

Abstract
Chronic lymphocytic leukemia (CLL) is a disease in which a single B-cell clone proliferates relentlessly in peripheral lymphoid organs, bone marrow, and blood. DNA sequencing experiments have shown that about 30% of CLL patients have stereotyped antigen-specific B-cell receptors (BCRs) with a high level of sequence homology in the variable domains of the heavy and light chains. These include many of the most aggressive cases that have IGHV-unmutated BCRs whose sequences have not diverged significantly from the germ line. This suggests a personalized therapy strategy in which a toxin or immune effector function is delivered selectively to the pathogenic B-cells but not to healthy B-cells. To execute this strategy, serum-stable, drug-like compounds able to target the antigen-binding sites of most or all patients in a stereotyped subset are required. We demonstrate here the feasibility of this approach with the discovery of selective, high affinity ligands for CLL BCRs of the aggressive, stereotyped subset 7P that cross-react with the BCRs of several CLL patients in subset 7p, but not with BCRs from patients outside this subset.
Keywords: antibody, cancer therapy, drug discovery, drug screening, immunoglobulin G (IgG), leukemia, Antigen Surrogate, B Cell Receptor, chronic lymphocytic leukemia, combinatorial chemistry
Introduction
Chronic lymphocytic leukemia (CLL)2 is the most common type of adult blood cancer leukemia with ∼15,000 new cases reported every year in the United States (1). CLL involves the relentless expansion of a single B-cell clone, arguing that this occurs in response to a particular antigen, although the nature of these antigens is largely unknown (2). Many cases of CLL follow a relatively indolent course, whereas others are highly aggressive.
There have been impressive advances in the treatment of CLL. In addition to standard chemotherapy, anti-CD20 antibodies such as Rituximab, Ofatumumab, and Obinotuzumab are now used in combination or alone in standard clinical practice (1, 3). Inhibitors of Burton's tyrosine kinase, such as ibrutinib (4), which blunt B-cell activation, also have shown considerable promise. Nonetheless, all drugs currently on the market or in clinical trials are general immunosuppressants that fail to distinguish the pathogenic CLL cells from healthy B-cells. Ideally, drug targets would be identified that allow selective suppression or killing of the malignant CLL cells without interfering with the normal function of the humoral immune system (5).
One such target is the antigen-specific B-cell receptor (BCR) itself. Indeed, the pathogenic B-cells in a CLL patient are virtually the only cells in the body that express this particular receptor. Thus, if compounds highly selective for the receptor, with little off-target binding, could be discovered, it is possible that they could be employed chronically, unlike general B-cell killers. Although the antigen-binding pocket of an antibody is not generally considered a drug target, screening studies using phage-displayed peptide libraries have demonstrated the feasibility of obtaining selective ligands for the antigen-binding sites of some CLL BCRs (6–8). Moreover, we demonstrated recently that high affinity, non-peptidic, and serum-stable ligands for a CLL BCR can be discovered via combinatorial library screening (9). These studies support the feasibility of targeting CLL BCR for the development of highly selective drugs.
However, there are at least two major questions that have yet to be addressed with respect to drugging CLL BCRs. First, would a BCR-targeted strategy necessitate the development of a unique compound for each patient? This is obviously impractical. Large-scale DNA sequencing studies of the variable regions of BCRs from thousands of CLL patients has revealed that about one-third of CLL cases are “stereotyped,” meaning that the complementarity determining region 3 (HCDR3) of the pathogenic BCRs has significant homology to the CDR3s of CLL BCRs from other patients (10, 11). Indeed, patients exist whose entire IGHV-D-J amino acid sequences are identical, and some of these have remarkably identical IGκ/λV-Jκ/λ rearrangements. Thus, a critical question is whether a single compound could be identified that would cross-react with the BCRs of most or all of the patients falling into a particular stereotyped subset.
Second, the clinically most aggressive cases of CLL, where the need for new therapies is greatest, usually display so-called “IGHV-unmutated” (U-CLL) BCRs, defined as having a specific Ig variable region gene (IGHV) sequence differing less than 2% from the germ line (2, 12). There is reason to believe that unmutated BCRs would be more challenging drug targets than BCRs that have undergone more extensive somatic mutation. This is because unmutated pockets are thought to be more flexible and dynamic (13, 14), which is consistent with reports that U-CLL BCRs exhibit significant polyreactivity (7, 15) and that previous attempts to identify peptide ligands for these receptors have only yielded low affinity molecules (7).
In this study, we probe these issues. We report the discovery of selective, high affinity synthetic ligands for the BCR of a patient from stereotyped subset 7P (10), which represents an aggressive, unmutated group of U-CLLs. These ligands were tested for binding to BCRs from other patients of the same stereotyped subset and BCRs from CLL patients outside of that subset. Gratifyingly, there is excellent cross-reactivity between the ligand and all of the 7P stereotype CLL BCRs, but not between the ligand and BCRs outside of that stereotyped subset. The CLL BCR ligands also do not bind detectably to healthy B-cells or T-cells. These data strongly support the idea that the development of BCR-targeted reagents for the treatment of most or all CLL patients of a given stereotyped subset is a tractable goal.
Experimental Procedures
Full details of all the experimental procedures not presented below are provided under supplemental “Materials and Methods.”
Design and Synthesis of the Combinatorial Library
The one bead one compound combinatorial library was synthesized on 90-μm TentaGel MB NH2 resin (0.28 mmol/g loading, ∼2.8 million beads/g, from Rapp Polymere, Germany). The general schematic design of the branched library, side chain diversity elements (primary amines), and carboxylic acids used are shown in Fig. 1. Details are provided in the supplementary information (supplemental Figs. S1 and S2). To determine efficient synthesis of each library member, 24 random beads were isolated from the library and treated individually with 20 μl of CNBr solution to release the compounds. The parent ion mass of the compounds was determined by MALDI-TOF and the sequence of the unknown compound was determined by multiple sequence-specific product ions obtained from ESI Linear Ion Trap Mass Spectrometer coupled to electron transfer dissociation (LTQ-ETD, ThermoFinnigan) and 4800Plus MALDI-Tof/Tof (ABSciex).
FIGURE 1.

