

Abstract
Lysophosphatidylcholine acyltransferase 3 (Lpcat3) is involved in phosphatidylcholine remodeling in the small intestine and liver. We investigated lipid metabolism in inducible intestine-specific and liver-specific Lpcat3 gene knock-out mice. We produced Lpcat3-Flox/villin-Cre-ERT2 mice, which were treated with tamoxifen (at days 1, 3, 5, and 7), to delete Lpcat3 specifically in the small intestine. At day 9 after the treatment, we found that Lpcat3 deficiency in enterocytes significantly reduced polyunsaturated phosphatidylcholines in the enterocyte plasma membrane and reduced Niemann-Pick C1-like 1 (NPC1L1), CD36, ATP-binding cassette transporter 1 (ABCA1), and ABCG8 levels on the membrane, thus significantly reducing lipid absorption, cholesterol secretion through apoB-dependent and apoB-independent pathways, and plasma triglyceride, cholesterol, and phospholipid levels, as well as body weight. Moreover, Lpcat3 deficiency does not cause significant lipid accumulation in the small intestine. We also utilized adenovirus-associated virus-Cre to deplete Lpcat3 in the liver. We found that liver deficiency only reduces plasma triglyceride levels but not other lipid levels. Furthermore, there is no significant lipid accumulation in the liver. Importantly, small intestine Lpcat3 deficiency has a much bigger effect on plasma lipid levels than that of liver deficiency. Thus, inhibition of small intestine Lpcat3 might constitute a novel approach for treating hyperlipidemia.
Keywords: cholesterol metabolism, gene knockout, lipid absorption, lipoprotein metabolism, lipoprotein secretion, phosphatidylcholine, phospholipid metabolism, triacylglycerol
Introduction
The majority of lipids on the cell membrane as well as the plasma lipoproteins are phospholipids, in particular phosphatidylcholines (PCs)2 (1, 2). Monounsaturated and saturated fatty acids are usually esterified at the sn-1 position of PCs, whereas polyunsaturated fatty acids are esterified at the sn-2 position (3). The asymmetric distribution of fatty acids at the sn-1 and sn-2 positions of PCs is maintained in part by a deacylation-reacylation process known as the Lands cycle or PC remodeling (3, 4). One key enzyme in this remodeling is lysophosphatidylcholine acyltransferase (Lpcat), which utilizes lyso-PC and polyunsaturated acyl-CoA to produce sn-2 polyunsaturated PCs. There are four isoforms of this enzyme (5), and Lpcat3 is the major isoform in the small intestine and liver (6–8).
Lpcat3 is one of the downstream targets of liver X receptor (8) and peroxisome proliferator-activated receptor α (6). Acute knockdown of Lpcat3 expression in the liver of genetically obese mice exacerbates lipid-induced endoplasmic reticulum (ER) stress (8). Moreover, global Lpcat3 knock-out (KO) mice exhibit neonatal lethality and have an abnormal small intestine (9, 10). Liver-specific deletion of Lpcat3 has no effect on ER stress but results in a reduction of plasma triglycerides and induction of hepatosteatosis under high fat feeding conditions (9). Because of neonatal lethality (non-inducible approach was utilized), only 1-week-old newborns were analyzed for the impact of intestine-specific Lpcat3 deficiency on lipid metabolism (9). Very recently, we found that the neonatal lethality of global Lpcat3 KO mice could be rescued by oral administration of PC plus olive oil. However, the KO mice had shorter and wider small intestinal villi and a longer and larger small intestine (11). Thus, the following remains to be investigated: 1) the impact of small intestine Lpcat3 deficiency in adult mice on lipid metabolism; and 2) the respective contribution of small intestine and liver Lpcat3 to plasma lipid levels.
In this study, we specifically ablated Lpcat3 in the small intestine of mice using an inducible Cre-LoxP approach. Our results indicate that this ablation reduces the levels of plasma cholesterol, triglycerides, and phospholipids. We also evaluate the effect of liver-specific Lpcat3 deficiency on plasma lipid metabolism. We found that small intestine but not liver Lpcat3 deficiency has a dominant effect on plasma lipid metabolism.
