of Gln-1870. Of note, in complexes B and C, the side chains of these two amino acids adopt different conformations. Finally, water-mediated hydrogen bonds with residues Asn-1900 and Thr-1868 complete the network of interactions. The second glucosyl unit (equivalent to Glc-1 in complex C) is weakly stabilized by a hydrogen bond between O5 and the side chain of Lys-1881.
The V-L Binding Pocket
Electron density corresponding to one d-glucopyranosyl unit in 4C1 conformation was found in the binding pocket V-L of complexes B and C (Fig. 7). The interaction network is similar to that described for the complex with d-glucose (Fig. 3A). Interestingly, residues Tyr-1914, Gln-1942, Gln-1951, Lys-1953, and Lys-1969 of the V-K pocket are conserved in the V-L pocket. The glucopyranosyl residue is bound so that the O1 and O6 atoms are pointing toward the solvent (Fig. 7). In this pocket, there is no tryptophan equivalent to Trp-1849 of V-K pocket that could stabilize additional glucosyl residues.
Discussion
In recent years, major advances have been made in thecomprehension of structure-function relationships of GH70 sucrose-active enzymes thanks to the three-dimensional structure resolution of GTF180-ΔN GTFA-ΔN, GTF-SI, and ΔN123-GBD-CD2 glucansucrases in the apo form (9, 35–37). These structures revealed the unique U-fold type of these enzymes organized in five structural domains. In addition, several structures in complex with sucrose donor, maltose acceptor, or acarbose (a known inhibitor of various glycoside-hydrolases) were solved that allowed further description of the −1, +1, +2, and +2′ subsites of these enzymes (35, 36). However, structural data are still missing to draw up the full scenario of polymerization at the molecular level and identify the structural determinants controlling substrate and product specificity. In particular, three-dimensional structures in complex with either isomalto-oligosaccharides or dextrans, the natural acceptors of these enzymes, were heavily lacking, thus preventing molecular descriptions of isomalto-oligosaccharide or dextran-binding sites. In particular, no complexes were previously disclosed in which carbohydrates were present in domain V, although this domain was biochemically proven to act as a glucan-binding domain in several GH70 enzymes (25, 31, 32) and was suggested to participate in the processivity of polymerization (26, 28, 29). Regarding the α-(1→2) branching sucrase (ΔN123-GBD-CD2) enzymes, structural data are even scarcer as only two structures in the apo form have been solved thus far (9).
In this context, solving the structure of the α-(1→2) branching sucrase ΔN123-GBD-CD2 in complex with a gluco-oligosaccharide acceptor was quite challenging, and we have obtained the first three-dimensional structures of this enzyme in complex with d-glucose, isomaltosyl, or isomaltotriosyl residues. The complex with d-glucose clearly showed that one glucose molecule is accommodated in −1 subsite in a conformation similar to that described for the GTF180-ΔN·sucrose complex, corroborating the α-retaining mechanism proposed for GBD-CD2. In addition, the glucose molecules occupying sites A2 and A3, in proximity of the catalytic center, both expose their O1 and O6 atoms to the solvent in a proper orientation to be engaged in α-(1→6) osidic linkages. Such positioning suggests that A2 and A3 sites may provide binding patches delineating a gorge capable of accommodating longer α-(1→6) linked gluco-oligosaccharides or dextrans. The question remains of whether binding sites A2 and A3 define acceptor subsites connected to −1 and +1 subsites of the catalytic center. Additionally, it is unknown if the binding sites could be connected to the other glucose-binding sites found in domains B, IV, and V. Only complexes with longer oligosaccharides could help answer this question (66). Interestingly, residues Glu-2265, Ser-2266, and Glu-2270 of binding site A2 are also conserved in the recently discovered α-(1→2) branching sucrase BRS-A, suggesting that they could be determinants of α-(1→2) branching specificity (10).