General strategy for the identification of synthetic ligands against an unmutated CLL B-cell receptor antibody. Design and synthesis of the combinatorial library used in this study. The general structure of the library is shown at the top left. The building blocks employed are shown below the structure. The theoretical diversity is about 960,000 compounds. Each molecule contains three different branches radiating from a 4-azido proline-derived central element. The library was synthesized on solid phase (90 μm TentaGel beads, 0.28 mmol/g loading) following sub-monomer methods and split and pool techniques, resulting in each bead displaying many copies of a single compound. The strategy employed for high-throughput bead-based screening against soluble monoclonal antibodies is shown on the right. The library beads were pre-screened against human-IgG and goat anti-human IgG secondary antibody conjugated to a red quantum dot (QD). Any bead that displayed a red halo under a low power fluorescence microscope was removed from the library pool manually. The remainder of the beads was then incubated with recombinant CLL 014 BCR IgG. The hit beads were collected and the structure of the compounds were revealed using MALDI-TOF/TOF and LTQ-ETD mass spectrometry (supplemental Fig. S2).
High-throughput Bead-based Screening of Combinatorial Library
The TentaGel beads displaying a 4-azidoproline-branched oligomeric library were pre-blocked with 1× TBST Starting Block containing 3% BSA before incubating with 100 nm pooled human IgG (Sigma) and goat F(ab′)2 anti-human IgG Qdot 655 (Life Technologies) for 45 min at room temperature. The general scheme of the multistep screening processes is shown in Fig. 1. The beads were visualized under a fluorescence microscope (Olympus BX-51) equipped with a 10× DAPI filter and any bead emitting red light was collected manually by a micropipette and discarded. The denuded COPA library beads were incubated with 100 nm purified CLL 014 mAb in binding buffer (Tris-buffered saline, pH 7.5, 0.05% Tween 20, 1% BSA) for 45 min at room temperature followed by incubating with a 1:500 dilution of goat F(ab′)2 anti-human IgG Qdot for 45 min at room temperature. A total of 83 beads emitting red fluorescent (brightly fluorescent) light were collected manually using a micropipette. Beads were stripped of any bound proteins and the screening process mentioned above was repeated. From the final rounds of the selection process a total of 19 re-validated hit beads were identified. The individual hit beads were treated with 20 μl of CNBr solution overnight to release the compounds and the sequence of the compounds were elucidated by MS and tandem MS/MS using 4800Plus MALDI-Tof Analyzer and LTQ-Orbitrap equipped with ETD (supplemental Figs. S2 and S3).
Biophysical Characterization of Initial Hit Compounds
Hit compounds were synthesized with a free cysteine molecule in the invariable linker (for fluorescein or biotin conjugation) and purified on HPLC using C18 reverse phase column (supplemental Figs. S4). A control compound with a similar branched backbone with random side chains was also synthesized and purified on HPLC (supplemental Fig. S4, bottom panel). Fluorescence polarization experiments were carried out using 50 nm of probe compound and serially diluted CLL 014 mAb in binding buffer (50 mm Tris, 100 mm NaCl, 0.05% Tween 20, pH 7.6, 1% BSA) in a 384-well microtiter plate. Fluorescence polarization (mP units) was measured on a Tecan Microplate Reader (Infinite 200Pro). For ELISA, biotin-tagged compounds and controls were immobilized on a streptavidin- or neutravidin-coated 96-well microtiter plate and titrated with increasing concentrations of CLL mAbs for 45 min at room temperature, followed by a 1:5,000 (1-Step Ultra TMB ELISA substrate) or 1:20,000 (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific) dilution of IgG1-conjugated HRP (Southern Biotech) for 30 min at room temperature. After developing plates, the data were recorded on a Tecan Microplate Reader (Infinite 200Pro).
Bio-layer Interferometry (BLI) Assay for Binding Affinity Measurements
Binding kinetics of various CLL IgGs with synthetic ligands were analyzed by BLI using an Octet RED96 system (Pall ForteBio). The highest affinity compounds, KMS31 and KMS32, were synthesized with biotin at the linker and immobilized on streptavidin sensors. The kinetic measurements were carried out by exposing sensors with serially diluted soluble CLL IgGs in binding buffer (1× PBS, pH 7.4, containing 1% BSA) in wells of a 96-well microtiter plate. The association and dissociation profiles of the compounds were monitored at 30 °C. Binding curves were analyzed by global fitting of sensorgrams to a bivalent binding model using data analysis software provided by Pall ForteBio.
Binding of Multimeric Synthetic Antigen Surrogates with CLL Cells from Subset 7P Patients
Cryopreserved PBMCs from CLL patients from stereotyped subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297) and from a other non-subset 7P patients were thawed and cultured in RPMI 1640 media supplemented with 10% FBS for 2 h at 37 °C in 5% CO2. Re-suspended PBMCs in 1× PBS were carefully passed through a density gradient using Ficoll-Paque Plus (from GE Healthcare) to remove dead cells. Approximately, 0.4 × 106 PBMCs from each CLL patient (CLL 014, CLL 529, CLL 675, CLL 1297, and CLL 3006) and from MEC1 cell line cells (DSMZ) were placed in each well of a 96-well microtiter plate for binding assays. The cells were pre-blocked with 3% BSA in 1× PBS containing 0.1% sodium azide and incubated with dextran-conjugated KMS31, KMS32, and control compounds (biotin-dextran, control compounds conjugated to dextran) in a 40 nm concentration for 40 min at 4 °C in binding buffer (1× PBS, pH 7.4, containing 1% BSA and 0.1% sodium azide). Following washing, cells were treated with SA-PE (to monitor binding), anti-human CD5-APC antibody, and anti-human CD19-FITC antibody (to identify CD5+/CD19+ CLL B-cell populations), and Po-Pro dye (for the staining of dead cells) for 30 min in the dark on ice. Selective staining of leukemic B-cells (CD5+/CD19+ populations) by multivalent compounds, dext-KMS31 and dext-KMS32, was detected by SA-PE signaling on flow cytometry (BD LSR II).
