

Abstract
Redox fluctuations within cells can be detrimental to cell function. To gain insight into how cells normally buffer against redox changes to maintain cell function, we have focused on elucidating the signaling pathways that serve to sense and respond to oxidative redox stress within the endoplasmic reticulum (ER) using yeast as a model system. Previously, we have shown that a cysteine in the molecular chaperone BiP, a Hsp70 molecular chaperone within the ER, is susceptible to oxidation by peroxide during ER-derived oxidative stress, forming a sulfenic acid (−SOH) moiety. Here, we demonstrate that this same conserved BiP cysteine is susceptible also to glutathione modification (−SSG). Glutathionylated BiP is detected both as a consequence of enhanced levels of cellular peroxide and also as a by-product of increased levels of oxidized glutathione (GSSG). Similar to sulfenylation, we observe glutathionylation decouples BiP ATPase and peptide binding activities, turning BiP from an ATP-dependent foldase into an ATP-independent holdase. We show glutathionylation enhances cell proliferation during oxidative stress, which we suggest relates to modified BiP's increased ability to limit polypeptide aggregation. We propose the susceptibility of BiP to modification with glutathione may serve also to prevent irreversible oxidation of BiP by peroxide.
Keywords: 70-kilodalton heat shock protein (Hsp70), endoplasmic reticulum (ER), glutathionylation, GRP78, molecular chaperone, oxidative stress, redox signaling, BiP, Kar2
Introduction
Cell homeostasis and numerous vital cell functions rely on the maintenance of a proper intracellular redox balance. Oxidative folding in the endoplasmic reticulum (ER)2 is one intracellular system that is prone to disruption upon alterations in the redox poise. Insufficient oxidizing capacity in the ER leads to the accumulation of proteins in reduced non-native states (1, 2). Alternatively, an overly oxidizing ER results in cellular toxicity, likely caused in part by the mis-pairing of protein cysteine residues resulting from a decreased capacity to reduce non-native disulfides (3, 4). Crucial for maintaining a continual flux of folding polypeptides through the ER are the systems that buffer against fluctuations in the ER redox environment.
Recently, several proteins have been demonstrated to act as sensors of redox fluctuations in the ER. In a common theme, these proteins sense changes in the ER redox environment and respond with beneficial alterations in their enzymatic activities. The enzyme Ero1 is a major source of oxidizing equivalents in the ER, and Ero1 activity is coupled to the ER redox environment. When the ER balance shifts to overly oxidizing conditions, regulatory cysteine residues in Ero1 become oxidized to disulfides, which decreases Ero1 oxidase activity to help restore redox balance (5–7). In addition, sensors that cope with the potential for protein folding defects have emerged. These redox-dependent chaperones do not directly impact the flux of oxidizing equivalents, like Ero1, but rather are activated to help limit polypeptide aggregation during unfavorable redox conditions. Oxidation of active site cysteines in the oxidoreductase PDI has been demonstrated to trigger a conformational change that exposes a hydrophobic patch to facilitate high chaperone activity (8). Similarly, oxidation of cysteine(s) in the Hsp70 chaperone BiP can augment BiP activity as a polypeptide holdase (9, 10).
Key to these described ER sensors are redox active cysteines. Cysteine oxidation is largely dependent on the species and concentrations of oxidants they contact, and post-translational oxidation of protein cysteine thiols offers a powerful means for cells to sense and offset redox changes. In the case of Ero1, the regulatory cysteines are maintained in a reduced state based on the cellular levels of reduced glutathione (GSH) and/or the reduced form of the disulfide oxidoreductase PDI (5–7, 11). Here, Ero1 senses the available capacity of ER reducing equivalents. Alternatively, BiP senses fluctuations in ER peroxide levels, which have the potential to cause oxidative damage to folding polypeptides. In mammalian cells, BiP oxidation (and the corresponding increase in holdase activity) is a consequence of a redox relay initiated by peroxide-mediated oxidation of the stress sensor GPx7 (NPGPx) (10). For yeast BiP (also known as Kar2), we have shown its single cysteine (Cys-63) is susceptible to direct modification by peroxide, forming a sulfenic acid adduct.
Although stable sulfenic acids in proteins have been observed, it is generally accepted that sulfenic acid is most often a metastable intermediate (12–14). We were intrigued by the prospect that the sulfenic acid adduct observed for yeast BiP may undergo further oxidation and that distinct biochemical activities for BiP may be observed that are dependent on specific redox modifications. Sulfenic acid is prone to further oxidation by thiols; GSH, which contains a free thiol, is abundant within the ER lumen (15). We anticipated yeast BiP might be susceptible to oxidation by glutathione (glutathionylation). Notably, precedent exists for Hsp70 glutathionylation. Human BiP (GRP78) was identified as one of 23 proteins glutathionylated during diamide-induced oxidative stress in ECV304 endothelial cells (16), and the cytosolic rat Hsp70 HSC70 was shown to be glutathionylated in retinal pigment epithelium cells (17). Human BiP and rat Hsc70 have two and four cysteines, respectively; it was not determined which cysteines were modified by glutathione in the detected glutathionylated proteins. How glutathione addition impacts BiP activity was not explored.
Here, we show that the single cysteine in yeast BiP is susceptible to glutathione modification within cells upon oxidative stress. We suggest glutathionylation occurs not only through interaction of reduced GSH with peroxide-oxidized BiP but also BiP can be directly modified by oxidized glutathione (GSSG). These data imply a capacity for BiP to sense increases in both peroxide and GSSG within the ER lumen. Similar to what we reported previously for sulfenylated BiP (9), glutathionylated BiP shows enhanced holdase activity, which we propose serves to prevent protein aggregation and promote cell survival during stress. By employing BiP alleles with an increased susceptibility to oxidation by glutathione, we observed an enhanced growth rate that correlates with the proportion of oxidized BiP in cells during stress.
Experimental Procedures
Strains and Growth Conditions
Saccharomyces cerevisiae strains were grown and genetically manipulated using standard techniques (18). YPD is a rich medium with 2% glucose. SMM, SMM Raf, and SMM Gal are synthetic minimal media containing 2% glucose, 2% raffinose, or 2% galactose. Uracil or leucine medium supplements were removed to select for plasmids as needed. All BiP proteins used in this study are derived from yeast BiP. Note, we use the standard yeast name KAR2 (instead of the common name BiP) for yeast strain genotypes and plasmid inserts. KAR2 is an essential gene.
CSY5 (MATa GAL2 ura3-52 leu2-3,112), CSY214 (MATa GAL2 ura3-52 leu2-3,112 kar2Δ::KanMX [pCS623]), CSY275 (MATa GAL2 ura3-52 leu2-3,112 kar2-C63A), CSY316 (MATa GAL2 ura3-52 leu2-3,112 kar2Δ::KanMX pep4Δ::NatMX [pCS757]), and CSY319 (MATa GAL2 ura3-52 leu2-3,112 kar2Δ::KanMX pep4Δ::NatMX [pCS760]) are described in Ref. 9. CSY318 and CSY693-CSY697 were generated by transformation of CSY214 with pCS757, pCS760, pJW22, pJW23, pJW36, or pJW37, respectively, followed by counter-selection of pCS623 on SMM with 5-fluoroorotic acid.
Plasmid Construction
Yeast expression plasmids pCS757 (CEN LEU2 KAR2-FLAG), pCS760 (CEN LEU2 kar2-C63A-FLAG), pCS681 (CEN LEU2 KAR2), pCS685 (CEN LEU2 kar2-C63A), pCS802 (CEN LEU2 kar2-C63D), pCS687 (CEN LEU2 kar2-C63F), and pCS688 (CEN LEU2 kar2-C63Y) are described in Ref. 9, and pCS452 (CEN URA3 PGAL1-ERO1*-myc) is described in Ref. 7. pCS703 (CEN URA3 KAR2) was made by cloning the KAR2 insert from pCS681 into pRS316 (19). pCS531 (CEN LEU2 PGAL1-ERO1*-myc) was constructed by ligating the PGAL1-ERO1*-myc fragment from pCS452 into pRS315 (19). pJW22 (CEN LEU2 kar2-D411K-FLAG), pJW23 (CEN LEU2 kar2-E412R-FLAG), pJW36 (CEN LEU2 kar2-C63A-D411K-FLAG), and pJW37 (CEN LEU2 kar2-C63A-E412R-FLAG) were mutagenized by QuikChange (Agilent Technologies, Santa Clara, CA) of pCS757. pCS958 (CEN LEU2 kar2-D411K), pCS959 (CEN LEU2 kar2-E412R), pCS960 (CEN LEU2 kar2-C63A-D411K), and pCS961 (CEN LEU2 kar2-C63A-E412R) were mutagenized from pCS681. pCS704 (CEN URA3 kar2-C63A), pCS964 (CEN URA3 kar2-C63D), pHS39 (CEN URA3 kar2-C63F), pCS965 (CEN URA3 kar2-C63Y), pCS962 (CEN URA3 kar2-D411K), pCS963 (CEN URA3 kar2-E412R), pCS967 (CEN URA3 kar2-C63A-D411K), and pCS968 (CEN URA3 kar2-C63A-E412R) were mutagenized from pCS703. pCS951 (CEN URA3 eroGFP-iE) was constructed by introduction of the GFP mutations F99S, H148S, M153T, V163A, I167T, and a glutamic acid insertion after GFP residue 147, in pPM28 (CEN URA3 eroGFP) (a gift from Feroz Papa; Addgene plasmid 20131) (20).
Plasmids for protein expression in bacteria, pCS630 (kar2-(40–668)-His6), pCS631 (kar2-(40–668)-C63A-His6), pCS817 (His6-kar2-(42–682)), pCS818 (His6-kar2-(42–682)-C63A), and pCS675 (GST-sec63J-(121–221)), are described in Ref. 9. To make pJO1 (His6-kar2-(48–428)), a sequence coding for the ATPase domain of Kar2 (residues 48–428) was cloned into pET-28a (EMD Millipore, Billerica, MA). pJW24 (His6-kar2-(48–428)-C63A), pJW29 (His6-kar2-(48–428)-D411K), pJW30 (His6-kar2-(48–428)-E412R), pCS956 (His6-kar2-(48–428)-C63A-D411K), and pCS957 (His6-kar2-(48–428)-C63A-E412R) were generated by mutagenesis of pJO1. GrxC expression plasmids pHS32 (grxC-C14S-C65Y- His6) and pJW1 (grxC-C11S-C14S-C65Y- His6) were created by amplification of the Escherichia coli grxC gene from XL1-Blue cells, ligation of the corresponding DNA into pET-28a, and site-directed mutagenesis to generate amino acid substitutions.
Recombinant Protein Purification
His-tagged BiP proteins (Kar2 residues 40–668 or 42–682) and GST-Sec63J protein were purified as described in Ref. 9. His-tagged BiP ATPase domain proteins (Kar2 residues 48–428) were induced and purified from BL21 (DE3) pLysS cells. Cells were grown overnight at 37 °C in LB medium containing 15 μg/ml kanamycin and 34 μg/ml chloramphenicol, and cells were diluted 1:75 in TB medium with antibiotics and grown to an A600 of 0.7 at 37 °C, and cultures were shifted to 18 °C for 1 h prior to protein induction at 18 °C with 0.5 mm isopropyl β-d-thiogalactopyranoside overnight at 18 °C. Purification conditions were as described previously (21) except tris(2-carboxyethyl)phosphine was absent from all buffers. GrxC proteins were induced and purified from BL21 (DE3) pLysS cells. Cell were grown overnight at 37 °C in LB medium containing 15 μg/ml kanamycin and 34 μg/ml chloramphenicol. Cells were diluted 1:20 in TB medium with antibiotics, grown to an A600 of 0.7, and protein expression was induced with 0.4 mm isopropyl β-d-thiogalactopyranoside at 37 °C for 5 h. Cells were solubilized in lysis buffer (50 mm Na2HPO4, 0.5 m NaCl, 10 mm imidazole, pH 8.0) containing 1 mm PMSF and 5 mm β-mercaptoethanol (BME), and cells were lysed by treatment with lysozyme followed by sonication. Insoluble material was removed by centrifugation at 16,000 × g for 20 min, and soluble material was loaded onto a HiTrap chelating column (GE Healthcare, Little Chalfont, UK) charged with nickel. The column was washed with 20 column volumes of lysis buffer, 20 column volumes of wash buffer (50 mm Na2HPO4, 0.3 m NaCl, 20 mm imidazole, pH 8.0), and GrxC was eluted with wash buffer containing a final concentration of 0.2 m imidazole. Fractions containing GrxC were dialyzed against 50 mm Na2HPO4, 0.1 m NaCl, 10% glycerol, pH 7.5. All proteins were flash-frozen in liquid nitrogen and stored at −80 °C. Protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific, Waltham, MA) using bovine serum albumin as a standard. GrxC protein activity was confirmed using the HEDS assay (22).