Structural analysis of complexes B and C also provide the first structural evidence of interactions between domain V of ΔN123-GBD-CD2 and glucose, as well as isomaltosyl and isomaltotriosyl groups. Surprisingly, comparison of complex B with the apo form revealed that loop 7 of the (β/α)8 barrel moves and opens a cleft toward subsite −1. Thus, it may be tempting to propose that such a rearrangement could assist oligodextran accommodation (or release) in (or from) the observed grooves and could play a role in the ping-pong bi-bi mechanism as well as in the previously described stochastic branching of short dextran chains (39, 67). In domain V of ΔN123-GBD-CD2, the amino acid residues of binding pockets V-K and V-L that interact with carbohydrates are well conserved, with the exception of Trp-1849 and Thr-1868, which are replaced by a threonine and alanine in the pocket V-L, respectively. These differences may be sufficient to modulate affinity and only permit glucose binding in pocket V-L. Furthermore, by inspecting the structures of GTFA-ΔN and GTF180-ΔN, we identified a pocket similar to the V-K and V-L binding pockets of ΔN123-GBD-CD2 in each (Fig. 9). These four pockets contain from 76 to 81 residues and are also constituted by a sequence of three super-secondary motifs that can be either β-hairpins or three-stranded antiparallel β-sheets (Figs. 9 and 10). Most of the conserved aromatic residues are mainly oriented toward the hydrophobic core of the pocket and belong to β-strands. The four pockets are exposed to the solvent and delineated by acidic or basic residues (Asp, Glu, Lys, and Arg), by aromatic residues (Trp or Tyr), and by polar residues (Thr, Ser, Gln, and Asn). They all contain one aromatic residue (Phe or Tyr) at the bottom, suggesting that they could also promote dextran binding. However, structural data obtained by Vujičić-Žagar et al. from GTF180-ΔN crystals soaked with d-glucose, isomaltose, panose, or nigerotriose did not reveal any sugar binding in domain V (35). It remains to be understood what subtle differences in the experimental conditions or in the putative binding pocket of GTF180-ΔN render this pocket non-functional. However, the complete truncation of domain V in GTF180-ΔN strongly reduces its ability to synthetize α-glucans from sucrose in favor of oligosaccharide synthesis (69). This suggests that domain V of GTF180-ΔN may have a positive impact on α-glucan synthesis, possibly by promoting the binding to α-glucan chains.
FIGURE 9.

Comparison of the sugar binding pockets found in the glucan-binding domains of ΔN123-GBD-CD2 (complex C), and putative sugar binding pockets of GTF180-ΔN (PDB entry 3KLK) and GTFA-ΔN (PDB entry 4AMC) glucansucrases. Secondary structure elements are shown in pale red in agreement with Fig. 9. Molecular surfaces are displayed in light gray. Residues delineating the binding pockets are shown as pale red sticks. Stacked sugars in the binding pockets of ΔN123-GBD-CD2 are represented in yellow lines.
FIGURE 10.

Sequence alignment and LOGO sequence of the GBD repeats proved to bind α-glucans of GH70 glucansucrases. GBD1A, GBD4RS, and GBD5 bind dextrans with Kd values of 0.6 μm, 2.5 μm, and 11.1 nm, respectively. Sequences are GBD1A_GTFI_rp# (repeats 1 to 4 of the GBD1A of GTFI from S. downei), GBD-4RS_GTFI_rp# (repeats 1 to 4 of the GBD-4RS of GFTI from S. sobrinus), GBD5_DSR-S_rp# (repeats 1 to 3 of the GBD5 of DSR-S from L. mesenteroides B-512F), GBD_GTF180_rp1 (repeat 1 of the GBD of GTF180-ΔN from Lactobacillus reuteri), GBD_GTFA_rp1 (repeat 1 of the GBD of GTFA-ΔN from L. reuteri 121), and GBD_DSR-E_rp# (repeats J to L of the GBD of ΔN123-GBD-CD2 from L. citreum NRRL-1299). Residues highlighted in red are well conserved, whereas those tending toward blue are not. Residues framed in gray delineate the sugar binding pockets and are pointed out in the LOGO sequence. Residues framed in dashed line on the LOGO sequence correspond to YG repeats. In the repeats of GBD1A, residues circled in white, were mutated by Shah et al. (25) (see ”Discussion“). Secondary structure elements, as determined by DSSP from PDB entries 3TTQ, 3KLK, and 4AMC, are displayed below the LOGO sequence. β-Strands, numbered according to Fig. 8, linked by dashed lines represent either β-hairpins or three-stranded β-sheets.