Results
Combinatorial Library Screening Provides Non-peptidic Ligands for an Unmutated, Stereotyped CLL BCR
We set as our target BCRs that belong to stereotype 7P, an example of U-CLL BCRs. After expressing the soluble form of the BCR from a patient in this sub-type (CLL 014) as an IgG in HEK 293T cells, the antibody was exposed to a one bead one compound library of molecules displayed on 90-μm TentaGel beads as a way to identify ligands for the antigen-specific binding site. The theoretical diversity of this library was just under one million compounds. As shown in Fig. 1, following a common linker, the variable region of the library contains two peptoid residues followed by a branch point derived from 4-azidoproline, from which two “arms” were constructed that contain units designed to impose conformational constraints (16, 17) (Fig. 1). The synthesis and characterization of this library are discussed in detail elsewhere.3
Screening was conducted largely as described previously (9). The library was first cleansed of beads that display ligands to the conserved regions of IgG antibodies or to antigen-binding sites of non-CLL antibodies. To do so, the beads were pre-blocked in 3% BSA and exposed to commercially available human total IgG, followed by anti-human IgG Fc antibody conjugated to red-emitting quantum dots. After thorough washing, the beads were examined under a fluorescent microscope and those that exhibited an obvious red halo were removed from the population manually using a micropipette (Fig. 1). The remainder of the library was then incubated with 100 nm soluble CLL 014 mAb. After another wash, the beads were incubated with labeled secondary antibody for visualization under the fluorescent microscope. A total of 83 brightly fluorescent beads were picked as putative hits.
Given the propensity of this procedure to yield false positives (19, 20), these beads were carried through another cycle of screening. First, the beads were washed with a denaturing buffer to remove any bound protein. After re-equilibration in a native buffer, the 83 beads were re-exposed to pooled human IgG and secondary antibody. Any beads that acquired a red fluorescent halo were discarded. The rest of the beads were incubated with CLL 014 mAb and subsequently with a secondary antibody. This final step of screening resulted in a total of 19 beads that fluoresced brightly. These beads were considered potential hits.
Next, these beads were placed in individual wells of a 96-well plate and treated with CNBr to release the compounds into solution via cleavage of the methionine residue in the conserved linker (Fig. 1). The molecules were analyzed by a combination of MALDI-TOF/TOF and ESI/LTQ-ETD mass spectrometry (21) (supplemental Fig. S2). From these data, the structures of 11 hits were elucidated unambiguously (supplemental Fig. S3). All of these compounds were re-synthesized and purified by HPLC (supplemental Fig. S4). In each case, a C-terminal cysteine was included to facilitate site-specific labeling with fluorescein or biotin for fluorescence polarization experiments or surface immobilization.
Hit Validation
To examine binding of the hits to the CLL 014 IgG, both fluorescence anisotropy and ELISAs were employed. Fluorescein or biotin was conjugated to the C-terminal cysteine and the molecules were HPLC purified. The anisotropy data (Fig. 2) showed that many of the compounds exhibited low to moderate affinity for CLL 014 IgG, but three of the compounds, KMS30, KMS31, and KMS32, demonstrated relatively robust binding. The KD values of the ligand-CLL 014 IgG complexes derived from these plots ranged from 70 to 200 nm with KMS31 being the highest affinity ligand (KD = 67 ± 11 nm). The ELISA experiments using the biotinylated compounds immobilized on streptavidin-coated plates showed the same trend (Fig. 2).
FIGURE 2.

Validation of binding of initial hit compounds. A, fluorescence polarization assay to validate the binding of hit compounds against recombinant CLL 014 IgG. Serially diluted CLL 014 mAb was incubated with 10 nm fluorescein-tagged compounds/control in 1× PBST containing 1% BSA at room temperature for 45 min in the dark before recording fluorescence polarization using a Tecan Plate Reader (Infinite M100Pro). B, ELISA of five highest affinity compounds and a control compound with the same linker and branched backbone but random side chains (as shown in Fig. 2). Biotinylated compound was immobilized on a streptavidin-coated 96-well ELISA plate and treated with increasing concentrations of CLL014 IgG followed by anti-human IgG1-HRP-conjugated secondary antibody. The plate was treated with TMB reagent and the binding data were recorded on a Tecan Plate Reader (Infinite M100Pro).
To further explore the association and dissociation properties of the complexes of CLL 014 IgG with KMS31 and KMS32, we carried out BLI experiments using the Octet Red96 System. Streptavidin sensors were coated with biotinylated KMS31 or KMS32 and these were dipped into wells containing varying concentrations of soluble CLL 014 Ig to monitor association and dissociation kinetics (Fig. 3). The binding affinities measured (KD) from the association and dissociation sensograms are shown in Table 1. KMS31 exhibited the highest affinity for CLL 014 Ig (KD = 51 ± 11 nm), which was in good agreement with the ELISA data. The kinetic association and dissociation constants were 2.44 × 104 m−1 s−1 and 1.25 × 10−3 s−1, respectively. Under these conditions, the half-life of the complex was ∼550 s.
FIGURE 3.
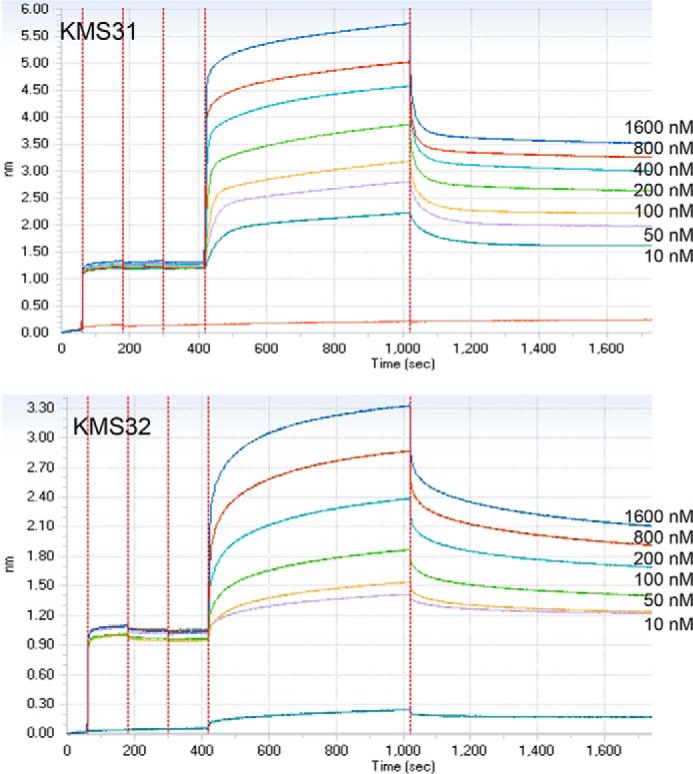
BLI assay for binding affinity measurements. Two highest affinity compounds, KMS31 and KMS32 (biotin tagged), were immobilized on streptavidin sensors. The kinetic measurements were carried out by exposing sensors with serially diluted soluble CLL 014 IgG in binding buffer (1× PBS, pH 7.4 containing 1% BSA) in wells of a 96-well microtiter plate. The association and dissociation profiles of the compounds was monitored at 30 °C using an Octet RED96 system (Pall ForteBio). Binding curves were analyzed by global fitting of sensorgrams to a 1:1 binding model using data analysis software provided by Pall ForteBio.