In Vitro Glutathionylation
Full-length BiP (Kar2 residues 40–668) (70 μm) was incubated with oxidants in TNE buffer (10 mm Tris-HCl, pH 7.4, 50 mm NaCl, 1 mm EDTA) in a 70-μl total volume. Proteins treated with 1 mm diamide and 2 mm GSH were incubated for 30 min at 30 °C. Proteins treated with 8 mm oxidized glutathione (GSSG), 2 mm cumene hydroperoxide (CHP) and 1 mm reduced glutathione (GSH), or 1 mm sodium hypochlorite (NaOCl) and 1 mm GSH were incubated for 60 min at 37 °C. NaOCl was prepared from Clorox germicidal bleach (The Clorox Co., Oakland, CA) assuming 8.25% NaOCl concentration. For experiments with a GSH/GSSG mixture, 70 μm BiP was reacted with a combination of GSH and GSSG (8 mm total) for 60 min at 37 °C. The ATPase domain of BiP (Kar2 residues 48–428) (31 μm) was glutathionylated by incubation with the indicated amount of GSSG in TN buffer (20 mm Tris-HCl, pH 7.4, 0.3 m NaCl) for 30 min at 30 °C. Unmodified BiP control proteins were prepared identically except TN buffer was substituted for oxidants. For immunodetection of BiP-glutathione adducts, an equal volume of 2× sample buffer (150 mm Tris-HCl, pH 6.8, 40% glycerol, 6% SDS, 0.02% bromphenol blue) containing 0.1 m N-ethylmaleimide (NEM) was added, and proteins were separated by SDS-PAGE. Proteins were transferred to nitrocellulose, and the membrane was incubated with TBST (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.05% Tween 20) containing 5% (w/v) nonfat dry milk and 5 mm NEM for 2 h at room temperature. Protein-glutathione adducts were detected with a mouse monoclonal anti-protein glutathione antibody (clone D8) (Virogen, Watertown, MA) and an HRP-conjugated anti-mouse secondary antibody. Chemiluminescent signal was detected using a ChemiDoc MP system (Bio-Rad). To monitor the extent of BiP cysteine oxidation, samples were treated with a 5-kDa maleimide-PEG (mal-PEG5K) (Laysan Bio Inc., Arab, AL), after small molecules were removed with a Bio-Spin P6 column (Bio-Rad) equilibrated with TNE buffer. Desalted, oxidized BiP was added to an equal volume of 2× sample buffer containing 10 mm mal-PEG5K diluted from a 0.1 m stock prepared in DMSO. Samples were incubated for 30 min at room temperature; 10% BME was added, and proteins were incubated for an additional 30 min at room temperature. Proteins were separated by SDS-PAGE and visualized with SYPRO Ruby stain (Life Technologies, Inc.).
ATPase activity and protein aggregation assays with glutathionylated full-length BiP proteins were completed essentially as described previously (9), except DTT was removed from all buffers, and BiP levels in the ATPase assay were increased to 1.5 μm, based on protein levels prior to desalting. Glutathionylated BiP used in the activity assays was prepared using diamide and GSH as described above, and prior to use in activity assays, small molecules were removed with a Bio-Spin P6 column equilibrated with TNE. ATPase activity assays for the BiP ATPase domain were completed with 2 μm BiP, 5 μm GST-Sec63J in ATPase buffer plus 0.25 m KCl, 50 μm cold ATP, and 0.1 μCi/μl [α-32P]ATP (PerkinElmer Life Sciences). Control ATPase reactions, containing all assay components but no recombinant BiP, showed no detectable ATP hydrolysis (data not shown).
Biotin-switch Assay
CSY316 and CSY319 containing pRS316 (19) or pCS452 were cultured overnight at 30 °C in SMM Raf, subcultured into SMM Gal the next morning, and grown for 6 h at 30 °C. Alternatively, mid-log cultures of CSY316 or CSY319 containing pRS316, CSY318, CSY694, or CSY695 grown in SMM at 30 °C were treated with 5 mm CHP or 10 mm diamide for 15 min prior to harvesting cells. When noted, dimedone was added to 0.1 m final concentration, from a 0.5 m dimedone stock prepared as a 0.5 m bis-tris and DMSO 1:1 (v/v) mixture. In the time course assays, medium from diamide-treated cells was removed by filtering; cells were resuspended in SMM containing 20 μg/ml cycloheximide, and cells were returned to 30 °C until the time of harvest. The biotin-switch assay was performed essentially as described previously (9) except GrxC was substituted for the previously used reductants (BME or sodium arsenite). Briefly, alkylated protein pellets from 10 A600 eq of cells were solubilized in a mixture of 20 μl of urea-containing cysteine modification buffer (0.1 m HEPES-NaOH, pH 7.4, 1% SDS, 10 mm diethylenetriaminepentaacetic acid, 6 m urea) and 300 μl of TE buffer (0.1 m Tris-HCl, 1 mm EDTA, pH 8.0). GrxC assay buffer (0.5 mm GSH, 1 mm NADPH, 0.25 units/ml glutathione reductase (Roche Applied Science, Switzerland)) (1 ml), and 60 μg of purified GrxC were added to the solubilized mixture, and samples were incubated for 15 min at 30 °C. After reduction, samples were quenched with trichloroacetic acid (TCA) and processed as described previously. Immunoblots were imaged and quantitated using a ChemiDoc MP system and associated ImageLab software (Bio-Rad).
Dimedone and DAz-2 Modification
CSY316 containing pRS316 was grown at 30 °C in SMM to mid-log at which time dimedone was added to a 0.1 m final concentration, from a 0.5 m dimedone stock prepared in a 0.5 m bis-tris and DMSO 1:1 (v/v) mixture. Control cells were treated with an equal volume of bis-tris/DMSO. After a 15-min incubation with dimedone, 5 mm CHP or 10 mm diamide was added, and cultures were incubated for an additional 15 min at 30 °C. Cells (10 A600 eq) were harvested by centrifugation, and pellets were flash-frozen in liquid nitrogen and stored at −80 °C. Cells were suspended in 40 μl of 10% TCA; zirconium beads were added, and cells were lysed in a FastPrep 24 instrument (MP Biomedical, Santa Ana, CA) with three 1-min pulses at speed 4 separated by 5-min ice rests. Samples were diluted with 1 ml of 10% TCA, and liquid was transferred to a new tube. Proteins were precipitated by centrifugation at 21,000 × g for 10 min at 4 °C, and pellets were washed once with ice-cold 5% TCA and once with ice-cold ethanol. Pellets were solubilized in 100 μl of urea-containing cysteine modification buffer, diluted with 1 ml of IP buffer (50 mm Tris-HCl, pH 7.4, 0.15 m NaCl, 1% Triton X-100), and incubated for 10 min at room temperature. Insoluble material was removed by centrifugation at 21,000 × g for 5 min at 4 °C, and 30 μl of 50% anti-FLAG M2 bead slurry (Sigma) was added to the soluble material. Samples were rotated at 4 °C for 1 h; beads were washed three times with IP buffer, and proteins solubilized in 2× sample buffer. Proteins were separated by SDS-PAGE, transferred to nitrocellulose, and probed with a rabbit polyclonal anti-cysteine sulfenic acid (anti-dimedone) antibody (catalogue number 07 -2139) (EMD Millipore) and an HRP-conjugated anti-rabbit IgG, or a mouse monoclonal anti-FLAG M2 antibody (catalogue number 200471) (Agilent Technologies) and an Alexa488-conjugated anti-mouse IgG. Chemiluminescent and fluorescent signals were detected using a ChemiDoc MP System (Bio-Rad). DAz-2 samples were processed as described previously (9). CSY316 and CSY319 containing pRS316 were grown to mid-log at 30 °C and treated with 5 mm CHP or 10 mm diamide for 15 min prior to harvest.
Glutathione Measurements
CSY316 and CSY319 containing pRS316 or pCS452 were cultured overnight at 30 °C in SMM Raf, subcultured into SMM Gal the next morning, and grown for 6 h at 30 °C. CSY316 and CSY319 containing pRS316 were grown in SMM at 30 °C to mid-log at which time cells were treated with 5 mm CHP or 10 mm diamide for 15 min. Cells (5 A600 eq) were harvested by centrifugation, and pellets were flash-frozen in liquid nitrogen and stored at −80 °C. Glutathione levels were measured using the 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB)-GSSG reductase recycling assay (23). Pellets were suspended in 40 μl of 1% 5-sulfosalicylic acid, and cells were lysed with zirconium beads in a FastPrep 24 instrument as described above. An additional 200 μl of 1% 5-sulfosalicylic acid was added, and samples were incubated on ice for 30 min, followed by centrifugation for 5 min at 21,000 × g, and soluble material was assayed for glutathione. Glutathione measurements were done in a 96-well microplate and read by a BioTek Synergy 2 reader. Total glutathione was measured by dilution of acidified lysate (20 μl) into 200 μl of assay mixture (0.2 m sodium phosphate, pH 7.5, 1 mm EDTA, 0.2 mm NADPH, 0.2 mm DTNB, and 0.1 unit/ml glutathione reductase). The change in absorbance at 405 nm was measured over 15 min. Standard curves were generated for each experiment using 0–20 μm GSSG solubilized in 1% 5-sulfosalicylic acid. Oxidized glutathione was measured by treating acidified lysate with 2-vinylpyridine, which modifies free thiols preventing reduced glutathione from being a glutathione reductase substrate. 2-Vinylpyridine was diluted with an equal volume of 25% triethanolamine, and the 2-vinylpyridine/triethanolamine mixture was added to lysates and GSSG standards at a ratio of 1 μl per 25 μl of sample, and samples were incubated at room temperature for 60 min prior to addition to the assay mixture.
Oxidation State of eroGFP-iE
CSY5 and CSY275 containing pCS951 were grown to mid-log in SMM and treated with 5 mm DTT, 10 mm diamide, or 5 mm CHP for 15 min at 30 °C prior to harvesting. CSY5 and CSY275 containing pCS951 and pCS531 (or empty vector, pRS315) were grown at 30 °C in SMM Raf overnight, subcultured into SMM Gal the next morning, and grown for 6 h prior to harvesting. Cells were suspended in 10% TCA; zirconium beads were added, and cells were lysed by agitation in a FastPrep 24 instrument. Protein pellets (from 3 A600 of harvested cells) were suspended in 25 μl of sample buffer with 0.1 m Tris-HCl, pH 8.0, and 15 mm 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) (Thermo Fisher Scientific), and samples were rotated for 30 min at room temperature, and unreacted AMS was quenched with 10% BME for 5 min. Samples were analyzed by immunoblotting with a mouse monoclonal anti-enhanced GFP antibody (catalogue 632569) (Clontech) and Alexa488-conjugated anti-mouse IgG, and fluorescent signal was detected using the ChemiDoc MP system.
Microscopy
An Eclipse E600 microscope (Nikon, Tokyo, Japan) equipped with a Clara Interline CCD camera (Andor Technology, Belfast, UK) and NIS-Elements Advanced Research (NIS-Elements AR) imaging software was used for fluorescence and differential interference contrast microscopy. CSY5 cells containing pCS951 were grown to mid-log phase in SMM, and cells were harvested and suspended in 10 mm Tris-HCl, pH 7.4, 50 mm NaCl. Cells were live imaged at room temperature using a ×60 oil immersion lens with a 1.4 numerical aperture and a fluorescein isothiocyanate filter and a differential interference contrast H objective. Autoquant X software (Media Cybernetics, Rockville, MD) was used to perform a three-dimensional deconvolution of a series of z-stacks (3.5 μm total distance with a step size of 0.5 μm).
Growth Curves
CSY275 cells containing pRS315, pCS681, pCS685, pCS802, pCS687, pCS688, or pCS750 plasmids, or CSY318, CSY693, CSY694, CSY695, CSY696, and CSY697 cells, were grown overnight at 30 °C in SMM and subcultured the next morning to an A600 of 0.2. To monitor growth in the presence of diamide, 1.1 or 1.2 mm diamide was added to the cultures at the time of subculturing. To monitor growth post-diamide removal, cultures were returned to 30 °C, and when the cells reached mid-log, 5 mm diamide was added. Cells were cultured for 1 h in diamide, at which time the cultures were filtered, and cells were suspended in fresh SMM at equal cell densities for all cultures. Cultures were sampled hourly for A600 measurement.