In addition, structural alignments with other domains V of several GH70 glucansucrases, which were experimentally proved to bind α-glucans, were performed with EXPRESSO to check the possible conservation of the residues delineating the binding pockets. The alignment reveals that several sequences putatively corresponding to sugar binding pockets are present. These putative binding pockets include four YG repeats, as defined by Giffard and Jacques (20), two complete repeats flanked by two partial ones (Fig. 10). The residues of the V-K pockets interacting with carbohydrates are conserved, i.e. the aromatic residues acting as a stacking platform (logo number 19 -Fig. 10), Trp-34, Gln-71, and Lys-73 (respectively, equivalent to Tyr-1834, Trp-1849, Gln-1879, and Lys-1881 in the V-K pocket). Then the alignment was extended to the entire GBD of DSR-E, from which ΔN123-GBD-CD2 is derived. As seen in Fig. 11, 10 sequences of putative binding pockets (named V-A to V-J), containing 46 to 84 residues each, aligned with the sequences of V-K and V-L pockets. As for the domains that were shown to bind dextrans, these sequences contained the residues involved in sugar binding in V-L and V-K pockets (i.e. aromatic residue 11, Trp-26, Gln-62, and Lys-64, LOGO sequence numbering, Fig. 11), indicating that these pockets are likely to be functional. In 2004, Shah et al. (25) carried out a site-directed mutagenesis study focusing on the conserved residues of GBD1A, a truncated domain V disconnected from the enzymatic core. A total of nine amino acids belonging to binding pocket repeats 1, 2, 3, and 4 were targeted, including Phe-1122, Trp-1163, Trp-1227, Trp-1292, Tyr-1294, Tyr-1314, Phe-1315, Gln-1321, and Lys-1323 (Fig. 11). Eight mutants lost their ability to bind biotinylated dextrans, whereas eight others retained this ability, showing that some positions are tolerant to mutations and others are more critical for this function (25).
FIGURE 11.

Sequence alignment of the 12 repeats identified in the GBD of DSR-E. Black highlighted residues are involved in sugar binding in repeats K and L, whereas gray highlighted residues are proposed to play the same role in repeat J. Framed residues would participate in sugar binding for repeats A to I. A LOGO sequence based on this alignment is shown. The YG repeats are framed in dashed lines on the LOGO sequence.
The sugar-binding sites identified in domain V are likely involved in the α-glucan binding ability of ΔN123-GBD-CD2, even if we cannot exclude that dextran binding also occurs in other domains. We determined Kd values for dextrans in the micromolar range. In comparison, the full-length DSR-S, a dextransucrase produced by a constitutive mutant of L. mesenteroides NRRL B-512F, was shown to bind 170-kDa dextran with a Kd value of 18 nm using a quartz crystal microbalance (68). The whole domain V of DSR-S, devoid of the catalytic domain, showed a Kd value for biotinylated dextran of 70 nm (32). Although the methods and the dextran used for determining the Kd value vary among these studies, our results suggest that the truncated branching sucrase ΔN123-GBD-CD2 has a weaker affinity for dextran than the full-length domain V of DSR-S dextransucrase, which seems to contain a higher number of putative binding pockets. Using isothermal titration calorimetry, Komatsu et al. (31) studied the binding of 25-kDa dextran to a set of independent truncated domain V derived from GTFI, including the truncated form GBD-4RS and the full-length GBD (Fig. 11). The exothermic binding of dextran to the full-length domain V gave a Kd value of 20 nm at 25 °C. The binding stoichiometry was as low as 0.12, which indicates that several molecules bind to stretches of dextran chains. Moreover, –ΔH0 and –ΔG0 were found to be proportional to the number of residues of the truncated domain V, which substantiated the fact that the binding sites are independent and similar. The affinity of this GBD with dextran was proposed to be the sum of weak interactions with different glucose-binding sites, which is consistent with our structural data.