TABLE 1.
Binding affinities and association and dissociation kinetic parameters determined by ELISA and Biolayer interferometry assay (Octet) for synthetic ligands (KmS31 and KmS32) against CLL 014 BCR Ig
| Ligands | ELISA KD | Octet |
||
|---|---|---|---|---|
| kon | koff | KD | ||
| nm | 1/ms | 1/s | nm | |
| KmS31 | 67 ± 13 | 2.44 × 104 | 1.25 × 10−3 | 51 ± 11 |
| KmS32 | 167 ± 39 | 7.98 × 103 | 1.13 × 10−3 | 141 ± 27 |
To examine the selectivity of these three ligands, the immobilized biotinylated species were titrated with CLL 014 IgG, or IgGs derived from CLL BCRs outside of subset 7P (CLL 068 and CLL 169) as well as total human IgG using the ELISA format. Note that CLL 068 uses the same unmutated IGHV as the selecting mAb CLL 014. As shown in Fig. 4, KMS31 and KMS32 exhibited good selectivity for binding to CLL 014 as opposed to other human antibodies. KMS30, on the other hand, showed comparatively higher “off target” binding to the other human IgGs, so further characterization efforts were focused on KMS31 and KMS32.
FIGURE 4.

Binding selectivity of the highest affinity CLL 014 ligands. Biotin-tagged KMS30–32 or a control molecule not reactive with CLL 014 IgG was immobilized on streptavidin-coated ELISA plates and titrated with CLL 014, CLL 169, or CLL 068 monoclonal IgGs or a mixture of non-CLL human IgGs. None of the other antibodies represent the subset 7P to which the CLL 014 IgG belongs. The structures of the molecules are shown on the left and the binding curves on the right. Data analysis and curve fitting were performed with GraphPad Prism 6.0 using a nonlinear regression method. Error bars indicate the standard deviation of data obtained from three independent experiments.
To determine whether a similar level of selectivity is observed in a more native-like environment where the IgG is displayed on a cell surface, CLL 014, CLL 068, CLL 169, and CLL 183 IgGs were expressed on cells as surface immunoglobulins (smIg) by inserting a transmembrane anchoring domain at the C terminus of the heavy chain using methods described previously (9). HEK 293T cells were co-transfected with heavy and light chain expression vectors and the expression levels of surface membrane IgGs were determined by staining the cells with anti-human IgG Fc-specific antibody conjugated to allophycocyanin (anti-huIgFc-APC). Flow cytometry analysis confirmed expression of all 4 BCR IgGs of subset 7P on HEK 293T cells (supplemental Fig. S5).
To assess binding of KMS31 and KMS32 to these cells, the ligands were conjugated to a biotinylated dextran polymer that displays an average of 20–30 ligands per polymer chain (9, 22). They were then incubated with cells and binding was assayed by flow cytometry after staining with PE-conjugated streptavidin (Fig. 5). The dextran conjugates are employed in this type of experiment because the relatively rapid dissociation rate of the monomeric ligands (Fig. 3) preclude the use of flow cytometry for analysis of binding, as discussed previously (9). The multivalent dextran conjugates provide strong avidity-driven, long-lived binding. As shown in Fig. 5, both of the dextran-conjugated compounds, dext-KMS31 and dext-KMS32, stained the cells only when they were expressing CLL 014 smIg. Much lower levels of binding were observed for cells expressing the other CLL smIgs not belonging to subset 7P, including CLL mAb068 that uses the same IGHV1–69.
FIGURE 5.

Selective binding of dextran-conjugated multimeric ligands on HEK 293T cells expressing CLL smIgs. Flow cytometry analysis of dextran-conjugated multimeric ligand binding to cells displaying CLL 014 smIg and other non-selecting control smIgs as controls on HEK 293T cells. The VH expression vectors were re-engineered by introducing a transmembrane domain at the C terminus of the CH3 constant region to express CLL BCR as smIgG. HEK 293T cells were transiently transfected with pIg γ-transmembrane and Igλ/κ plasmid pairs for CLL 014, CLL 068, CLL 169, and CLL 183 monoclonal Igs. The expression levels (bottom row) of the smIgs were determined by staining cells with anti-human Ig Fc-conjugated to allophycocyanin (anti-huIg Fc-APC) and analyzed on flow cytometry. The cells expressing smIgs were treated with bt-dext-KMS31 or bt-dext-KMS32 followed by staining with PE-conjugated streptavidin (sa-PE) before analysis by flow cytometry. Both of these multimeric ligands were selectively binding to cells only when they were expressing CLL 014 smIg. As shown, no significant staining was observed when cells were expressing non-selecting control CLL smIgs. bt-dex, biotinylated dextran.
KMS31 and KMS32 Recognize Multiple BCRs within Subset 7P
With selective ligands for the CLL 014 BCR in hand, we turned to the critical question of whether these compounds would recognize other BCRs in subset 7P. Biotinylated KMS31 or KMS32 were immobilized on a neutravidin-coated 96-well ELISA plate or streptavidin sensors (for Octet) and titrated with increasing amounts of soluble monoclonal IgGs that represent other BCRs from subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297; see Fig. 6 for the sequences of the CDR3s) or BCRs from CLL patients outside of subset 7P (CLL 169 and CLL 068). CLL 169 is a mutated, non-stereotyped CLL mAb, whereas CLL 068 is an unmutated CLL mAb from a different subset (subset 6) that nevertheless uses the same IGHV1–69 (8). A mixture of commercially available human IgG was used as an additional control. As shown in Fig. 7, the Octet and ELISA data show clearly that KMS31 and KMS32 recognized the subset 7P CLL IgGs to a much greater extent than IgGs outside of this subset. Quantitatively, the KD values of the complexes between KMS31 and the other subset 7P IgGs are ∼4- to 7-fold higher (weaker binding) compared with the KD of the KMS31-CLL 014 IgG complex (Fig. 8 and Table 2). For KMS32, the range was 2- to 3.5-fold. Most importantly, no significant binding was observed for either compound against the other non-subset 7P control U-CLL or M-CLL Igs and against polyclonal human IgG.