Materials and Methods
Generation of Intestine-specific Lpcat3-deficient Mice
Lpcat3-Flox mice (prepared by inGenious Targeting Laboratory) were crossed with Villin-Cre-ERT2 transgenic mice. A 9-kb segment of the regulatory region of the mouse Villin gene drives expression of Cre recombinase fused to a mutated ligand-binding domain of the human estrogen receptor. Cre recombination is inducible with tamoxifen treatment in epithelial cells all along the crypt-villus axis and in undifferentiated progenitor cells in the crypt region. We established Lpcat3-Flox/Villin-Cre-ERT2 mice according to the strategy shown in Fig. 1A. The genotype was confirmed by polymerase chain reaction (PCR) (Fig. 1B). To delete the gene in the small intestine, tamoxifen (2 mg/mouse, dissolved in 200 μl of corn oil (Sigma C8267)) was injected intraperitoneally on four alternate days. Lpcat3-Flox mice injected with tamoxifen were used as controls. Both male and female 12-week-old mice on a C57BL6J background were used in this study. All studies were approved by the Institutional Animal Care and Use Committee of State University of New York Downstate Medical Center and conformed with the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health (publication 85-23, revised in 1996).
FIGURE 1.

Generation and characterization of inducible intestine-specific Lpcat3 KO mice. A, strategy used to disrupt mouse Lpcat3 in the small intestine. B, mouse genotyping. Two-month-old wild-type (WT) and Lpcat3 KO female mice were treated with tamoxifen. C, Lpcat3 mRNA in different tissues was measured by real time PCR at day 9. D, Lpcat mRNA levels in the small intestine on day 9 post-tamoxifen treatment. E, total Lpcat activity in the small intestine measured on day 9. F, hematoxylin and eosin staining of the small intestine. Values are means ± S.D., n = 7, *, p < 0.01.
Liver-specific Lpcat3-deficient Mouse Preparation
We injected (intraperitoneally) AAV-Cre (1 × 1012) into the Lpca3-Flox mice. Because the Cre recombinase expression was driven by the liver-specific thyroxine-binding globulin promoter (12), the AAV approach resulted in sustained Cre expression in the liver for at least 20 weeks (13).
mRNA Measurement
Mice were sacrificed by cervical dislocation. The jejunum and liver were dissected, and total RNA was extracted from each tissue using the RNeasy mini kit (Qiagen). cDNA was synthesized with the SuperscriptTM III first-strand synthesis kit (Invitrogen). PCR was performed in a total volume of 20 μl with the SYBR Green kit from Applied Biosystems. The amplification program was as follows: activation at 95 °C for 10 min followed by 40 amplification cycles of 95 °C for 15 s and 60 °C for 60 s. Each sample was assayed in triplicate. The genes encoding 18S rRNA and GAPDH were used as internal controls. Relative gene expression is expressed as the mean ± S.D. Mouse Lpcat3 primers forward, TTTCTGGTTCCGCTGCATGT, and reverse, CCGACAGAATGCACACTCCTTC, and 18SrRNA primers forward, AGTCCCTGCCCTTTGTACACA, and reverse, GATCCGAGGGCCTCACTAAAC, were used.
Lipoprotein/Apolipoprotein Analysis and Lipid Absorption
Lipoproteins and apolipoproteins (apoA-I, apoB, and apoE) in mice (both sexes) were measured as described (7). Cholesterol absorption studies were performed with male mice using the conventional fecal dual-isotope ratio method (14). Absorption of triglycerides and phospholipids was measured with male mice using our reported method (15).
Primary Enterocyte Preparation and Plasma Membrane Isolation
Primary enterocytes were isolated according to two previous reports (14, 16). The plasma membrane was isolated from primary enterocytes according to our protocol (17).
Measuring Total Lpcat Activity and PC Subspecies
Total Lpcat activity was measured as described (7). PC subspecies were measured with liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) as described (7).
Western Blotting
Primary enterocyte homogenates were subjected to Western blotting as described (11) with antibodies against NPC1L1 (Niemann-Pick C1-like 1) (a gift from Dr. Bao Liang Song, Chinese Academy of Sciences), CD36 (Abcam), ATP-binding cassette transporter 8 (ABCG8; Novus Biologicals), ABCA1 (Novus Biologicals), MTP (BD Transduction Laboratories), and fatty acid transport protein 4 (FATP4; Santa Cruz Biotechnology, and SAR1B (Proteintech). GAPDH was used as a loading control.
Hematoxylin and Eosin Staining
The small intestine was dissected out and fixed with 4% formalin. The tissue was embedded in paraffin and then sliced (5 μm thick). Each slice was deparaffinized and stained with hematoxylin and eosin.
Immunohistochemical Staining
Prior to antibody staining, the sections were deparaffinized in xylene, rehydrated in a gradient series of ethanol, and then subjected to high temperature antigen retrieval in 50 mm Tris-HCl (pH 9.0) containing 1 mm EDTA. Sections were permeabilized in 50 mm Tris buffer (pH 7.4) containing 0.5% (w/v) Triton X-100 and 5% horse serum. The sections were incubated with anti-NPC1L1 or anti-villin overnight at 4 °C. The sections were then incubated with fluorescently labeled secondary antibodies.