Competition Assay
Strains used for the yeast growth competition assays are outlined in Tables 1 and 2. Plasmid-covered kar2Δ strains with URA3-marked plasmids were generated by transformation of a diploid MATa/α ura3-52/ura3-52 leu2-3,112/leu2-3,112 KAR2/kar2Δ::KanMX strain with the indicated plasmid, followed by sporulation and selection of MATa Ura+ KanMX+ segregants. Plasmid-covered kar2Δ strains with LEU2-marked plasmids were generated by transformation of CSY214 with the indicated plasmid, followed by counter-selection of pCS623 on SMM with 5-fluoroorotic acid. Cells containing a URA3- or LEU2-marked plasmid were grown to mid-log phase in selective media. Co-cultures of the desired pairs of strains (one containing a URA3-marked and one containing a LEU2-marked plasmid; see Tables 1 and 2) were prepared at a 1:1 ratio in SMM (containing both leucine and uracil) at a final A600 of 0.1. Co-cultures were incubated at 30 °C and were diluted every 7–12 h to maintain cells in an exponential growth phase. At the indicated times, cells (1 × 10−4 A600 eq) were plated onto SMM-ura and SMM-leu plates. After 2 days of growth at 30 °C, the relative proportion of URA3 and LEU2 plasmid-containing strains in each co-culture was determined by counting the colony-forming units (cfu) on each selective plate using the ImageQuant TL software package (GE Healthcare). The fraction of the co-culture containing each plasmid was expressed relative to the total cfu for each time point.
TABLE 1.
Yeast strain pairs used for growth competition assays described in Figs. 8, D and F, and 9, A and C
Each row indicates a co-cultured strain pair. Shaded rows highlight pairs of co-cultures with the same two plasmid-borne BiP alleles on reciprocally marked plasmids, which are depicted together in a single graph with the indicated symbols.
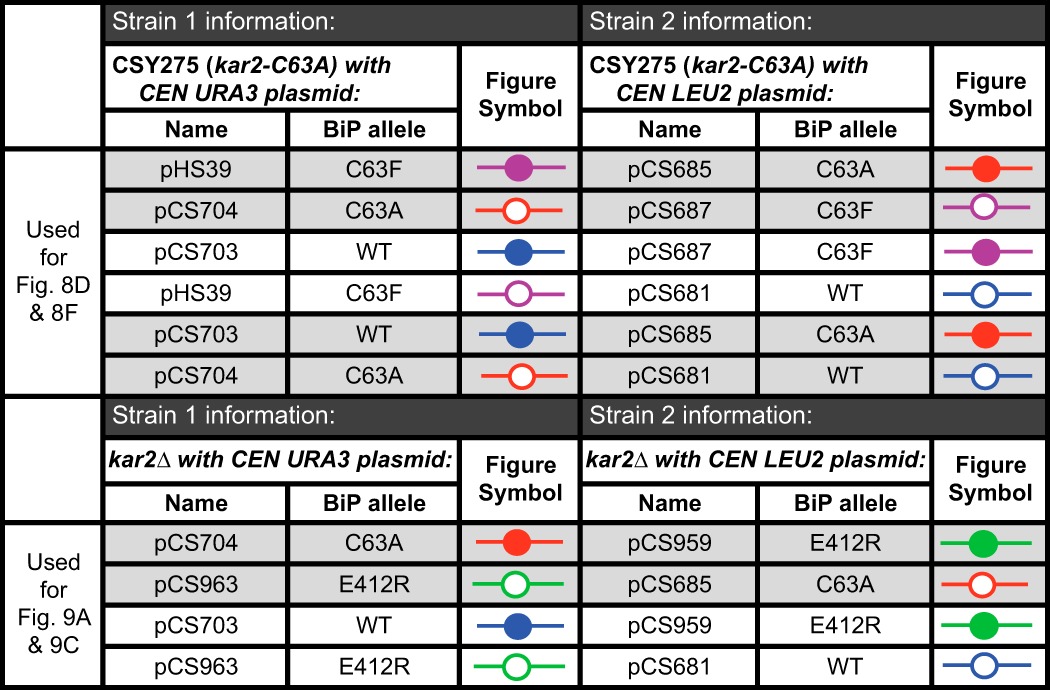
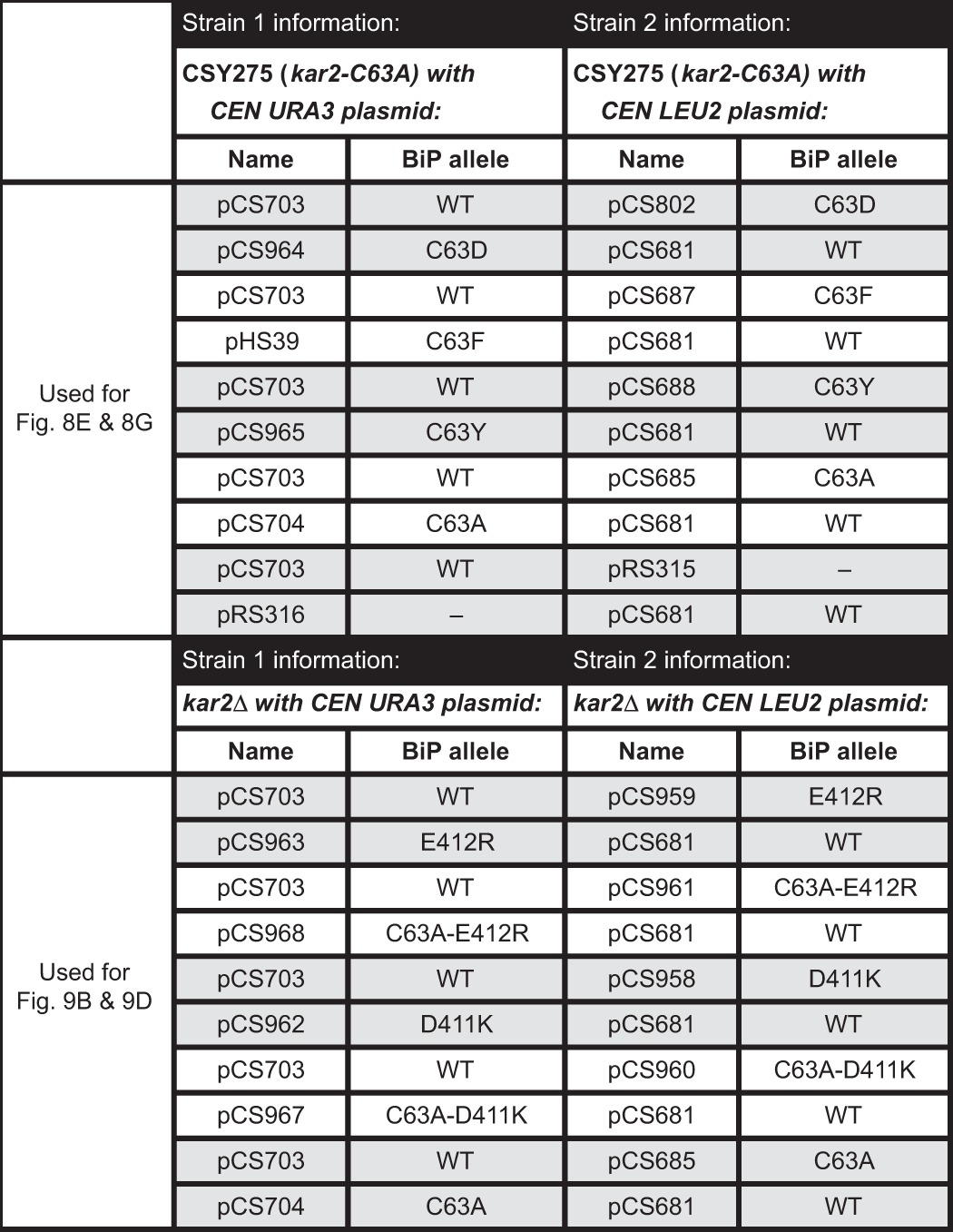
TABLE 2.
Yeast strain pairs used for growth competition assays described in Figs. 8, E and G, and 9, B and D
Each row represents a co-cultured strain pair. Shaded rows highlight pairs of co-cultures with the same two plasmid-borne BiP alleles on reciprocally marked plasmids; data from these paired strains were averaged and depicted in the indicated figure panel.
Determination of Cysteine pKa
Purified BiP ATPase domain proteins were used to determine the Cys-63 pKa value by reaction with DTNB. Protein (21 μm) was reacted with 30 μm DTNB in 80 μl total of TN buffer, which was adjusted to a range of pH values between 5.8 and 9.2 using HCl or NaOH as appropriate. The pH-dependent absorbance change at 412 nm was monitored over 10 min with 10-s readings. Data were fit to a non-linear first-order reaction to extrapolate the maximal absorbance at 412 nm (2-nitro-5-thiobenzoic (TNB) acid release) at each pH; maximal release of TNB upon oxidation of BiP should correlate with the proportion of BiP with a deprotonated thiol at each pH. The TNB release values were used in a second-order fit to obtain the Cys-63 pKa using Equation 1,
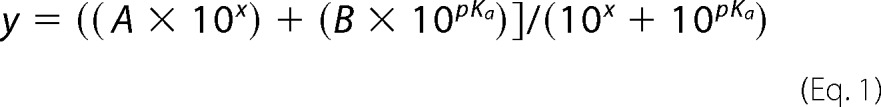 |
where y is the absorbance value for TNB release; x is the pH value, and A and B are the limiting values of y (the plateaus) at high and low pH, respectively.
Results
BiP Is Glutathionylated during Oxidative ER Stress
We employed the biotin-switch assay to determine whether yeast BiP (Kar2) is glutathionylated in cells in response to increased ER oxidation (oxidative stress). In its general form, the biotin-switch assay involves three steps as follows: 1) alkylation of free thiols in cell lysates; 2) reduction of oxidized cysteines; and 3) tagging of the uncovered sulfhydryl groups of initially oxidized thiols with biotin-maleimide (24). To selectively detect glutathionylated proteins, a recombinant mutant E. coli glutaredoxin (GrxC-C14S-C65Y), engineered for enhanced and selective deglutathionylation of glutathione-protein mixed disulfides, was used as the reductant in step 2 (Fig. 1A) (16, 25, 26). To induce oxidative stress within the ER, we overexpressed a hyperactive allele of the Ero1 protein (Ero1*), which we have characterized previously as an effective means to facilitate ER oxidative stress (7, 9). Cells overproducing Ero1* were lysed and treated with the thiol-alkylating reagent N-ethylmaleimide (NEM). Glutathionylated proteins were reduced by GrxC, and thiols of the initially glutathionylated proteins were labeled with maleimide-biotin. FLAG-tagged BiP was isolated from the lysates with anti-FLAG beads and separated by SDS-PAGE, and biotinylated BiP was detected by Western blotting. Cells overexpressing Ero1* showed a 4-fold increase in the fraction of biotinylated BiP relative to non-stressed cells (Fig. 1B, lanes 1 and 2), suggesting an enhanced level of glutathionylated BiP during increased ER oxidation. A BiP-biotin signal was absent in a strain containing a cysteine-less variant of BiP (Fig. 1A, lane 4), confirming biotinylation of the BiP cysteine.
FIGURE 1.

BiP is glutathionylated during oxidizing ER conditions. A, outline of the biotin-switch procedure used in B–D. B, lysates from cells overexpressing a galactose-inducible hyperactive Ero1 allele (Ero1*) were assayed for glutathionylated BiP using the biotin-switch protocol with a mutant GrxC as the reductant. A catalytically inactive GrxC was used for the no GrxC control reactions. BiP was enriched by immunoprecipitation, and total and glutathionylated BiP were identified by Western blotting with anti-BiP serum or an avidin probe. The relative proportion of BiP with an oxidized cysteine was determined by comparing the intensity of the BiP-biotin signal relative to the total level of BiP. The signal ratio was set to 1.0 for wild-type cells not exposed to any stressor. C and D, cells were treated with 5 mm CHP or 10 mm diamide for 15 min, and lysates were subject to the biotin-switch protocol as outlined in B.
To ensure that the BiP-biotin signal reflected GrxC-mediated deglutathionylation activity, we also employed a catalytically dead GrxC in the biotin-switch assay (GrxC-C11S-C14S-C65Y). Lysate treated with catalytically dead GrxC showed a decreased biotinylated BiP signal, relative to that observed with catalytically active GrxC, confirming a GrxC-dependent BiP-biotin signal reflecting glutathionylation of BiP (Fig. 1B, lane 3 versus 2). However, a measurable, ∼2-fold, BiP-biotin signal was also observed in samples prepared with the catalytically dead GrxC (Fig. 1B, lane 3). GrxC activity is facilitated by a regeneration mixture of glutathione reductase, reduced glutathione (GSH), and NADPH, and all biotin-switch samples were completed in regeneration mixture plus the indicated GrxC protein (catalytically active or inactive GrxC). The BiP-biotin signal observed with the catalytically dead GrxC suggests the regeneration mixture allows for some reduction (and biotinylation) of BiP. It is possible some of the GrxC-independent signal may reflect reduction of protein-glutathione disulfides by glutathione reductase; however, glutathione disulfide (GSSG) (not protein-glutathione disulfide) is the characterized glutathione reductase substrate. The GrxC-independent BiP-biotin signal may also indicate GSH-catalyzed reduction of non-glutathione adducts, such as sulfenic acid previously identified on BiP under similar growth conditions (9). Together, these data demonstrate an enhanced glutathionylation of BiP during stress; these data do not exclude the presence of additional modifications on BiP.