FIGURE 6.

VH and VL CDR3 sequence homology of BCRs in subset 7P. Protein sequence alignments and sequence logos of the variable heavy and light chain CDR3s of U-CLL Igs from stereotyped subset 7P.
FIGURE 7.
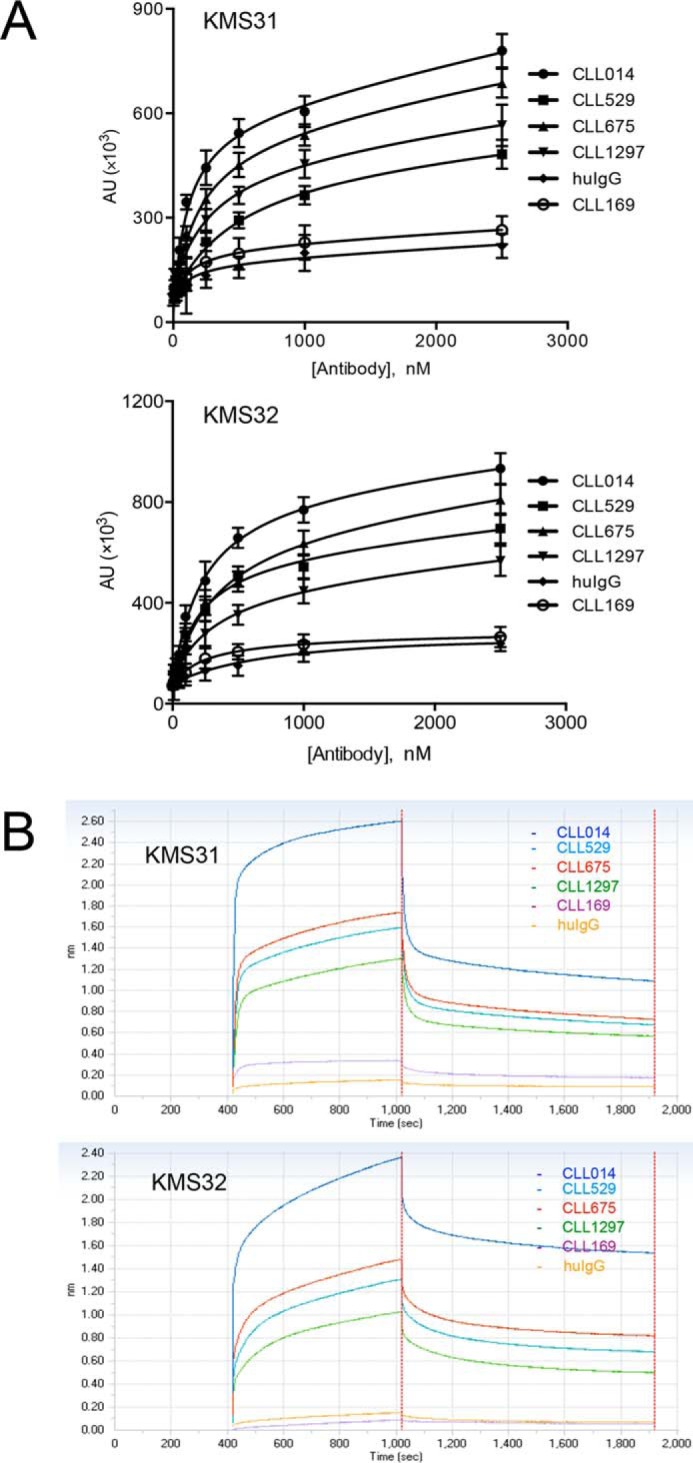
Reactivity of CLL 014 ligands with other CLL IgGs within same stereotypic subset. A, biotinylated KMS31 and KMS32 immobilized on neutravidin-coated ELISA plate were treated with CLL 014 IgG and other CLL IgGs belonging to the same stereotyped subset 7P as CLL 014 to determine their binding affinities. Data analysis and curve fitting were performed with GraphPad Prism 6.0 using a nonlinear regression method. Error bars indicate the standard deviation of data obtained from three independent experiments. B, BLI kinetic binding sensograms for association/dissociation of KMS31 and KMS32 to stereotyped CLL IgGs from U-CLL subset 7P. Biotinylated compounds were immobilized on streptavidin sensors and the sensograms were recorded using the Octet Red96 system. Both of these experiments exhibited similar binding profiles with varying degree of reactivity of CLL 014 ligands (KMS31 and KMS32) toward CLL 529, CLL 675, and CLL 1297 IgGs. No significant binding was observed with CLL 169 IgG, which is outside of subset 7P, or with a mixture of human IgGs. The KD values derived from association and dissociation curves are listed in Table 2.
FIGURE 8.

BLI assay for binding reactivity determination. Kinetic measurements of CLL014 ligands (KMS31 and KMS32) against other U-CLL IgGs from the same stereotyped subset (subset 7P) were carried out using BLI. The biotinylated compounds were immobilized on streptavidin sensors and the kinetic measurements were carried out by exposing sensors with serially diluted soluble monoclonal IgGs (CLL 014, CLL 529, CLL 675, and CLL 1297) in binding buffer (1× PBS, pH 7.4, containing 1% BSA) in wells of a 96-well microtiter plate. The association and dissociation profiles of the compounds was monitored at 30 °C using an Octet RED96 system (Pall ForteBio). Binding curves were analyzed by global fitting of sensorgrams to a 1:1 binding model using data analysis software provided by Pall ForteBio.
TABLE 2.