Our findings reveal for the first time that binding of isomalto-oligosaccharides to domain V occurs via sugar binding pockets comprising between 60 and 81 residues. In the V-K and V-L pockets, disclosed herein, eight residues are interacting with the sugar rings. Only four of them, one aromatic residue acting as a stacking platform, one tryptophan, one glutamine, and one lysine, are conserved in the putative binding pockets of other domain V shown to bind α-glucans. It remains to be understood what specific features in these binding pockets confer, or do not confer, a strong affinity for α-glucans. The x-ray structures of other domain V, in complex with longer isomalto-oligosaccharides and containing variable numbers of sugar pockets, could help to elucidate the possible synergy between the binding pockets and its influence on glucan binding strength. In addition, glucan binding in domain V probably contribute to increasing the local concentration of carbohydrate chains in the vicinity of the active site and may play a key role in presenting these chains to the active site for their branching or elongation. This assumption is corroborated by the fact that DSR-S and ASR glucansucrases partially truncated of their domain V synthesized glucans of lower molar mass than those synthesized by the entire enzymes (29). Efforts should be pursued to solve the structure of glucansucrases or branching sucrases with short dextran chains bound in the active site as well as in domains B, IV, and V and combine structural analyses to mutagenesis and detailed biochemical characterization to get further insight in these mechanisms.
Author Contributions
Y. B., M. R. S., and S. T. designed the study. Y. B. purified, crystallized the enzyme, and solved the x-ray structures. Y. M. purified the enzyme for affinity gel electrophoresis. G. C. realized all molecular dynamics calculations. Y. B., G. C., L. M., M. R. S., and S. T. wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We greatly acknowledge the staff from the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and the Integrated Screening Platform of Toulouse (PICT, Toulouse, France) for providing access to macromolecular crystallography equipment.
The authors declare that they have no conflict of interest with the content of this article.
- GH
- glycoside hydrolase
- GBD
- glucan-binding domain
- GBD-CD2
- GBD-catalytic domain 2
- Glcp
- glucopyranose
- Glc
- glucose
- r.m.s.
- root mean square.
References
- 1. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., and Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leemhuis H., Pijning T., Dobruchowska J. M., van Leeuwen S. S., Kralj S., Dijkstra B. W., and Dijkhuizen L. (2013) Glucansucrases: three-dimensional structures, reactions, mechanism, α-glucan analysis and their implications in biotechnology and food applications. J. Biotechnol. 163, 250–272 [DOI] [PubMed] [Google Scholar]
- 3. Leemhuis H., Dijkman W. P., Dobruchowska J. M., Pijning T., Grijpstra P., Kralj S., Kamerling J. P., and Dijkhuizen L. (2013) 4,6-α-Glucanotransferase activity occurs more widespread in Lactobacillus strains and constitutes a separate GH70 subfamily. Appl. Microbiol. Biotechnol. 97, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Côté G. L., and Leathers T. D. (2005) A method for surveying and classifying Leuconostoc spp. glucansucrases according to strain-dependent acceptor product patterns. J. Ind. Microbiol. Biotechnol. 32, 53–60 [DOI] [PubMed] [Google Scholar]
- 5. Naessens M., Cerdobbel A., Soetart W., and Vandamme E. J. (2005) Leuconostoc dextransucrase and dextran: production, properties and applications. J. Chem. Technol. Biotechnol. 80, 845–860 [Google Scholar]
- 6. Falconer D. J., Mukerjea R., and Robyt J. F. (2011) Biosynthesis of dextrans with different molecular weights by selecting the concentration of Leuconostoc mesenteroides B-512FMC dextransucrase, the sucrose concentration, and the temperature. Carbohydr. Res. 346, 280–284 [DOI] [PubMed] [Google Scholar]