Glutathionylation of the BiP cysteine was also detected in cells exposed to exogenous oxidant (Fig. 1, C and D). We have shown previously that addition of 5 mm CHP to cells impacts the ER redox environment and induces sulfenylation of BiP (9). We observe also addition of 5 mm CHP to cells for 15 min resulted in the recovery of biotinylated BiP using the biotin-switch assay with GrxC, indicative of glutathionylation of the BiP cysteine (Fig. 1C, lane 2). Prior studies have shown treatment of yeast cells with a wide range of diamide concentrations (1–20 mm) for 30 min alters the redox potential inside the cell, including creating a more oxidizing cytoplasmic, mitochondrial, and peroxisomal redox environment (27). We observed treatment with an intermediate amount of diamide (10 mm diamide) for 15 min also alters the ER redox status, resulting in glutathionylation of the BiP cysteine (Fig. 1D, lane 2). Biotinylated BiP was dependent on both the presence of the BiP cysteine and oxidant treatment (Fig. 1, C and D). Overall, a 3–4-fold induction of glutathionylation was observed upon increased ER oxidation; approximately half of the biotinylation signal observed upon treatment with the GrxC mixture is independent of GrxC activity (Fig. 1, B–D).
BiP Glutathionylation Can Be Mediated by Oxidized or Reduced Glutathione
Several pathways for protein glutathionylation have been proposed (Fig. 2A). Reduced glutathione can modify cysteines primed by other oxidants; for example, alterations in cellular reactive oxygen species (ROS) can promote interaction of thiols with GSH through a protein-sulfenic acid intermediate (Fig. 2A, pathway b). Similarly, oxidation of GSH by ROS can result in oxidized glutathione adducts (e.g. glutathione sulfenic acid or glutathione disulfide S-oxide) that can react directly with protein thiols to form protein-glutathione adducts (Fig. 2A, pathway c). Alternatively, under extreme stress conditions, an accumulation of oxidized glutathione (GSSG) may trigger protein glutathionylation via thiol-disulfide exchange between a reduced protein thiol and GSSG (Fig. 2A, pathway a). To investigate whether BiP is susceptible to multiple pathways for glutathionylation, we incubated recombinant BiP with the following: (i) a molar excess of GSSG; (ii) a mixture of GSH and the thiol-oxidant diamide; or (iii) a mixture of GSH and peroxide (CHP) or bleach (NaOCl), which are ROS known to oxidize thiols to form sulfenic acid or chloride adducts, respectively (14, 28). Oxidation of the BiP cysteine was monitored by addition of mal-PEG5K. Oxidized BiP will not react with mal-PEG5K, due to unavailability of the cysteine; reduced BiP will react with mal-PEG5K, resulting in a slower mobility on SDS-PAGE. All four glutathione mixtures resulted in BiP oxidation, as evident by the faster migrating BiP species observed post-oxidant incubation (Fig. 2B). To confirm the inaccessibility of the BiP cysteine to modification with mal-PEG5K reflected oxidation of the BiP cysteine with glutathione, samples were probed by Western blotting with an anti-protein glutathione (anti-PSSG) antibody. All samples showed a positive signal by immunoblotting that was dependent on both oxidant addition and the presence of the BiP cysteine (Fig. 2C). The anti-PSSG antibody showed some signal that did not coincide with the major recombinant BiP protein band (detected by Ponceau S) (Fig. 2C); we anticipate these bands are glutathionylated bacterial proteins present in the recombinant BiP samples and/or degradation products of glutathionylated recombinant BiP, consistent with the absence for some of these smaller bands in the lanes containing the BiP-C63A mutant. Mass spectrometry of the major recombinant BiP species post-incubation with GSSG (the equivalent to the ∼72-kDa band in Fig. 2C) further confirmed the Cys-63 residue was modified by glutathione; two cysteine-containing peptides with a 305-Da mass increase consistent with a glutathione adduct were identified by LC-MS/MS and confirmed by combined collision-induced dissociation-electron transfer dissociation (CID-ETD) analysis (data not shown).
FIGURE 2.

BiP is susceptible to oxidation by reduced or oxidized glutathione. A, potential routes for protein glutathionylation (P-SSG) formation, as supported by biochemical and cellular studies (63, 64). Asterisks denote especially labile species. ROS, reactive oxygen species. B, recombinant BiP was incubated with a molar excess of oxidized glutathione (GSSG), or reduced glutathione (GSH) plus diamide, CHP, or bleach (NaOCl). Small molecules were removed by desalting, and proteins were reacted with mal-PEG5K prior to separation by SDS-PAGE and visualization with a fluorescent protein strain. Oxidized BiP denotes protein unmodified by mal-PEG5K due to the oxidation of BiP by the glutathione mixture. C, recombinant wild-type BiP or a BiP-C63A mutant was treated with oxidants as in B, separated by non-reducing SDS-PAGE, and transferred to nitrocellulose. Total BiP and glutathione-BiP adducts were detected by Ponceau S stain and anti-PSSG antibody, respectively. Note a high molecular weight band, consistent with an intermolecular BiP disulfide-bonded species, was also observed after oxidant treatment (asterisk). D, dimedone addition limits the recovery of glutathionylated BiP from peroxide-treated cells. Cells were pre-incubated with dimedone (0.1 m) for 15 min prior to the addition of 5 mm CHP. After 15 min of CHP treatment, cells were harvested, and lysates were prepared using the biotin-switch protocol. The signal ratio was set to 1.0 for cells containing wild-type BiP not exposed to any stressor. E, cells pre-treated with 0.1 m dimedone were incubated with the oxidants CHP (5 mm) or diamide (10 mm) for 15 min. BiP was enriched by immunoprecipitation; proteins were separated by SDS-PAGE, and BiP modification by dimedone was monitored using an anti-dimedone antibody. F, cell lysates from cells treated with CHP (5 mm) or diamide (10 mm) for 15 min were incubated with the dimedone-analog DAz-2. BiP was immunoprecipitated, and a Staudinger ligation reaction with phosphine-biotin was performed, allowing detection of DAz-2 with an avidin probe.
Ponceau S staining of the nitrocellulose from the glutathione-treated BiP samples prior to immunoblotting revealed a minor high molecular weight protein species in wild-type BiP samples treated with oxidant (Fig. 2C, asterisks). This band did not appear upon oxidant treatment of the cysteine-less BiP (Fig. 2C), was not recognized by the anti-PSSG antibody (Fig. 2C), and was resolved upon addition of reductant (data not shown), suggesting the presence of a disulfide bond between BiP and another protein or molecule larger than glutathione. Notably, the high molecular weight species is consistent in size with a BiP dimer. Given that purified recombinant BiP was used for these experiments, and is the major protein species present in the protein samples, we anticipate this band reflects a BiP-BiP disulfide-bonded species. Consistent with this speculation, mass spectrometry analysis of this higher molecular weight band confirmed that recombinant BiP was the major protein component of this band (supplemental Table S1). However, several bacterial proteins (expected contaminants from the recombinant protein preparation) were also identified in this high molecular weight band (supplemental Table S1), and we cannot exclude that this band represents a disulfide bond between BiP and a bacterial protein contaminant from the recombinant BiP purification process. The relevance of this potential BiP-BiP species to oxidation of BiP by glutathione in living cells is currently unclear.
BiP's susceptibility to glutathionylation by several oxidants in vitro led us to explore how glutathionylation of BiP is mediated in cells. We reported previously the addition of exogenous peroxide (CHP) to cells results in sulfenylated BiP (9). To determine whether BiP glutathionylation observed upon CHP treatment (Fig. 1C) is mediated through a sulfenic acid adduct (Fig. 2A, pathway b), we added dimedone to cells prior to assaying for glutathionylation with the biotin-switch protocol. Dimedone specifically reacts with sulfenic acid to form a covalent bond, which prevents reactivity with other oxidants (e.g. GSH). We observed a decrease in glutathionylated BiP upon dimedone addition, suggesting sulfenic acid primes BiP for glutathione modification (Fig. 2D, lane 3 versus 2). Note, the level of BiP glutathionylation induced upon CHP treatment (reflected by the BiP-biotin signal) was decreased relative to that observed in Fig. 1C (a 1.5-fold versus 3.1-fold increase in BiP-biotin levels). We anticipate the lower BiP-biotin signal upon CHP treatment here relates to the high level of DMSO (10%) added to cells during the dimedone (and mock) treatment, which likely impacts cell viability and the response to CHP. The modest 1.5-fold signal difference in glutathionylation upon CHP treatment with or without dimedone addition (Fig. 2D, lanes 2 versus 3) was small yet reproducible; signals averaged over four independent experiments showed an average 1.7-fold increase in BiP glutathionylation in response to CHP (equivalent of Fig. 2D, lane 2 versus 1) and an average 1.4-fold reduction in signal when cells were pretreated with dimedone prior to CHP (equivalent of Fig. 2D, lane 3 versus lane 2) (data not shown). To confirm that the effect of dimedone was due to dimedone addition to sulfenylated BiP, we probed for the BiP-dimedone adduct with an anti-dimedone antibody. A BiP-dimedone signal was observed that was dependent both on dimedone addition and CHP treatment (Fig. 2E). A BiP sulfenic acid adduct induced upon CHP addition was observed also with the dimedone-based probe DAz-2, which can be visualized using an avidin probe after Staudinger ligation of phosphine biotin to the azido group of DAz-2 (Fig. 2F, lane 3 versus 1) (29). Collectively these data are consistent with a pathway for glutathionylation of BiP in cells through a sulfenic acid intermediate. These data also lend further evidence for the sulfenylation of BiP upon CHP treatment (9).
A BiP-dimedone adduct was not observed in cells treated with the thiol-specific oxidant diamide, suggesting that diamide-induced glutathionylation in cells may occur through a sulfenic acid-independent mechanism. Pretreatment of cells with dimedone prior to diamide addition did not result in a detectable BiP-dimedone adduct using the anti-dimedone antibody (Fig. 2E). Similarly, no induction of a BiP sulfenic acid adduct by diamide was observed with the DAz-2 probe (Fig. 2F, lane 2 versus lane 1). A modest cysteine-dependent BiP-DAz-2 signal was observed in untreated cells, which is consistent with a basal level of sulfenylated BiP in cells, as reported previously (Fig. 2F, lane 1) (9). The basal BiP sulfenic acid signal observed with the DAz-2 was not enhanced upon diamide addition (Fig. 2F, lane 1 versus lane 2). Of note, the basal BiP sulfenic acid signal was barely detectable with the anti-dimedone antibody (Fig. 2E, lane 1); the stronger signal with the DAz-2 probe (Fig. 2F, lane 1) underscores an advantage in our system for dimedone detection using an avidin probe and a biotin handle, relative to recognition of dimedone using the anti-dimedone antibody. The absence of an increase in BiP sulfenylation upon diamide treatment is consistent with a previous study, which observed protein sulfenates are not elevated in rat ventricular myocytes treated with diamide (30).