Binding affinities determined by ELISA and Biolayer interferometry assay (Octet) for synthetic ligands (KmS31 and KmS32) against stereotyped BCR Igs from U-CLL patients in subset 7P
| Subset 7p IgG |
KmS31 |
KmS32 |
||
|---|---|---|---|---|
| ELISA KD | Octet KD | ELISA KD | Octet KD | |
| nm | ||||
| CLL014 | 67 ± 13 | 51 ± 11 | 167 ± 39 | 141 ± 37 |
| CLL529 | 331 ± 63 | 264 ± 57 | 391 ± 67 | 347 ± 59 |
| CLL675 | 239 ± 041 | 197 ± 36 | 237 ± 51 | 271 ± 33 |
| CLL1297 | 425 ± 73 | 340 ± 66 | 529 ± 90 | 489 ± 86 |
Again, these biochemical assays were supplemented by analyzing binding of the KMS31- and KMS32-dextran conjugates to HEK 293T cells expressing the subset 7P smIgs (CLL 014, CLL 529, CLL 675, and CLL 1297) and other control smIgs using the assay depicted in Fig. 9A. Subsequent staining with PE-conjugated streptavidin for flow cytometry analysis revealed that both KMS31 and KMS32 dextran conjugates showed significant staining of the cells expressing stereotyped CLL 529, CLL 675, and CLL 1297 smIgs, but not cells expressing smIgs outside subset 7P (Fig. 9, B and C).
FIGURE 9.

Flow cytometry analysis of ligand binding to cells displaying stereotyped CLL smIgs on HEK 293T cells. A, general scheme of the cell binding assay with dextran-conjugated multimeric ligands. CLL mAbs from CLL 014, CLL 529, CLL 675, and CLL 1297 and other control CLL IgG were transiently expressed on the HEK 293T cell surface. The cells were incubated with a biotinylated dextran polymer displaying 20–30 copies of the KMS31 or KMS32 and controls followed by staining with phycoerythrin-conjugated streptavidin (sa-PE). B, overlaid flow cytometry histograms showing the binding of dext-KMS31 on the cells expressing stereotyped smIgs from subset 7P and controls. C, overlaid flow cytometry histograms showing the binding of dext-KMS32 on cells expressing stereotyped smIgs from subset 7P and controls.
Selective Binding of KMS31 and KMS32 to Primary CLL Cells from U-CLL “Subset 7P” Patients
CLL cells are known to express BCRs at lower density compared with healthy B-cells. Thus, whereas it is encouraging that the dextran conjugates of KMS31 and KMS32 bind to HEK 293T cells expressing subset 7P smIgs, it is important to analyze binding to bona fide patient-derived CLL B-cells. Frozen PBMCs from patients belonging to subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297) as well as CLL patients outside subset 7P were available in the collection at The Feinstein Institute for Medical Research. The frozen PBMCs were thawed and dead cells were removed by density gradient separation using Ficoll-Paque. The populations enriched for living cells were allowed to recover for 2 h before further manipulation. First, the isotype and expression levels of BCR Igs were confirmed by staining subset 7P and control cells with anti-human IgM (Fc5μ fragment-specific) antibody (Fig. 10).
FIGURE 10.

Flow cytometry analysis of B-cell receptor expression on patient-derived leukemic CLL B-cells. Expression of B-cell receptor, IgM antibodies, on leukemic CLL B-cells from stereotyped subset 7P patients (CLL 014, CLL 529, CLL 675, and CLL 1297), control B-cells from a random CLL patient and MEC1 IgM positive B-cells. The cells were stained with R-phycoerythrin AffiniPure F(ab′)2 fragment donkey anti-human IgM, Fc5μ-fragment specific, and analyzed by flow cytometry.
With expression of the BCR established, the cells were incubated with biotin-tagged dextran conjugates (KMS31, KMS32, or a control compound, not selected as a CLL 014 ligand, as shown in Figs. 4 and 11A). After washing, the cells were incubated with Po-Pro dye (to stain dead cells), as well as streptavidin-conjugated PE, anti-CD5-APC antibody, and anti-CD19-FITC antibody. CLL cells and T cells, but not healthy B-cells, express CD5 (23), whereas only B-cells (CLL and normal cells), but not T cells, express CD19, allowing these three cell types to be distinguished by these two markers. Double positive CD5+/CD19+ CLL cells (Fig. 11B, quadrant 2 (Q2)) were gated and monitored for staining by the synthetic molecule-dextran-streptavidin-conjugated PE by flow cytometry. Similarly, other populations of the blood lymphocytes (CD5+/CD19− T cells, CD5−/CD19+, and CD5−/CD19− cells; Q1 and Q3/Q4, respectively, in Fig. 11B) in PBMCs from the same CLL patient were also monitored for dextran conjugate interaction, which would reflect off target binding.
FIGURE 11.

Binding of CLL 014 ligands to lymphocytes. A, schematic representation of the assay employed. KMS31, KMS32, or a control compound was conjugated to a biotinylated dextran polymer. The construct was then mixed with PBMCs from CLL patients belonging to stereotyped subset 7P and binding was analyzed by flow cytometry. A PE-conjugated streptavidin (SA-PE) was used for monitoring binding, anti-human CD5-APC and CD19-FITC antibodies for selecting leukemic B-cells, and Po-Pro dye to label dead cells. PBMCs from a CLL patient outside of subset 7P and MEC1 cells (known to express IgM antibody as the surface BCR) were used as controls. B, dot plot to display populations of different lymphocytes in PBMCs including CLL B (CD5+/CD19+) cells in quadrant Q2. C, dext-KMS31 binding to lymphocytes. SA-PE staining of CD5+/CD19+/Po-Pro− cells from CLL patients from subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297) (left), T cells (Q1, center), and non-CLL B-cells (Q3/Q4, right). D, dext-KMS32 binding to lymphocytes. SA-PE staining of CD5+/CD19+/Po-Pro− cells from CLL patients from subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297) (left), T-cells (Q1, center), and non-CLL B-cells (Q3/Q4, right). E, binding of the dext-control compound to CD5+/CD19+/Po-Pro− cells from CLL patients from subset 7P (CLL 014, CLL 529, CLL 675, and CLL 1297). F, binding of dext-KMS31 and dext-KMS32 to the MEC1 B-cell line.