Statistical Analysis
Data are expressed as the mean ± S.D. Data between two groups was analyzed by the unpaired, two-tailed Student's t test. A p value of less than 0.05 was considered as significant.
Results
Production of Intestine-specific Inducible Lpcat3 KO Mice
To prepare intestine-specific Lpcat3-deficient mice, tamoxifen (2 mg/mouse), dissolved in corn oil, was injected intraperitoneally into female mice at days 1, 3, 5, and 7. Tamoxifen-treated Lpcat3-Flox mice were used as controls. We collected plasma and tissues on day 9 after the first treatment and measured Lpcat3 mRNA in the small intestine, liver, kidney, and adipose tissue. Compared with controls, Lpcat3 mRNA level was decreased by 90% in the small intestine but no other tissues (Fig. 1C). The levels of the remaining Lpcat isoforms (Lpcat1, -2, and -4) did not change in these Lpcat3-deficient mice (Fig. 1D). We then measured total Lpcat activity in the small intestine homogenate and found it was decreased by 85% in the Lpcat3-deficient mice compared with controls (Fig. 1E). Moreover, we did not observe morphological changes in the small intestine of Lpcat3-deficient mice (Fig 1F). Similar results were obtained with male mice (data not shown).
Lpcat3 Deficiency in the Small Intestine Significantly Decreases Body Weight and Plasma Lipid Levels
Lpcat3-deficient female mice underwent a significant loss of body weight compared with the controls (Fig. 2A) at day 9 after tamoxifen treatment. We next measured fasting plasma lipid levels in the Lpcat3 KO and control female mice. Lpcat3 deficiency significantly decreased plasma levels of total cholesterol (47%, p < 0.01), total phospholipids (51%, p < 0.01), and triglycerides (81%, p < 0.01) as compared with controls (Fig. 2, B–D). The lipid reduction was also observed at day 12 after tamoxifen treatment (Fig. 2, B–D). Similar results were obtained with male mice (data not shown).
FIGURE 2.

Body weight and plasma lipoprotein levels in wild-type and Lpcat3-deficient female mice. Two-month-old control and Lpcat3-Flox/Villin-Cre-ERT2 female mice were treated with tamoxifen. At day 9 and day 12 after treatment, the following parameters were measured. A, body weight. B–D, total cholesterol (B), phospholipid (C), and triglyceride (D) levels in plasma. E, fluorogram and quantitation of plasma apolipoprotein levels by Western blotting at day 9 after tamoxifen treatment. Proteins in plasma (0.2 μl) were separated with SDS-PAGE (4–15% gradient gel) and immunoblotted with polyclonal antibodies against apoA-I, apoB, apoE, and albumin. F, plasma lipoprotein distribution at day 9 after tamoxifen treatment assessed by FPLC. G, small intestine lipid levels. TG, triglyceride; Chol, total cholesterol; SM, sphingomyelin. Values are means ± S.D., n = 7, *, p < 0.01.
Assessment of plasma apolipoprotein levels in Lpcat3 KO and control female mice by Western blotting revealed that Lpcat3 deficiency (day 9 after tamoxifen treatment) in the gut significantly reduced plasma apoB48 (86%, p < 0.001), apoB100 (55%, p < 0.01), and apoA-I (75%, p < 0.001) levels compared with the controls (Fig. 2E). There was no significant difference in plasma apoE levels (Fig. 2E).
Plasma lipid distributions in pooled plasma were examined by fast protein liquid chromatography (FPLC). Cholesterol level was dramatically decreased in both high density lipoprotein (HDL) and non-HDL fractions from the Lpcat3-deficient mice compared with controls (Fig. 2F). Triglyceride in non-HDL was dramatically decreased in the deficient mice (Fig. 2F). Similar results were obtained with male mice (data not shown). We also measured small intestine lipids, including triglyceride, total cholesterol, phosphatidylcholine, and sphingomyelin, and we did not find significant changes (Fig. 2G).
To evaluate the effect of Lpcat3 deficiency on plasma polyunsaturated PCs, we used LC-MS/MS to measure PCs in the mice at day 9 after tamoxifen treatment. The sn-2 polyunsaturated PCs (16:0/18:2, 18:1/18:2, 18:1/18:3, and 18:0/20:4) were significantly decreased (p < 0.01; Table 1) in Lpcat3-deficient mice, suggesting that small intestine Lpcat3 contributes significantly to the spectrum of circulating polyunsaturated PCs.
TABLE 1.