We anticipate diamide facilitates BiP glutathionylation by increasing the levels of cellular GSSG. Consistent with this idea, we observed a 10-fold increase in the cellular GSSG/GSH ratio in diamide-treated cells relative to untreated cells (Fig. 3A). CHP treatment and overproduction of Ero1* both also impacted the cellular GSSG/GSH ratio, albeit to a lesser extent than diamide (Fig. 3, A and B). To confirm alterations observed in the total cellular GSSG/GSH ratio reflected changes in the ER glutathione pool, we utilized a redox-sensitive GFP (roGFP) sensor targeted to the ER lumen. roGFP contains a pair of engineered cysteines, and the redox state of these cysteines (thiol or disulfide bonded) is influenced by the cellular GSH-GSSG level. Of note, the response of roGFP to glutathione is facilitated by endogenous glutaredoxins (31), and two glutaredoxins are present in the S. cerevisiae ER (32). A roGFP sensor targeted to the ER of yeast (eroGFP) has been described previously (20); however, at steady state this eroGFP is fully oxidized, which limits its utility as a tool to monitor for further oxidation of the glutathione pool upon oxidant addition. Therefore, we generated an eroGFP variant, eroGFP-iE, which contains a glutamic acid insertion that confers a lower redox potential (33). Fluorescence microscopy showed the same characteristic ER localization pattern for eroGFP-iE as observed previously for eroGFP (20), suggesting the additional amino acid changes in eroGFP-iE did not disrupt the folding or targeting of eroGFP to the ER (Fig. 3C). The redox state of roGFP can be followed by monitoring formation of the roGFP disulfide bond or by taking advantage of the different fluorescence properties of the oxidized and reduced roGFP. Consistent with the literature, the eroGFP-iE reporter showed a weak fluorescent signal that limited our ability to generate high quality images of the spectral shifts that occur in roGFP upon increased cellular oxidation (34). Thus, we followed oxidation of the roGFP disulfide bond by taking advantage of the slower mobility of reduced eroGFP-iE after treatment with the thiol-modifying agent AMS. The lower redox potential for eroGFP-iE was evident based on its partial oxidation under steady-state growth conditions (Fig. 3, D and E). Notably, addition of exogenous diamide resulted in a more oxidized (faster migrating) eroGFP-iE species, consistent with an increase in the GSSG/GSH ratio in the ER lumen (Fig. 3D). Conversely, addition of peroxide (CHP) to cells had no notable impact on the eroGFP-iE redox state (Fig. 3D), suggesting that although CHP impacts the cellular glutathione redox state (Fig. 3A), the primary effect of CHP is likely on the cytosolic, not the ER, glutathione pool. Overproduction of Ero1* has previously been suggested to increase ER GSSG levels (7); here, we also observed a shift in eroGFP-iE to a more oxidized state upon Ero1* overproduction (Fig. 3E). Altogether, these data are consistent with increased levels of GSSG being the principal mediator for glutathionylation of BiP upon diamide treatment. We anticipate CHP and Ero1* can both facilitate glutathionylation of BiP through a BiP-sulfenic acid species; the observed shifts in the total oxidized glutathione level and the redox state of eroGFP-iE sensor upon Ero1* treatment suggests the possibility that Ero1* overexpression may also impact BiP glutathionylation through GSSG.
FIGURE 3.

Diamide treatment alters the GSSG/GSH ratio in cells. A, wild-type or a cysteine-less BiP (kar2-C63A) yeast strain was treated with CHP (5 mm) or diamide (10 mm) for 15 min, and lysates were analyzed for intracellular glutathione levels. Glutathione levels are plotted as a ratio of GSSG/GSH. B, wild-type and kar2-C63A strains, containing a plasmid-borne ERO1* allele or an empty vector, were grown for 6 h in the presence of galactose (to induce Ero1*), and intracellular glutathione levels were analyzed as in A. For each treatment condition in A and B, data are plotted from four independent experiments ± S.E. C, representative fluorescent microscopy images of wild-type yeast expressing eroGFP-iE. D, wild-type and kar2-C63A yeast with the eroGFP-iE reporter were treated with DTT (5 mm), diamide (10 mm), or CHP (5 mm) for 15 min. Lysates were treated with the thiol-modifying agent AMS; samples were resolved by SDS-PAGE, and eroGFP-iE was visualized by immunodetection with an anti-GFP antibody. DTT treatment served as a mobility standard for reduced eroGFP-iE. E, wild-type and kar2-C63A strains, containing a plasmid-borne ERO1* allele, or an empty vector, and the eroGFP-iE reporter were harvested after 6 h of growth in the presence of galactose. Lysates were processed and eroGFP-iE was visualized as in D. The results from D and E were reproduced in two independent assays; the smear, which extends into the reduced probe mobility range, observed here for eroGFP-iE in the kar2-C63A strain upon diamide or Ero1* treatment was not observed in the independent replicates. F, recombinant BiP was reacted with a molar excess of glutathione. Total glutathione levels were kept constant, whereas the ratio of oxidized and reduced glutathione was varied. Glutathione was removed by desalting, and proteins were reacted with mal-PEG5K prior to separation by SDS-PAGE and visualization with a with a fluorescent protein strain. DIC, differential interference contrast.
To further probe the potential impact of alterations in the ER glutathione redox state on BiP oxidation, we monitored the susceptibility of BiP to oxidation in vitro dependent on the GSSG/GSH ratio. Our measurements of the total cellular glutathione pool show a steady-state GSSG/GSH ratio of 1:20 to 1:40, dependent on whether the cells were grown in glucose (Fig. 3A) or galactose (Fig. 3B) medium; these measurements reflect the total cellular glutathione content, after the cells are lysed and all intracellular glutathione pools are mixed together. Organelle-specific measurements suggest distinct GSSG/GSH ratios in different cellular compartments. Specifically, the resting ER GSSG/GSH ratio has been measured as 1:1 or 1:3, whereas the cytoplasmic ratio has been placed at 1:30 to 1:100 (15). To mimic the ER redox environment, we incubated BiP in a 1:3 and 1:1 mixture of GSSG/GSH, conditions similar to those measured within a non-stressed ER (15). Under these conditions, we observed the majority of BiP remained reduced, consistent with a lack of extensive glutathionylation of BiP under resting ER redox conditions (Fig. 3F). Notably, shifting the ratio of GSSG/GSH in favor of GSSG by even 3-fold (3:1) increased the recovery of oxidized BiP (Fig. 3F), suggesting that subtle increases in the cellular GSSG level may facilitate some BiP glutathionylation. This modest increase is in keeping with the observed increase in the total GSSG/GSH ratio we observed upon Ero1* induction (Fig. 3B). Unlike the addition of exogenous oxidants, overproduction of Ero1* is anticipated to have its primary impact on the ER glutathione pool, and we expect that changes in the total cellular glutathione pool observed upon Ero1* overexpression principally reflect changes in the ER lumen. Ero1* overproduction showed an ∼4-fold increase in the GSSG/GSH ratio (Fig. 3B), which would be analogous to a new ER-specific GSSG/GSH ratio of 4:1.
Strains containing wild-type or the cysteine-less BiP showed similar increases in the GSSG/GSH ratio upon oxidant treatment, suggesting that BiP oxidation does not mediate the observed increase in the GSSG/GSH ratio (Fig. 3). However, it worth noting that an intriguing trend toward a slightly increased mean GSSG/GSH ratio in the cysteine-less BiP strain (relative to the wild-type strain) was observed under all three oxidant treatments (Fig. 3, A and B). A larger dataset and more robust analysis will be required to determine whether a more subtle level of feedback regulation exists, wherein BiP oxidation limits the increase in the GSSG/GSH ratio.
Glutathionylation Decouples BiP ATPase and Peptide Binding Activities
We previously reported oxidation of the BiP cysteine by peroxide decouples the allostery between the ATPase domain and peptide-binding domain, abolishing BiP's ATPase activity yet enhancing BiP's ability to prevent protein aggregation (9). We suggest BiP glutathionylation results in a similar alteration in BiP activities. Similar to sulfenylated BiP, recombinant glutathionylated BiP showed no measurable ATPase activity (Fig. 4A). A cysteine-less BiP mutant treated with a GSH and diamide mixture showed a similar ATPase activity to that observed for untreated wild-type BiP, demonstrating that the effect of glutathione was linked to cysteine modification (Fig. 4A). Similar to what we observed for sulfenylated BiP, glutathionylated BiP maintained its chaperone activity. Chemically denatured rhodanese and IgY are prone to aggregate upon dilution from denaturant; aggregation can be followed spectroscopically as an increase in light scattering over time. Glutathionylated BiP lessened the observed light scattering associated with rhodanese and IgY aggregation; specifically, glutathionylated BiP was more effective than wild-type BiP in minimizing aggregation (Fig. 4, B and C). Relative to wild-type BiP, treatment of a BiP-C63A mutant with a diamide/GSH mixture did not appreciably enhance its ability to lessen aggregation of either rhodanese or IgY (Fig. 4, B and C), suggesting that the enhanced capacity to limit aggregation is linked to oxidation of the BiP cysteine. Consistent with our observations, glutathionylation of the mammalian cytosolic Hsp70 family member Hsc70 has also been shown to augment chaperone activity (17).
FIGURE 4.
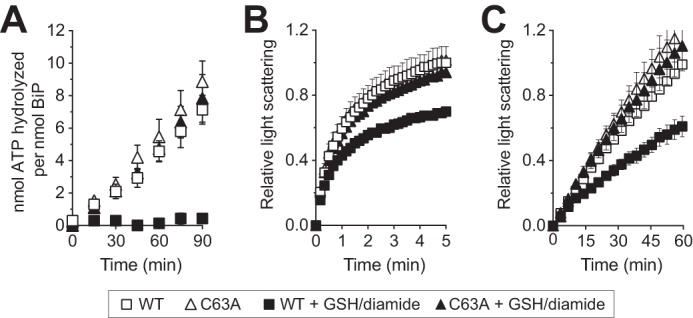
Glutathionylation alters BiP ATPase and peptide binding activities. Glutathionylated BiP was prepared by incubation of recombinant BiP with diamide and GSH. A, ATP hydrolysis was assessed by determining the fraction of [α-32P]ATP converted to [α-32P]ADP. Data represent the means ± S.E. of three independent assays. B and C, ability of BiP to suppress polypeptide aggregation was measured by following light scattering of aggregation-prone polypeptides. Denatured rhodanese (B) was diluted away from denaturant into a solution containing a 4-fold molar excess of BiP; rhodanese aggregation was followed by monitoring light scattering at 320 nm. Denatured IgY (C) was diluted into a non-denaturing solution containing an equimolar amount of BiP and heated to 45 °C; IgY aggregation was monitored by following light scattering at 360 nm. Data in B and C represent the mean ± S.E. for two and three independent assays, respectively.
BiP Glutathionylation Correlates with Enhanced Cell Protection during Oxidative Stress
We previously suggested that BiP oxidation enhances cell survival during oxidative stress; we showed addition of an exogenous BiP allele that phenotypically mimics BiP oxidation (e.g. BiP-C63F) facilitates cell survival of a yeast strain sensitized to oxidative stress (a strain with BiP-C63A as the only cellular BiP) (9). To provide more direct evidence connecting the physical oxidation of the BiP cysteine and protection against oxidative ER stress, we generated BiP mutants that showed greater susceptibility to oxidation by glutathione. We asked whether these alleles further enhance cell survival beyond the increased cell viability observed with oxidized wild-type BiP.
Redox active cysteines are often flanked in space by basic amino acid residues, which increase cysteine reactivity by lowering the pKa value of the cysteine sulfhydryl group and increasing the fraction of cellular protein with a deprotonated reactive cysteine thiol. Conversely, negative charges from acidic amino acid side chains electrostatically disfavor a depressed pKa. BiP contains two acidic residues in close proximity to the redox active BiP cysteine; the Cβ atoms of the BiP Asp-411 and Glu-412 side chains are located 6.6 and 5.8 Å, respectively, from the Cβ atom of BiP Cys-63 (Fig. 5A). We reasoned that mutation of these acidic residues should decrease the BiP cysteine pKa, rendering BiP more susceptible to oxidation. Consistent with our expectation, the charge reversal mutants BiP-D411K and BiP-E412R decreased the apparent BiP cysteine pKa to 6.7 ± 0.1 and 6.8 ± 0.2, respectively, relative to a measured pKa for wild-type BiP of 7.6 ± 0.1 (Fig. 5B). Notably, a cysteine residue unaffected by its environment is anticipated to have a pKa value of ∼8.5–9.0, and the lower pKa value observed for wild-type BiP is consistent with a protein cysteine more susceptible to oxidation. Importantly, the lowered pKa value of BiP-D411K and BiP-E412R correlated with an increased modification of the BiP cysteine by glutathione in vitro and in vivo. The ATPase domain from the mutants BiP-D411K or BiP-E412R was more susceptible to oxidation by GSSG in vitro relative to wild-type BiP (Fig. 5C). Note, the higher salt buffer conditions used in these experiments likely accounts for the less pronounced oxidation of the ATPase domain of wild-type BiP in these experiments (relative to the oxidation observed in Fig. 3F); higher salt was used to maintain solubility of the BiP D411K and E412R mutants. Using the biotin-switch protocol, we observed an enhanced level of glutathionylation for both BiP-D411K and BiP-E412R in vivo; a 2-fold increase in glutathionylated BiP was recovered from diamide-treated cells containing the BiP mutants relative to the cells containing wild-type BiP (Fig. 5D).
FIGURE 5.