The results are shown in Fig 11. The dextran-KMS31 (Fig. 11C) and dextran-KMS32 (Fig. 11D) conjugates bound to the CLL 014 cells robustly, but the control dextran conjugate did not (Fig. 11E). In each case, the CLL 014 cells were slightly better with the dextran conjugates than the other subset 7P cells, CLL 529, CLL 675, and CLL 1297, presumably reflecting the somewhat lower affinity of the ligands for these BCRs. Nonetheless, all of the CLL subset 7P cells were well separated from the control populations in experiments employing KMS31 and KMS32. The KMS31- and KMS32-dextran conjugates did not stain CLL cells from patients that do not belong to subset 7P (Fig. 11, C and D, left panels) nor did they stain T cells or CD5-B-cells from any of the patients (Fig. 11, C and D, middle and right panels). In addition, no binding was observed when the conjugates were mixed with the IgM-expressing B-cell line derived from a distinct CLL patient, called MEC1 (24) (Fig. 11F). These data show clearly that the novel synthetic ligands KMS31 and KMS32 bind robustly to bona fide CLL cells that have stereotyped subset 7P BCRs and that binding is highly selective with little off target interactions with other B-cells or T cells.
Discussion
A major goal in translational cancer research is to develop effective therapies that have little or no toxicity to healthy cells. In this paper, we explore a novel strategy of this sort for the development of therapeutics for CLL. As mentioned above, CLL involves the accumulation of a single antigen-specific B-cell clone, arguing for antigen stimulation. In theory because of the tremendous diversity in the structure of antigen-binding sites on Ig that recombination, N-addition, and somatic mutation impart, no other cell in the body displays this receptor, making these pathogenic BCRs attractive targets for therapy.
Moreover, most CLL B-cells are frozen at the smIg stage of B-cell development and therefore do not differentiate into plasma cells. Hence, the blood of CLL patients does not contain high levels of antibodies corresponding to the antigen-specific BCR displayed on the CLL cells. Therefore, interception of the “drug” by secreted Ig before it reaches the target cell is much less likely than for other B-cell malignancies for which the malignant cell is at a later stage of B-cell maturation and the levels of soluble Ig are likely higher, e.g. Hairy Cell Leukemia and non-Hodgkins lymphoma.
However, many hurdles must be surmounted to execute this strategy. First, the antigen sites of BCRs are not generally considered druggable targets. There are a few reports of the identification of peptide ligands for CLL BCRs (6–8) but, prior to our work (9), no examples of non-biological, proteolytically stable CLL BCR ligands. Although, the peptide ligands against CLL 014 U-CLL BCR identified by Chiorazzi and co-workers (15) were selective, they bound with low affinity. Indeed the binding was too weak to measure a binding constant, and highlight the major improvement realized with the ligands reported here. Moreover, the BCRs from highly aggressive U-CLL patients are highly polyreactive (15), which is thought to be a reflection of a “looser” and more dynamic antigen-binding pocket (13, 14) because these receptors have not undergone somatic hypermutation. This could make them more difficult targets for the development of highly selective and potent drugs (7). In addition, there is the question of whether or not BCR-targeted drugs would be absolutely patient-specific. However, sequencing studies have revealed the existence of several CLL stereotyped subsets that account for about one-third of all CLL cases (10, 25). The high level of sequence homology between the BCRs of patients within one of these stereotyped subsets suggests that a compound isolated against a given stereotyped BCR might exhibit considerable binding affinity for other Igs in the same subset.
The data reported here show that relatively high affinity, non-peptidic, serum-stable (Fig. 12) ligands for a stereotyped U-CLL BCR can indeed be discovered through combinatorial library screening. We identified several molecules from a library of almost one million bead-displayed targets that bound a soluble, IgG version of a CLL BCR from stereotyped subset 7P (8). Two of these, KMS31 and KMS32, proved to have high affinity and selectivity for the CLL 014 IgG. Thorough in vitro characterization of the binding properties of these two molecules showed that they did indeed bind well to three other CLL IgGs from patients in subset 7P, with only a modest 2- to 7-fold reduction in affinity, and with little or no binding to IgGs outside of this subset. Experiments using dextran conjugates of these molecules and HEK 293T cells expressing CLL smIgGs provided similar results. Most importantly, as shown in Fig. 11, the KMS31- and KMS32-dextran targets stained patient-derived primary CLL cells if and only if they displayed a stereotyped subset 7P BCR. These data provide strong support that it should indeed be feasible to develop BCR-targeted drugs applicable to the treatment of most or all CLL patients in a given stereotyped subset.
FIGURE 12.

Serum stability of non-peptidic branched oligomer ligands. Synthetic ligands KMS31 and KMS32 were incubated in human serum at 37 °C. Aliquot of samples were taken out at specific time intervals as shown, mixed with internal standard, and purified on a C18-spin column before analysis by ESI/LC-MS. Total ion chromatograms of KMS31 and KMS32 at different time intervals are shown.
KMS31 and KMS32 are primary screening hits from a completely unbiased combinatorial library (Fig. 1). Thus it will be important, and should be quite feasible, to affinity mature them into much higher potency BCR-targeting agents. We have shown recently that this kind of hit improvement can be achieved through the construction of “derivative libraries” that explore chemical space around the structure of the primary hit in hopes of optimizing the fit of the molecule in the protein pocket (18). Another important step will be to tether KMS31, KMS32, or improved derivatives to some type of toxic moiety or immunological effector function to kill the CLL target cells. This work is underway.
Author Contributions
M. S., C. R., N. C., and T. K. designed the research; M. S., Y. L., J. Q., H. P., and J. M. performed the research; M.S., C.R., N. C., and T. K. wrote the paper.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants 1 DP3 DK094309 (to T. K.) and CA081554 (to N. C.), and a contract from the NHLBI, Stanford Proteomics Center, NO1-HV-00242 (to T. K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S5 and “Materials and Methods.”
M. Sarkar and T. Kodadek, unpublished data.
- CLL
- chronic lymphocytic leukemia
- BCR
- B-cell receptor
- BLI
- bio-layer interferometry
- PBMC
- peripheral blood mononuclear cell
- smIG
- surface immunoglobulin
- PE
- phycoerythrin.