- 7. Vettori M. H. P. B., Blanco K. C., Cortezi M., De Lima C. J. B., and Contiero J. (2012) Dextran: effect of process parameters on production, purification and molecular weight and recent applications. Diálogos Ciênc. 2012, 171–186 [Google Scholar]
- 8. Bowen W. H., and Koo H. (2011) Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 45, 69–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brison Y., Pijning T., Malbert Y., Fabre É., Mourey L., Morel S., Potocki-Véronèse G., Monsan P., Tranier S., Remaud-Siméon M., and Dijkstra B. W. (2012) Functional and structural characterization of α-(1→2) branching sucrase derived from DSR-E glucansucrase. J. Biol. Chem. 287, 7915–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Passerini D., Vuillemin M., Ufarté L., Morel S., Loux V., Fontagné-Faucher C., Monsan P., Remaud-Siméon M., and Moulis C. (2015) Inventory of the GH70 enzymes encoded by Leuconostoc citreum NRRL B-1299: identification of three novel α-transglucosylases. FEBS J. 282, 2115–2130 [DOI] [PubMed] [Google Scholar]
- 11. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mooser G., and Wong C. (1988) Isolation of a glucan-binding domain of glucosyltransferase (1,6-α-glucan synthase) from Streptococcus sobrinus. Infect. Immun. 56, 880–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferretti J. J., Gilpin M. L., and Russell R. R. (1987) Nucleotide sequence of a glucosyltransferase gene from Streptococcus sobrinus MFe28. J. Bacteriol. 169, 4271–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banas J. A., Russell R. R., and Ferretti J. J. (1990) Sequence analysis of the gene for the glucan-binding protein of Streptococcus mutans Ingbritt. Infect Immun. 58, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilmore K. S., Russell R. R., and Ferretti J. J. (1990) Analysis of the Streptococcus downei gtfS gene, which specifies a glucosyltransferase that synthesizes soluble glucans. Infect. Immun. 58, 2452–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abo H., Matsumura T., Kodama T., Ohta H., Fukui K., Kato K., and Kagawa H. (1991) Peptide sequences for sucrose splitting and glucan binding within Streptococcus sobrinus glucosyltransferase (water-insoluble glucan synthetase). J. Bacteriol. 173, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giffard P. M., Simpson C. L., Milward C. P., and Jacques N. A. (1991) Molecular characterization of a cluster of at least two glucosyltransferases genes in Streptococcus salivarius ATCC 25975. J. Gen. Microbiol. 137, 2577–2593 [DOI] [PubMed] [Google Scholar]
- 19. Giffard P. M., Allen D. M., Milward C. P., Simpson C. L., and Jacques N. A. (1993) Sequence of the gtfK gene of Streptococcus salivarius ATCC 25975 and evolution of the gtf genes of oral streptococci. J. Gen. Microbiol. 139, 1511–1522 [DOI] [PubMed] [Google Scholar]
- 20. Giffard P. M., and Jacques N. A. (1994) Definition of a fundamental repeating unit in streptococcal glucosyltransferase glucan-binding regions and related sequences. J. Dent. Res. 73, 1133–1141 [DOI] [PubMed] [Google Scholar]
- 21. Fernández-Tornero C., López R., García E., Giménez-Gallego G., and Romero A. (2001) A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat. Struct. Biol. 8, 1020–1024 [DOI] [PubMed] [Google Scholar]
- 22. Greco A., Ho J. G., Lin S.-J., Palcic M. M., Rupnik M., and Ng K. K. (2006) Carbohydrate recognition by Clostridium difficile toxin A. Nat. Struct. Mol. Biol. 13, 460–461 [DOI] [PubMed] [Google Scholar]
- 23. Maestro B., Santiveri C. M., Jiménez M. A., and Sanz J. M. (2011) Structural autonomy of a β-hairpin peptide derived from the pneumococcal choline-binding protein LytA. Protein Eng. Des. Sel. 24, 113–122 [DOI] [PubMed] [Google Scholar]
- 24. Olvera C., Fernández-Vázquez J. L., Ledezma-Candanoza L., and López-Munguía A. (2007) Role of the C-terminal region of dextransucrase from Leuconostoc mesenteroides IBT-PQ in cell anchoring. Microbiology 153, 3994–4002 [DOI] [PubMed] [Google Scholar]