BiP mutants with a lowered pKa are more susceptible to oxidation by glutathione. A, ribbon diagram of the yeast BiP ATPase domain in complex with Mg-ADP (Protein Data Bank code 3QFU) (65). Cys-63, Asp-411, Glu-412, and ADP are shown as sticks. Magnesium is represented as an orange sphere. B, pH dependence of the reactivity of BiP Cys-63 was measured using DTNB. TNB released was followed at 412 nm at different pH values. Fitted data result in a pKa of 7.6 for wild-type BiP, which was lowered to a pKa of 6.8 and 6.7 for the BiP-E412R and BiP-D411K mutants, respectively. Data show an R2 value above 0.95 for the fit, and the error in the fit is reported as S.E. C, recombinant BiP (ATPase domain; 31 μm) was incubated for 30 min at 30 °C with increasing amounts of GSSG. Glutathione was removed by desalting, and proteins were reacted with mal-PEG5K prior to separation by SDS-PAGE and visualization with a fluorescent protein strain. D, yeast strains deleted for endogenous BiP (kar2Δ) containing plasmids encoding FLAG-tagged wild-type BiP, BiP-E412R, or BiP-D411K were treated with 5 mm diamide for 15 min, and lysates were prepared using the biotin-switch protocol with GrxC as the reductant. The signal ratio was set to 1.0 for the cells containing wild-type BiP. E, ATP hydrolysis was assessed by determining the fraction of [α-32P]ATP converted to [α-32P]ADP. Data represent the means ± S.E. of three independent assays.
Given the proximity of Asp-411 and Glu-412 to the site of nucleotide binding, we also determined whether altering these residues impacted BiP ATPase activity. The BiP-E412R mutant showed an ATP hydrolysis rate similar to that observed for wild-type BiP (Fig. 5E). In contrast, the BiP-D411K mutant exhibited a decreased ATP hydrolysis rate relative to both wild-type and BiP-E412R proteins, demonstrating this mutation affects more than just the BiP Cys-63 pKa (Fig. 5E). We have shown that wild-type BiP and a BiP-C63A mutant both show similar ATPase activity under non-oxidizing conditions (Figs. 4A and 5E) (9). Consistent with these data, mutation of Cys-63 to Ala in BiP-D411K and BiP-E412R did not further alter the ATPase activity of either mutant (Fig. 5E).
Previously, we reported that a strain containing wild-type BiP allowed for more robust growth in the presence of oxidant than a cysteine-less BiP mutant strain, suggesting a role for cysteine oxidation in cell proliferation during oxidative stress (9). Here, we observed a BiP-E412R mutant, which shows enhanced cysteine glutathionylation during stress (Fig. 5D), exhibits an even more robust growth than wild-type BiP in the presence of diamide (Fig. 6). Consistent with increased oxidation of the BiP cysteine being the contributing factor for the enhanced growth rate, a double C63A/E412R BiP mutant showed a growth pattern identical to that observed for BiP-C63A (Fig. 6). These data provide additional support for a role for oxidized BiP in increased cell viability during oxidative stress. These data also suggest BiP oxidation can be further stimulated to enhance cell growth beyond what is observed with endogenous wild-type BiP. The BiP-D411K mutant showed a similar growth trend as the BiP-E412R mutant; mutation of D411K resulted in a substantially enhanced resistance to diamide, and the observed resistance to diamide was diminished when Cys-63 was replaced with alanine (Fig. 6). Yet the growth rate was significantly faster for all D411K mutants relative to their counterpart alleles. BiP-D411K showed a more robust growth rate than wild-type or BiP-E412R (Fig. 6). Proliferation of a BiP-C63A/D411K mutant, although slowed relative to BiP-D411K, was faster than a BiP-C63A mutant (Fig. 6). We anticipate the cysteine-independent growth enhancement observed for the strain with BiP-C63A/D411K during diamide-induced stress may relate to other changes in BiP function upon introduction of the D411K mutation, such as its observed decreased ATPase rate (Fig. 5E). We suggest that enhanced modification of the cysteine combined with changes in catalytic activity for the BiP-D411K mutant may together contribute to robust growth for the BiP-D411K mutant observed during conditions of oxidative stress (Fig. 6). The increased viability during conditions of stress that we observed here for our alleles with increased propensity for modification (BiP-D411K and BiP-E412R) parallels the enhanced cell proliferation we previously characterized for alleles that phenotypically mimic constitutive oxidation BiP (9). In the absence of stressor, the BiP-D411K- and BiP-E412R-expressing strains showed no growth differences relative to a wild-type strain (Fig. 6).
FIGURE 6.

BiP with a lowered cysteine pKa enhances protection during oxidative stress. A, yeast strains deleted for endogenous BiP (kar2Δ) kept alive by the indicated plasmid-encoded BiP allele were spotted on SMM plates with or without diamide, and plates were incubated at 30 °C for 2 days. B, growth curves of the strains described in A. Cells were grown to mid-log at 30 °C in SMM, subcultured to the same density in SMM with or without diamide (1.2 mm) (t = 0 h), and sampled hourly for absorbance measurements at 600 nm.
Cysteine Oxidation Mimetic Alleles Suggest Constitutive Oxidation of BiP Is Detrimental for Cell Proliferation during Non-stress Conditions
It is generally accepted that the utility of protein glutathionylation as a regulatory mechanism stems from its reversibility. As the environment becomes more reducing, when activity changes associated with protein glutathionylation are no longer beneficial, glutathione is removed, and “normal” activity is restored. To confirm and explore the dynamics for reversibility of glutathionylated BiP, we monitored glutathionylated BiP levels after removal of oxidant. Cells were exposed to 10 mm diamide for 15 min; oxidant was removed, and medium was replaced. Cycloheximide was added at the time of diamide removal to prevent an artifactually perceived loss of the glutathione adduct due to increased total BiP levels from new synthesis. The 3-fold induction of glutathionylated BiP observed upon stress was reversed to non-stress levels by 30 min (Fig. 7A). A fit of the normalized BiP-biotin signal to a one-phase decay equation revealed a half-time for glutathionylated BiP post-oxidant of ∼10 min, demonstrating efficient removal of glutathione from BiP once stress conditions cease (Fig. 7B). The total BiP level appeared constant over time, suggesting that the decreased glutathionylated BiP signal reflects reduction versus degradation of the glutathionylated protein (Fig. 7A). Mutant proteins BiP-E412R and BiP-D411K, which both show an increased BiP-biotin level upon diamide addition (relative to wild-type BiP), also showed a restoration of pre-stress BiP-biotin signal within 30–45 min post-oxidant removal with a half-time of ∼5–10 min (Fig. 7). These data suggest alterations in the cysteine pKa impact modification during stress but do not significantly alter reversibility of the BiP glutathione adduct.
FIGURE 7.

BiP cysteine oxidation is reversible. A, yeast strains deleted for endogenous BiP (kar2Δ) containing plasmids encoding FLAG-tagged wild-type BiP, BiP-E412R, or BiP-D411K were treated with 5 mm diamide for 15 min; diamide was removed by filtration, and cells were resuspended in SMM containing cycloheximide. Cells were collected a various times post-diamide removal, and levels of glutathionylated BiP were monitored using the biotin-switch protocol. For each strain, the signal ratio was set to 1.0 for cells grown in the absence of diamide. B, plot of the quantified data from A. The relative intensity of BiP-biotin signal (relative to the total level of BiP) over time shows a return to a basal level of oxidized BiP by 45 min, with a half-time of less than 10 min.
If reversibility is an important feature for glutathionylation, we anticipate irreversible oxidation of BiP will be detrimental to cells once stress subsides. To date, we have not characterized BiP alleles or cellular machinery that result in slow (or no) deglutathionylation of BiP post-stress. Yet, we reasoned our previously characterized oxidation mimetic alleles (BiP-C63D, BiP-C63F, and BiP-C63Y) could be used as proxies for constitutively irreversible oxidation of the BiP cysteine (9). All three mutants as the only copy of BiP show temperature-sensitive phenotypes that could complicate data interpretation (9); thus, for these experiments we transformed plasmids encoding these BiP cysteine mutants into a sensitized cysteine-less BiP yeast strain (kar2-C63A) to avoid any confounding growth phenotypes unrelated to oxidative stress. We reported previously that ectopic expression of these oxidation mimetic alleles promotes cell growth during Ero1*-induced oxidative stress (9). Here, we observed the BiP-C63D, BiP-C63F, and BiP-C63Y alleles also show an improved growth rate in diamide-treated cells relative to a strain with ectopic addition of an unmodified BiP-C63A or cells containing an empty vector (Fig. 8, A and B). We suggest the improved growth rate observed for the strains containing the BiP-C63F and BiP-C63Y alleles reflects the protective effect of oxidized BiP during oxidative stress, which is phenotypically mimicked by the BiP-C63F, BiP-C63Y, and oxidized BiP proteins (Fig. 8, A and B) (9). Notably, the growth enhancement observed during stress for strains containing the oxidation mimetic alleles was not observed post-stress. Indeed, when cell growth was monitored after stress removal, it was wild-type BiP (now reduced) and the allele that phenotypically copies reduced BiP (BiP-C63A) that showed the most robust growth (Fig. 8C). The strains with BiP-C63D, BiP-C63F, and BiP-C63Y exhibited a growth delay post-oxidant removal relative to strains containing either wild-type BiP or BiP-C63A (Fig. 8C). We suggest the slow growth post-stress, observed in the strains containing the oxidation mimetic alleles, relates to the inability to restore normal BiP ATPase and folding activity. All ectopic alleles show similar growth rates in liquid culture under non-stress conditions (Fig. 8C).
FIGURE 8.

BiP cysteine oxidation mimetic mutants confer enhanced growth during oxidative stress but negatively impact growth under non-stress conditions. A, a cysteine-less BiP strain (kar2-C63A) was transformed with an additional plasmid-encoded BiP allele or empty vector as indicated. Cells were grown to mid-log at 30 °C in SMM, subcultured to the same density in SMM with or without diamide (1.1 mm) (t = 0 h), and sampled hourly for absorbance measurements at 600 nm. B, strains described for A were spotted on SMM plates with or without diamide, and plates were incubated at 30 °C for 2 days. C, liquid cultures of the strains described in A were adjusted to the same cell density prior to incubation with 5 mm diamide (or mock treatment) for 1 h at 30 °C. Diamide was removed by filtration (t = 0 h), and cells were sampled hourly for absorbance measurements at 600 nm. D, relative fitness of the yeast strains described in A was assessed by co-culturing strains over a period of 77 h in the presence of 1 mm diamide. Graphs depict the relative proportion of each strain in the co-culture over time. Solid and open circles reflect the flipped plasmid-markers for each co-cultured pair of BiP alleles. (See “Experimental Procedures” and Table 1 for details.) E, relative fraction of each strain in the indicated co-culture grown in 1 mm diamide is plotted at 0 and 77 h post-mixing of the two strains. The data are plotted as an average of the indicated BiP allele co-cultures from strains with the swapped plasmid markers. Error bars reflect the range in values for the two averaged co-cultures (See “Experimental Procedures” and Table 2 for details.) F, strains as described in D, grown in the absence of diamide. G, strains depicted in E, grown in the absence of diamide.
To further explore the growth changes observed with the oxidation mimetic alleles during and post-stress, we performed pairwise competitive growth assays (fitness assays). We anticipated that a growth competition assay should amplify the growth enhancement observed with the individual yeast strains during oxidative stress (Fig. 8, A and B). We speculated that the constitutive disruption of BiP ATPase activity for the mimetic alleles may also confer a subtle growth disadvantage during non-stress conditions, which might be more readily visualized under the competitive growth conditions and would be consistent with the modest growth defect we observed post-stress (Fig. 8C). Indeed, in keeping with our prior growth assays, we observed that during oxidative stress conditions (growth in the presence of 1 mm diamide) strains containing the constitutive oxidation mimetic alleles BiP-C63F, BiP-C63D, and BiP-C63Y outcompeted strains containing either wild-type BiP or the BiP-C63A mutant (Fig. 8, D and E). The growth advantage for the BiP-C63F allele was more pronounced relative to a strain with a BiP allele that could not be oxidized (BiP-C63A) versus a strain with wild-type BiP (Fig. 8D). These data are consistent with wild-type BiP becoming glutathionylated in cells upon diamide treatment, which confers a growth advantage relative to an unmodified BiP-C63A allele. In keeping with this interpretation, a strain with wild-type BiP also outcompeted a BiP-C63A mutant strain when these strains were co-cultured in diamide (Fig. 8D). Strikingly, under non-stress conditions, the strains with the constitutive-oxidation mimetic alleles (BiP-C63F, BiP-C63D, and BiP-C63Y) showed a decreased fitness relative to strains with either wild-type BiP (now predominantly reduced) or a BiP-C63A allele (Fig. 8, F and G). Notably, no growth distinction was apparent under non-stress conditions for the strains containing wild-type BiP and BiP-C63A (Fig. 8, F and G). Together, these data suggest continual oxidation (here mimicked by the BiP-C63F, BiP-C63D, and BiP-C63Y alleles) has a negative impact on cell proliferation under non-stress conditions. We propose the growth disadvantage during non-stress conditions reflects the detrimental impact for continual oxidation of BiP.