References
- 1. Esencay M., and Hamad B. (2015) The chronic lymphocytic leukaemia market. Nat. Rev. Drug Discov. 14, 381–382 [DOI] [PubMed] [Google Scholar]
- 2. Chiorazzi N., Rai K. R., and Ferrarini M. (2005) Chronic lymphocytic leukemia. N. Engl. J. Med. 352, 804–815 [DOI] [PubMed] [Google Scholar]
- 3. Jaglowski S. M., Alinari L., Lapalombella R., Muthusamy N., and Byrd J. C. (2010) The clinical application of monoclonal antibodies in chronic lymphocytic leukemia. Blood 116, 3705–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byrd J. C., Furman R. R., Coutre S. E., Flinn I. W., Burger J. A., Blum K. A., Grant B., Sharman J. P., Coleman M., Wierda W. G., Jones J. A., Zhao W., Heerema N. A., Johnson A. J., Sukbuntherng J. et al. (2013) Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 369, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J., Baskar S., Kwong K. Y., Kennedy M. G., Wiestner A., and Rader C. (2011) Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS ONE 6, e21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buhl L., Szecsi P. B., Gisselø G. G., and Schafer-Nielsen C. (2002) Surface immunoglobulin on B lymphocytes as a potential target for specific peptide ligands in chronic lymphocytic leukaemia. Br. J. Haematol. 116, 549–554 [DOI] [PubMed] [Google Scholar]
- 7. Liu Y., Higgins C. D., Overstreet C. M., Rai K. R., Chiorazzi N., and Lai J. R. (2013) Peptides that bind specifically to an antibody from a chronic lymphocytic leukemia clone expressing unmutated immunoglobulin variable region genes. Mol. Med. 19, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seiler T., Woelfle M., Yancopoulos S., Catera R., Li W., Hatzi K., Moreno C., Torres M., Paul S., Dohner H., Stilgenbauer S., Kaufman M. S., Kolitz J. E., Allen S. L., Rai K. R. et al. (2009) Characterization of structurally defined epitopes recognized by monoclonal antibodies produced by chronic lymphocytic leukemia B cells. Blood 114, 3615–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarkar M., Liu Y., Morimoto J., Peng H., Aquino C., Rader C., Chiorazzi N., and Kodadek T. (2014) Recognition of antigen-specific B cell receptors chronic lymphocytic leukemia patients by synthetic “antigen surrogates”. Chem. Biol. 21, 1670–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agathangelidis A., Darzentas N., Hadzidimitriou A., Brochet X., Murray F., Yan X. J., Davis Z., van Gastel-Mol E. J., Tresoldi C., Chu C. C., Cahill N., Giudicelli V., Tichy B., Pedersen L. B., Foroni L., et al. (2012) Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood 119, 4467–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agathangelidis A., Vardi A., Baliakas P., and Stamatopoulos K. (2014) Stereotyped B-cell receptors in chronic lymphocytic leukemia. Leuk. Lymphoma 55, 2252–2261 [DOI] [PubMed] [Google Scholar]
- 12. Chiorazzi N., and Ferrarini M. (2011) Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood 117, 1781–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adhikary R., Yu W., Oda M., Walker R. C., Chen T., Stanfield R. L., Wilson I. A., Zimmermann J., and Romesberg F. E. (2015) Adaptive mutations alter antibody structure and dynamics during affinity maturation. Biochemistry 54, 2085–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adhikary R., Yu W., Oda M., Zimmermann J., and Romesberg F. E. (2012) Protein dynamics and the diversity of an antibody response. J. Biol. Chem. 287, 27139–27147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Catera R., Silverman G. J., Hatzi K., Seiler T., Didier S., Zhang L., Hervé M., Meffre E., Oscier D. G., Vlassara H., Scofield R. H., Chen Y., Allen S. L., Kolitz J., Rai K. R. et al. (2008) Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol. Med. 14, 665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aquino C., Sarkar M., Chalmers M. J., Mendes K., Kodadek T., and Micalizio G. C. (2012) A biomimetic polyketide-inspired approach to small-molecule ligand discovery. Nat. Chem. 4, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao Y., and Kodadek T. (2013) Synthesis and screening of stereochemically diverse combinatorial libraries of peptide tertiary amides. Chem. Biol. 20, 360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao Y., Amar S., Pahwa S., Fields G., and Kodadek T. (2015) Rapid lead discovery through iterative screening of one bead one compound libraries. ACS Comb. Sci. 17, 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lian W., Upadhyaya P., Rhodes C. A., Liu Y., and Pei D. (2013) Screening bicyclic peptide libraries for protein-protein interaction inhibitors: discovery of a tumor necrosis factor-α antagonist. J. Am. Chem. Soc. 135, 11990–11995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doran T. M., Gao Y., Mendes K., Dean S., Simanski S., and Kodadek T. (2014) The utility of redundant combinatorial libraries in distinguishing high and low quality screening hits. ACS Comb. Sci. 16, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarkar M., Pascal B. D., Steckler C., Aquino C., Micalizio G. C., Kodadek T., and Chalmers M. J. (2013) Decoding split and pool combinatorial libraries with electron-transfer dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 24, 1026–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morimoto J., Sarkar M., Kenrick S., and Kodadek T. (2014) Dextran as a generally applicable multivalent scaffold for improving immunoglobulin-binding affinities of peptide and peptidomimetic ligands. Bioconjug. Chem. 25, 1479–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yan X. J., Albesiano E., Zanesi N., Yancopoulos S., Sawyer A., Romano E., Petlickovski A., Efremov D. G., Croce C. M., and Chiorazzi N. (2006) B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 103, 11713–11718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stacchini A., Aragno M., Vallario A., Alfarano A., Circosta P., Gottardi D., Faldella A., Rege-Cambrin G., Thunberg U., Nilsson K., and Caligaris-Cappio F. (1999) MEC1 and MEC2: two new cell lines derived from B-chronic lymphocytic leukaemia in prolymphocytoid transformation. Leuk. Res. 23, 127–136 [DOI] [PubMed] [Google Scholar]
- 25. Messmer B. T., Albesiano E., Efremov D. G., Ghiotto F., Allen S. L., Kolitz J., Foa R., Damle R. N., Fais F., Messmer D., Rai K. R., Ferrarini M., and Chiorazzi N. (2004) Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J. Exp. Med. 200, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.