- 25. Shah D. S., Joucla G., Remaud-Simeon M., and Russell R. R. (2004) Conserved repeat motifs and glucan binding by glucansucrases of oral streptococci and Leuconostoc mesenteroides. J. Bacteriol. 186, 8301–8308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kralj S., van Geel-Schutten G. H., van der Maarel M. J., and Dijkhuizen L. (2004) Biochemical and molecular characterization of Lactobacillus reuteri 121 reuteransucrase. Microbiology 150, 2099–2112 [DOI] [PubMed] [Google Scholar]
- 27. Fabre E., Bozonnet S., Arcache A., Willemot R. M., Vignon M., Monsan P., and Remaud-Simeon M. (2005) Role of the two catalytic domains of DSR-E dextransucrase and their involvement in the formation of highly α1,2-branched dextran. J. Bacteriol. 187, 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joucla G., Pizzut S., Monsan P., and Remaud-Simeon M. (2006) Construction of a fully active truncated alternansucrase partially deleted of its carboxy-terminal domain. FEBS Lett. 580, 763–768 [DOI] [PubMed] [Google Scholar]
- 29. Moulis C., Joucla G., Harrison D., Fabre E., Potocki-Veronese G., Monsan P., and Remaud-Simeon M. (2006) Understanding the polymerization mechanism of glycoside-hydrolase family 70 glucansucrases. J. Biol. Chem. 281, 31254–31567 [DOI] [PubMed] [Google Scholar]
- 30. Lis M., Shiroza T., and Kuramitsu H. K. (1995) Role of C-terminal direct repeating units of the Streptococcus mutans glucosyltransferase-S in glucan binding. Appl. Environ. Microbiol. 61, 2040–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Komatsu H., Katayama M., Sawada M., Hirata Y., Mori M., Inoue T., Fukui K., Fukada H., and Kodama T. (2007) Thermodynamics of the binding of the C-terminal repeat domain of Streptococcus sobrinus glucosyltransferase-I to dextran. Biochemistry 46, 8436–8444 [DOI] [PubMed] [Google Scholar]
- 32. Suwannarangsee S., Moulis C., Potocki-Veronese G., Monsan P., Remaud-Simeon M., and Chulalaksananukul W. (2007) Search for a dextransucrase minimal motif involved in dextran binding. FEBS Lett. 581, 4675–4680 [DOI] [PubMed] [Google Scholar]
- 33. Mori T., Asakura M., and Okahata Y. (2011) Single-molecule force spectroscopy for studying kinetics of enzymatic dextran elongations. J. Am. Chem. Soc. 133, 5701–5703 [DOI] [PubMed] [Google Scholar]
- 34. Kingston K. B., Allen D. M., and Jacques N. A. (2002) Role of the C-terminal YG repeats of the primer-dependent streptococcal glucosyltransferase, GtfJ, in binding to dextran and mutan. Microbiology 148, 549–558 [DOI] [PubMed] [Google Scholar]
- 35. Vujicic-Zagar A., Pijning T., Kralj S., López C. A., Eeuwema W., Dijkhuizen L., and Dijkstra B. W. (2010) Crystal structure of a 117 kDa glucansucrase fragment provides insight into evolution and product specificity of GH70 enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 21406–21411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ito K., Ito S., Shimamura T., Weyand S., Kawarasaki Y., Misaka T., Abe K., Kobayashi T., Cameron A. D., and Iwata S. (2011) Crystal structure of glucansucrase from the dental caries pathogen Streptococcus mutans. J. Mol. Biol. 408, 177–186 [DOI] [PubMed] [Google Scholar]
- 37. Pijning T., Vujičić-Žagar A., Kralj S., Dijkhuizen L., and Dijkstra B. W. (2012) Structure of the α-1,6/α-1,4-specific glucansucrase GTFA from Lactobacillus reuteri 121. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1448–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pijning T., Vujičić-Žagar A., Kralj S., Dijkhuizen L., and Dijkstra B. W. (2014) Flexibility of truncated and full-length glucansucrase GTF180 enzymes from Lactobacillus reuteri 180. FEBS J. 281, 2159–2171 [DOI] [PubMed] [Google Scholar]
- 39. Brison Y., Laguerre S., Lefoulon F., Morel S., Monties N., Potocki-Véronèse G., Monsan P., and Remaud-Simeon M. (2013) Branching pattern of gluco-oligosaccharides and 1.5-kDa dextran grafted by the α-1,2 branching sucrase GBD-CD2. Carbohydr. Polym. 94, 567–576 [DOI] [PubMed] [Google Scholar]