Consistent with this model, strains containing BiP-E412R, which is oxidized during stress yet readily reduced post-stress (Figs. 5 and 7), showed a competitive growth advantage during oxidative stress (Fig. 9A) but did not show any fitness defects under non-stress conditions (Fig. 9C). Strains with BiP-E412R demonstrated a competitive growth advantage in the presence of diamide relative to a strain with wild-type BiP or a BiP-C63A mutant (Fig. 9A). The competitive advantage observed with the BiP-E412R allele during oxidative stress was lost when the BiP cysteine was replaced with alanine (Fig. 9B). The modest growth disadvantage for the BiP-C63A/E412R strain relative to a wild-type BiP strain during stress is consistent with an increased fitness conferred through cysteine oxidation (Fig. 9B). In contrast, the BiP-E412R mutant strain did not show any fitness difference under non-stress conditions relative to a strain containing wild-type BiP or a BiP-C63A allele (Fig. 9C), in keeping with an absence of BiP-E412R cysteine oxidation under non-stress conditions. A BiP-D411K mutant also demonstrated a competitive advantage during stress relative to a wild-type strain (Fig. 9B). However, consistent with our prior observations, this growth advantage was not solely dependent on oxidation of the BiP cysteine, as evident from the growth advantage observed also with the BiP-C63A/D411K mutant under non-stress conditions (Fig. 9B).
FIGURE 9.

Distinct from the constitutive oxidation mimetic alleles, the reversibly oxidized BiP alleles do not exhibit decreased fitness in the absence of stress. A, yeast deleted for endogenous BiP (kar2Δ) and containing the indicated plasmid-encoded BiP alleles were co-cultured to assess their relative fitness during oxidative stress. Strains were co-cultured over a period of 77 h in the presence of 1 mm diamide, and graphs show the relative proportion of each strain in the co-culture over time. Solid and open circles reflect the flipped plasmid markers for each co-culture. (See “Experimental Procedures” and Table 1 for details.) B, relative level of each co-cultured strain is plotted at 0 and 77 h post-mixing of the two strains in the presence of 1 mm diamide. The data are plotted as an average of the co-cultured strains carrying swapped plasmid markers. Error bars reflect the range in values for the two averaged co-cultures (See “Experimental Procedures” and Table 2 for details.) C, strains from A were grown in the absence of diamide. Data are represented as in A. D, strains were grown and analyzed as in B, except in the absence of diamide.
Discussion
We have demonstrated a conserved cysteine thiol in BiP is susceptible to oxidation by peroxide (9) and glutathione (Fig. 1) during oxidative stress conditions. Biochemical assays indicate that cysteine modification by either peroxide or glutathione similarly alters BiP chaperone activity, resulting in an enhanced ability of BiP to limit polypeptide aggregation (Fig. 4) (9). We suggest the augmented holdase activity observed for oxidized BiP helps the ER to maintain folding homeostasis during hyperoxidizing conditions by limiting accumulation of polypeptide aggregates (9). Employing BiP alleles with an increased susceptibility to oxidation by glutathione (Fig. 5), we observed that the degree of growth during stress scales with the proportion of oxidized BiP in cells during stress (Figs. 6 and 9). These data indicate BiP oxidation is not an “all-or-nothing” switch, wherein saturating levels of oxidized BiP are present in cells during stress. Instead, our data imply that cells can tune oxidized BiP to maintain ER folding homeostasis under a range of redox conditions.
A single mechanism for oxidation of a cysteine thiol by peroxide has been proposed; nucleophilic attack of a cysteine thiolate anion on peroxide generates a cysteine sulfenic acid adduct. Alternatively, several pathways for cysteine oxidation by glutathione have been put forward; the two most often suggested mechanisms involve (i) the reaction of reduced glutathione with a sulfenic acid moiety and (ii) oxidation of thiols via a thiol-disulfide exchange reaction between a protein thiol and GSSG. Our data suggest the possibility that both mechanisms can mediate BiP oxidation by glutathione in cells. We observed peroxide added exogenously or produced endogenously by overproduction of a hyperactive Ero1 mutant (Ero1*) results in formation of glutathionylated BiP (Figs. 1 and 2). Notably, glutathionylation stimulated by peroxide was prevented in the presence of the sulfenic acid-trapping agent dimedone, implying BiP glutathionylation under these conditions occurs through a sulfenic acid intermediate (Fig. 2). Our data suggest that exogenous addition of the thiol-specific oxidant diamide does not generate sulfenylated BiP (Fig. 2); instead, we propose diamide likely stimulates glutathionylation of BiP through GSSG accumulation within cells, in keeping with the observed increase in the cellular GSSG to GSH ratio upon diamide treatment (Fig. 3). Notably, the relevance of GSSG as a physiological oxidant has been a point of debate in the literature; specifically, it has been noted that given a typical cysteine pKa, a substantial ∼100-fold change in the ratio of GSSG to GSH in cells would need to occur for GSSG to mediate significant thiol oxidation (35, 36). However, these arguments center primarily on the requirements for glutathionylation of cytoplasmic proteins, where the resting GSSG/GSH ratio has been measured at 1:30 to 1:100 (15). In the ER, where the GSSG/GSH ratio approximates 1 (15), it is more likely that pronounced changes in the GSSG/GSH ratio in favor of GSSG could facilitate glutathionylation through GSSG-mediated thiol-disulfide exchange. In keeping with a potential role for GSSG as a potential oxidant for BiP in the ER lumen, we could recapitulate oxidation of BiP by glutathione in vitro with a modest shift in the GSSG/GSH ratio (Fig. 3). Given that high exogenous diamide levels are a non-physiological stress, a GSSG thiol-disulfide exchange route for BiP glutathionylation may not be a common cellular pathway. Nonetheless, we maintain that here, and throughout the literature, use of exogenous oxidants like diamide has served as a valuable tool to highlight potential means in which cellular oxidative stress can influence redox signaling.
Why does BiP undergo oxidation by more than one type of molecule? Precedents exist for distinct biochemical activities (for a given protein) that are dependent on specific redox modifications. The catalytic cysteine of cytosolic glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is susceptible to a variety of reversible thiol modifications, including sulfenylation, glutathionylation, nitrosylation, and sulfhydration (37–41); as a consequence of distinct modification inputs, GAPDH transduces alternative signals for adaptive adjustment of metabolism or cell death (42). Yet, for BiP, we observe similar activity changes for glutathionylated and sulfenylated BiP. At a very simplistic level, cellular ROS levels and the cellular GSSG/GSH ratio are both considered key indicators of redox homeostasis; direct modification of BiP in the presence of increased peroxide or GSSG may allow cells to sense and respond to multiple indicators of redox changes within the ER. In parallel, we propose glutathionylation that occurs via a sulfenic acid intermediate may serve to maintain reversibility of BiP modification in the presence of excess ROS. Sulfenic acid is prone to further oxidation by peroxide to form irreversible sulfinic (−SO2H) and/or sulfonic (−SO3H) acid adducts. Our data obtained using oxidation mimetic alleles as a proxy for irreversible BiP oxidation suggest continual (irreversible) modification of BiP is detrimental to cells under non-stress conditions (Fig. 8). We anticipate the slower recovery post-stress (Fig. 8C) and the fitness disadvantage under non-stress conditions (Fig. 8, F and G), observed with the oxidation mimetic alleles, relate to an inability of oxidized BiP proteins to release bound polypeptides, which should impact protein flux through the ER and disrupt folding homeostasis. The capacity of BiP for glutathionylation may serve to limit irreversible oxidation of BiP by peroxide, which, in turn, preserves the capacity for BiP cysteine reduction once hyperoxidizing conditions subside.
We observed rapid reversibility of the BiP-glutathione adduct in cells (Fig. 7). Given the efficient removal of glutathione from BiP post-stress, we anticipate an ER-localized enzyme catalyzes the deglutathionylation reaction. Glutaredoxins are widely considered the primary cellular catalysts for protein deglutathionylation (43, 44). Notably, two glutaredoxins (Grx6 and Grx7) localize to the ER/Golgi region in yeast, and these enzymes have the potential to facilitate deglutathionylation of BiP (32, 45). However, our preliminary data show no change in the kinetics of BiP-glutathione adduct reduction in the glutaredoxin-deficient grx6Δ grx7Δ strain (data not shown). These data indicate that if Grx6 and Grx7 mediate BiP deglutathionylation, then other cellular enzymes can compensate for the loss of Grx6/7 activity. Like glutaredoxin proteins, members of the PDI family also show deglutathionylation activity in vitro, although with a considerably decreased turnover relative to glutaredoxins (46–48). Yeast contain five ER-localized PDI family members, and it will be exciting to explore a possible role for PDI proteins in BiP deglutathionylation. PDI itself is susceptible to glutathionylation, which decreases PDI chaperone and isomerase activities (49–51). It is appealing to consider that glutathionylation of PDI could limit PDI deglutathionylation activity during oxidative stress. Regulation of PDI activity through cysteine oxidation could be an effective means to allow for oxidized BiP accumulation during stress and to facilitate BiP reduction when stress subsides. Yet, although intriguing on one level, such a mechanism is complicated by a potential need for additional redox players to facilitate deglutathionylation of PDI (and increased PDI deglutathionylation activity) post-stress.
To date, an impressive number of post-translational modifications have been identified on BiP orthologues. Similar to the oxidative modifications we have described for yeast BiP, human BiP cysteines are susceptible to oxidation by glutathione (16), peroxide (10), and prone to formation of an intramolecular disulfide bond (10). As we observed for yeast BiP, oxidation of human BiP augments its polypeptide binding activity (10). However, the range of modifications detected on BiP orthologues extends beyond cysteine oxidation. Post-translational modifications on numerous amino acids distributed throughout the BiP polypeptide sequence have been identified, including phosphorylation (52, 53), ADP-ribosylation (54–57), AMPylation (58, 59), aldehyde adducts (60), and arginylation (61). Intriguingly, in contrast to the BiP cysteine modifications, phosphorylation, ADP-ribosylation, and AMPylation of BiP are observed during conditions of decreased folding burden within the ER (e.g. starvation or cycloheximide treatment) (56, 58, 62); for phosphorylation and ADP-ribosylation, it has been proposed that these modifications partition BiP into inactive oligomers that can be reactivated quickly when secretory load increases (55, 56). Given the plethora of modifications identified on BiP, and the potential for conflicting biological outcomes, it will be exciting to determine the relationship between BiP post-translational modifications. An understanding of the combinatorial effects of specific BiP modifications, as well as the epistasis for modifications, will undoubtedly uncover a new layer in our understanding of the regulatory systems and signaling pathways that maintain ER homeostasis.
Author Contributions
C. S. S. conceived and coordinated the study. J. W. acquired the data, and C. S. S. provided assistance in the generation of plasmids and yeast strains. C. S. S. and J. W. analyzed and interpreted the data and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank John O'Donnell and Heather Sickles for assistance with plasmid construction and technical expertise. We also thank Dante Lepore for help with fluorescence microscopy and Dr. Ruth Collins for the use of her lab microscope. We acknowledge the Cornell Proteomics and Mass Spectrometry Core Facility for processing and analysis of samples, and we thank the director of the core facility, Dr. Sheng Zhang, for providing the methods describing the mass spectrometry procedures. The proteomics core facility was supported by National Institutes of Health Grant 1S10OD017992-01, which provided instrument support.
This work was supported by Cornell University and National Institutes of Health Grant GM105958 (to C. S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1.
- ER
- endoplasmic reticulum
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid)
- SMM
- synthetic minimal medium
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BME
- β-mercaptoethanol
- AMS
- 4-acetamido-4′-maleimidylstilbene2,2′-disulfonic acid
- CHP
- cumene peroxide
- mal-PEG5K
- maleimide conjugated to polyethylene glycol of 5 kDa
- TNB
- 2-nitro-5-thiobenzoic acid
- ROS
- reactive oxygen species
- roGFP
- redox-sensitive GFP
- eroGFP
- roGFP sensor targeted to ER of yeast
- NEM
- N-ethylmaleimide
- PDI
- protein-disulfide isomerase.
References
- 1. Frand A. R., and Kaiser C. A. (1998) The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1, 161–170 [DOI] [PubMed] [Google Scholar]
- 2. Pollard M. G., Travers K. J., and Weissman J. S. (1998) Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1, 171–182 [DOI] [PubMed] [Google Scholar]
- 3. Chakravarthi S., and Bulleid N. J. (2004) Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 279, 39872–39879 [DOI] [PubMed] [Google Scholar]
- 4. Molteni S. N., Fassio A., Ciriolo M. R., Filomeni G., Pasqualetto E., Fagioli C., and Sitia R. (2004) Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 279, 32667–32673 [DOI] [PubMed] [Google Scholar]
- 5. Appenzeller-Herzog C., Riemer J., Christensen B., Sørensen E. S., and Ellgaard L. (2008) A novel disulphide switch mechanism in Ero1α balances ER oxidation in human cells. EMBO J. 27, 2977–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baker K. M., Chakravarthi S., Langton K. P., Sheppard A. M., Lu H., and Bulleid N. J. (2008) Low reduction potential of Ero1α regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 27, 2988–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sevier C. S., Qu H., Heldman N., Gross E., Fass D., and Kaiser C. A. (2007) Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129, 333–344 [DOI] [PubMed] [Google Scholar]
- 8. Wang C., Yu J., Huo L., Wang L., Feng W., and Wang C. C. (2012) Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a′. J. Biol. Chem. 287, 1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J., Pareja K. A., Kaiser C. A., and Sevier C. S. (2014) Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. Elife 3, e03496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei P. C., Hsieh Y. H., Su M. I., Jiang X., Hsu P. H., Lo W. T., Weng J. Y., Jeng Y. M., Wang J. M., Chen P. L., Chang Y. C., Lee K. F., Tsai M. D., Shew J. Y., and Lee W. H. (2012) Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 48, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim S., Sideris D. P., Sevier C. S., and Kaiser C. A. (2012) Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J. Cell Biol. 196, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kettenhofen N. J., and Wood M. J. (2010) Formation, reactivity, and detection of protein sulfenic acids. Chem. Res. Toxicol. 23, 1633–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Devarie-Baez N. O., Silva Lopez E. I., and Furdui C. M. (2016) Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 172–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta V., and Carroll K. S. (2014) Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 1840, 847–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hwang C., Sinskey A. J., and Lodish H. F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 [DOI] [PubMed] [Google Scholar]
- 16. Lind C., Gerdes R., Hamnell Y., Schuppe-Koistinen I., von Löwenhielm H. B., Holmgren A., and Cotgreave I. A. (2002) Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem. Biophys. 406, 229–240 [DOI] [PubMed] [Google Scholar]
- 17. Hoppe G., Chai Y. C., Crabb J. W., and Sears J. (2004) Protein S-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp. Eye Res. 78, 1085–1092 [DOI] [PubMed] [Google Scholar]
- 18. Adams A., Gottschling D. E., Kaiser C. A., and Stearns T. (1998) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 19. Sikorski R. S., and Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Merksamer P. I., Trusina A., and Papa F. R. (2008) Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 135, 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisniewska M., Karlberg T., Lehtiö L., Johansson I., Kotenyova T., Moche M., and Schüler H. (2010) Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70–2, HSPA6/Hsp70B', and HSPA5/BiP/GRP78. PLoS ONE 5, e8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holmgren A., and Aslund F. (1995) Glutaredoxin. Methods Enzymol. 252, 283–292 [DOI] [PubMed] [Google Scholar]
- 23. Anderson M. E. (1985) Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 113, 548–555 [DOI] [PubMed] [Google Scholar]
- 24. Jaffrey S. R., and Snyder S. H. (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, pl1. [DOI] [PubMed] [Google Scholar]
- 25. Lind C., Gerdes R., Schuppe-Koistinen I., and Cotgreave I. A. (1998) Studies on the mechanism of oxidative modification of human glyceraldehyde-3-phosphate dehydrogenase by glutathione: catalysis by glutaredoxin. Biochem. Biophys. Res. Commun. 247, 481–486 [DOI] [PubMed] [Google Scholar]
- 26. Nordstrand K., slund F., Holmgren A., Otting G., and Berndt K. D. (1999) NMR structure of Escherichia coli glutaredoxin 3-glutathione mixed disulfide complex: implications for the enzymatic mechanism. J. Mol. Biol. 286, 541–552 [DOI] [PubMed] [Google Scholar]
- 27. Ayer A., Fellermeier S., Fife C., Li S. S., Smits G., Meyer A. J., Dawes I. W., and Perrone G. G. (2012) A genome-wide screen in yeast identifies specific oxidative stress genes required for the maintenance of sub-cellular redox homeostasis. PLoS ONE 7, e44278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hawkins C. L., Pattison D. I., and Davies M. J. (2003) Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 25, 259–274 [DOI] [PubMed] [Google Scholar]
- 29. Leonard S. E., Reddie K. G., and Carroll K. S. (2009) Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 4, 783–799 [DOI] [PubMed] [Google Scholar]
- 30. Charles R. L., Schröder E., May G., Free P., Gaffney P. R., Wait R., Begum S., Heads R. J., and Eaton P. (2007) Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol. Cell. Proteomics 6, 1473–1484 [DOI] [PubMed] [Google Scholar]
- 31. Meyer A. J., Brach T., Marty L., Kreye S., Rouhier N., Jacquot J. P., and Hell R. (2007) Redox-sensitive GFP in Arabidopsis thaliana is a quantitative biosensor for the redox potential of the cellular glutathione redox buffer. Plant J. 52, 973–986 [DOI] [PubMed] [Google Scholar]
- 32. Izquierdo A., Casas C., Mühlenhoff U., Lillig C. H., and Herrero E. (2008) Saccharomyces cerevisiae Grx6 and Grx7 are monothiol glutaredoxins associated with the early secretory pathway. Eukaryot. Cell 7, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lohman J. R., and Remington S. J. (2008) Development of a family of redox-sensitive green fluorescent protein indicators for use in relatively oxidizing subcellular environments. Biochemistry 47, 8678–8688 [DOI] [PubMed] [Google Scholar]
- 34. Birk J., Meyer M., Aller I., Hansen H. G., Odermatt A., Dick T. P., Meyer A. J., and Appenzeller-Herzog C. (2013) Endoplasmic reticulum: reduced and oxidized glutathione revisited. J. Cell Sci. 126, 1604–1617 [DOI] [PubMed] [Google Scholar]
- 35. Berndt C., Lillig C. H., and Flohé L. (2014) Redox regulation by glutathione needs enzymes. Front. Pharmacol. 5, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shelton M. D., Chock P. B., and Mieyal J. J. (2005) Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 7, 348–366 [DOI] [PubMed] [Google Scholar]
- 37. Molina y Vedia L., McDonald B., Reep B., Brüne B., Di Silvio M., Billiar T. R., and Lapetina E. G. (1992) Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J. Biol. Chem. 267, 24929–24932 [PubMed] [Google Scholar]
- 38. Grant C. M., Quinn K. A., and Dawes I. W. (1999) Differential protein S-thiolation of glyceraldehyde-3-phosphate dehydrogenase isoenzymes influences sensitivity to oxidative stress. Mol. Cell. Biol. 19, 2650–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mohr S., Hallak H., de Boitte A., Lapetina E. G., and Brüne B. (1999) Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 274, 9427–9430 [DOI] [PubMed] [Google Scholar]
- 40. Maller C., Schröder E., and Eaton P. (2011) Glyceraldehyde-3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid. Redox Signal. 14, 49–60 [DOI] [PubMed] [Google Scholar]
- 41. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hildebrandt T., Knuesting J., Berndt C., Morgan B., and Scheibe R. (2015) Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub. Biol. Chem. 396, 523–537 [DOI] [PubMed] [Google Scholar]
- 43. Lillig C. H., and Berndt C. (2013) Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 18, 1654–1665 [DOI] [PubMed] [Google Scholar]
- 44. Lillig C. H., Berndt C., and Holmgren A. (2008) Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 45. Mesecke N., Mittler S., Eckers E., Herrmann J. M., and Deponte M. (2008) Two novel monothiol glutaredoxins from Saccharomyces cerevisiae provide further insight into iron-sulfur cluster binding, oligomerization, and enzymatic activity of glutaredoxins. Biochemistry 47, 1452–1463 [DOI] [PubMed] [Google Scholar]
- 46. Peltoniemi M. J., Karala A. R., Jurvansuu J. K., Kinnula V. L., and Ruddock L. W. (2006) Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein-disulfide isomerase catalyzed reactions are defined by the γ-linkage present in glutathione. J. Biol. Chem. 281, 33107–33114 [DOI] [PubMed] [Google Scholar]
- 47. Lundström-Ljung J., Vlamis-Gardikas A., Aslund F., and Holmgren A. (1999) Reactivity of glutaredoxins 1, 2 and 3 from Escherichia coli and protein-disulfide isomerase towards glutathionyl-mixed disulfides in ribonuclease A. FEBS Lett. 443, 85–88 [DOI] [PubMed] [Google Scholar]
- 48. Xiao R., Lundström-Ljung J., Holmgren A., and Gilbert H. F. (2005) Catalysis of thiol/disulfide exchange. Glutaredoxin 1 and protein-disulfide isomerase use different mechanisms to enhance oxidase and reductase activities. J. Biol. Chem. 280, 21099–21106 [DOI] [PubMed] [Google Scholar]
- 49. Halloran M., Parakh S., and Atkin J. D. (2013) The role of S-nitrosylation and S-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 797914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiong Y., Manevich Y., Tew K. D., and Townsend D. M. (2012) S-Glutathionylation of protein disulfide isomerase regulates estrogen receptor α stability and function. Int. J. Cell Biol. 2012, 273549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uys J. D., Xiong Y., and Townsend D. M. (2011) Nitrosative stress-induced S-glutathionylation of protein disulfide isomerase. Methods Enzymol. 490, 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hendershot L. M., Ting J., and Lee A. S. (1988) Identity of the immunoglobulin heavy-chain-binding protein with the 78,000-dalton glucose-regulated protein and the role of posttranslational modifications in its binding function. Mol. Cell. Biol. 8, 4250–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Welch W. J., Garrels J. I., Thomas G. P., Lin J. J., and Feramisco J. R. (1983) Biochemical characterization of the mammalian stress proteins and identification of two stress proteins as glucose- and Ca2+-ionophore-regulated proteins. J. Biol. Chem. 258, 7102–7111 [PubMed] [Google Scholar]
- 54. Carlsson L., and Lazarides E. (1983) ADP-ribosylation of the Mr 83,000 stress-inducible and glucose-regulated protein in avian and mammalian cells: modulation by heat shock and glucose starvation. Proc. Natl. Acad. Sci. U.S.A. 80, 4664–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chambers J. E., Petrova K., Tomba G., Vendruscolo M., and Ron D. (2012) ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. J. Cell Biol. 198, 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Freiden P. J., Gaut J. R., and Hendershot L. M. (1992) Interconversion of three differentially modified and assembled forms of BiP. EMBO J. 11, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ledford B. E., and Leno G. H. (1994) ADP-ribosylation of the molecular chaperone GRP78/BiP. Mol. Cell. Biochem. 138, 141–148 [DOI] [PubMed] [Google Scholar]
- 58. Ham H., Woolery A. R., Tracy C., Stenesen D., Krämer H., and Orth K. (2014) Unfolded protein response-regulated Drosophila Fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J. Biol. Chem. 289, 36059–36069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanyal A., Chen A. J., Nakayasu E. S., Lazar C. S., Zbornik E. A., Worby C. A., Koller A., and Mattoo S. (2015) A novel link between Fic (filamentation induced by cAMP)-mediated adenylylation/AMPylation and the unfolded protein response. J. Biol. Chem. 290, 8482–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Galligan J. J., Fritz K. S., Backos D. S., Shearn C. T., Smathers R. L., Jiang H., MacLean K. N., Reigan P. R., and Petersen D. R. (2014) Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: functional independence of ATPase activity and chaperone function. Free Radic. Biol. Med. 73, 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cha-Molstad H., Sung K. S., Hwang J., Kim K. A., Yu J. E., Yoo Y. D., Jang J. M., Han D. H., Molstad M., Kim J. G., Lee Y. J., Zakrzewska A., Kim S. H., Kim S. T., Kim S. Y., et al. (2015) Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat. Cell Biol. 17, 917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leno G. H., and Ledford B. E. (1989) ADP-ribosylation of the 78-kDa glucose-regulated protein during nutritional stress. Eur. J. Biochem. 186, 205–211 [DOI] [PubMed] [Google Scholar]
- 63. Dalle-Donne I., Rossi R., Colombo G., Giustarini D., and Milzani A. (2009) Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci. 34, 85–96 [DOI] [PubMed] [Google Scholar]
- 64. Grek C. L., Zhang J., Manevich Y., Townsend D. M., and Tew K. D. (2013) Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 288, 26497–26504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yan M., Li J., and Sha B. (2011) Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem. J. 438, 447–455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.