- 40. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 43. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C. et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kozlikova B., Sebestova E., Sustr V., Brezovsky J., Strnad O., Daniel L., Bednar D., Pavelka A., Manak M., Bezdeka M., Benes P., Kotry M., Gora A., Damborsky J., and Sochor J. (2014) CAVER Analyst 1.0: graphic tool for interactive visualization and analysis of tunnels and channels in protein structures. Bioinformatics 30, 2684–2685 [DOI] [PubMed] [Google Scholar]
- 49. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salomon-Ferrer R., Götz A. W., Poole D., Le Grand S., and Walker R. C. (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs: 2. explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 [DOI] [PubMed] [Google Scholar]
- 51. Salomon-Ferrer R., Case D. A., and Walker R. C. (2013) An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210 [Google Scholar]
- 52. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 53. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 54. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 55. Jayaram B., Sprous D., and Beveridge D. L. (1998) Solvation free energy of biomacromolecules: parameters for a modified generalized born model consistent with the AMBER force field. J. Phys. Chem. B 102, 9571–9576 [Google Scholar]
- 56. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bozonnet S., Dols-Laffargue M., Fabre E., Pizzut S., Remaud-Simeon M., Monsan P., and Willemot R.-M. (2002) Molecular characterization of DSR-E, an α-1,2 linkage-synthesizing dextransucrase with two catalytic domains. J. Bacteriol. 184, 5753–5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Szklarczyk R., and Heringa J. (2004) Tracking repeats using significance and transitivity. Bioinformatics 20, i311–317 [DOI] [PubMed] [Google Scholar]
- 59. Armougom F., Moretti S., Poirot O., Audic S., Dumas P., Schaeli B., Keduas V., and Notredame C. (2006) Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 34, W604–W608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.-M., Taly J.-F., and Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Crooks G. E., Hon G., Chandonia J.-M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vuillemin M., Malbert Y., Laguerre S., Remaud-Siméon M., and Moulis C. (2014) Optimizing the production of an α-(1→2) branching sucrase in Escherichia coli using statistical design. Appl. Microbiol. Biotechnol. 98, 5173–5184 [DOI] [PubMed] [Google Scholar]
- 63. Takeo K. (1995) Advances in affinity electrophoresis. J. Chromatogr. A 698, 89–105 [Google Scholar]
- 64. Tomme P., Boraston A., Kormos J. M., Warren R. A., and Kilburn D. G. (2000) Affinity electrophoresis for the identification and characterization of soluble sugar binding by carbohydrate-binding modules. Enzyme Microb. Technol. 27, 453–458 [DOI] [PubMed] [Google Scholar]
- 65. Abbott D. W., and Boraston A. B. (2012) Quantitative approaches to the analysis of carbohydrate-binding module function. Methods Enzymol. 510, 211–231 [DOI] [PubMed] [Google Scholar]
- 66. Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brison Y., Fabre E., Moulis C., Portais J.-C., Monsan P., and Remaud-Siméon M. (2010) Synthesis of dextrans with controlled amounts of α-1,2 linkages using the transglucosidase GBD–CD2. Appl. Microbiol. Biotechnol. 86, 545–554 [DOI] [PubMed] [Google Scholar]
- 68. Nihira T., Mori T., Asakura M., and Okahata Y. (2011) Kinetic studies of dextransucrase enzyme reactions on a substrate- or enzyme-immobilized 27 MHz quartz crystal microbalance. Langmuir 27, 2107–2111 [DOI] [PubMed] [Google Scholar]
- 69. Meng X., Dobruchowska J. M., Pijning T., Gerwig G. J., Kamerling J. P., and Dijkhuizen L. (2015) Truncation of domain V of the multidomain glucansucrase GTF180 of Lactobacillus reuteri 180 heavily impairs its polysaccharide-synthesizing ability. Appl. Microbiol. Biotechnol. 99, 5885–5894 [DOI] [PubMed] [Google Scholar]