Measurement of PC subspecies by LC-MS/MS
Values are means ± S.D.; n = 5; *, p < 0.05; **, p < 0.01.
| 16:0/16:0 | 16:0/18:2 | 18:0/18:0 | 18:0/18:1 | 18:1/18:1 | 18:1/18:2 | 18:1/18:3 | 18:0/20:4 | |
|---|---|---|---|---|---|---|---|---|
| Plasma (μg/ml) | ||||||||
| Control | 5 ± 1 | 250 ± 51 | 5 ± 2 | 33 ± 7 | 216 ± 20 | 78 ± 10 | 67 ± 11 | 69 ± 9 |
| Lpcat3 KO | 4 ± 1 | 53 ± 9** | 4 ± 1 | 27 ± 6 | 187 ± 39 | 29 ± 5** | 16 ± 3** | 19 ± 3** |
| Enterocyte plasma membrane (ng/mg protein) | ||||||||
| Control | 113 ± 18 | 5663 ± 519 | 92 ± 17 | 81 ± 13 | 4539 ± 634 | 612 ± 75 | 1815 ± 328 | 2203 ± 339 |
| Lpcat3 KO | 164 ± 22* | 2852 ± 331** | 316 ± 57** | 388 ± 41** | 5954 ± 851 | 396 ± 63* | 788 ± 93** | 723 ± 88** |
| Small intestine homogenate (ng/mg protein) | ||||||||
| Control | 163 ± 29 | 7663 ± 990 | 66 ± 11 | 132 ± 19 | 6231 ± 701 | 832 ± 91 | 2123 ± 425 | 1959 ± 271 |
| Lpcat3 KO | 209 ± 42* | 4568 ± 289** | 196 ± 31** | 299 ± 28** | 6602 ± 981 | 501 ± 79* | 932 ± 120** | 559 ± 65** |
| Liver homogenate (ng/mg protein) | ||||||||
| Control | 268 ± 38 | 8172 ± 1076 | 106 ± 19 | 340 ± 55 | 8782 ± 917 | 718 ± 73 | 3324 ± 551 | 2223 ± 385 |
| Lpcat3 KO | 298 ± 27 | 7392 ± 923 | 129 ± 33 | 389 ± 42 | 9021 ± 1102 | 750 ± 90 | 3267 ± 422 | 2390 ± 343 |
| Kidney homogenate (ng/mg protein) | ||||||||
| Control | 815 ± 105 | 2765 ± 370 | 202 ± 32 | 166 ± 26 | 4519 ± 434 | 591 ± 57 | 2538 ± 318 | 1678 ± 220 |
| Lpcat3 KO | 933 ± 87 | 2983 ± 289 | 251 ± 20 | 180 ± 30 | 4705 ± 378 | 606 ± 71 | 2498 ± 263 | 1543 ± 291 |
Lpcat3 Deficiency Significantly Decreases Lipid Absorption in the Small Intestine
A defect in lipid uptake could be one of the major reasons for the observed lower plasma lipid profiles. We thus measured cholesterol absorption using a conventional fecal dual-isotope ratio method at day 9 after tamoxifen treatment. This involved the gavage of a single bolus of 0.1 μCi of [14C]cholesterol and 0.2 μCi of [3H]sitostanol in 20 μl of olive oil, with feces being collected after 48 h. We found that the Lpcat3 KO mice absorbed significantly less cholesterol than controls (Fig. 3A). Moreover, there was much less [14C]cholesterol in the plasma of Lpcat3 KO mice (Fig. 3B). We also measured triglyceride and PC absorption using [3H]triolein and [14C]arachidonyl-PC. The mice were gavaged with both radiolabeled lipids, and blood radioactivity was monitored 2, 4, and 8 h later. Lpcat3 deficiency significantly decreased [3H]glycerolipids (Fig. 3C) and [14C] glycerolipids in blood (Fig. 3D).
FIGURE 3.

Lipid absorption in Lpcat3 KO and control mice. Lpcat3-Flox/Villin-Cre-ERT2 and control female mice were treated with tamoxifen. At day 9 post-treatment, the mice were gavaged with 0.1 μCi of [14C]cholesterol and 0.2 μCi of [3H]sitostanol in 20 μl of olive oil. A, feces were collected at 48 h post-treatment, and lipids were extracted for counting. B, [14C]cholesterol in blood. C and D, female mice were gavaged with 0.2 μCi of [3H]triolein and 0.1 μCi of [14C]PC in 20 μl of olive oil. Blood was collected over 8 h and assayed for the presence of [3H]glycerolipids (C) and [14C]glycerolipid (D). Values are means ± S.D., n = 7, *, p < 0.01.
We hypothesized that Lpcat3 deficiency might decrease polyunsaturated PCs in the enterocyte apical membrane, resulting in diminished receptor density or membrane-associated transporter levels, thus reducing dietary lipid uptake and plasma lipid levels. We analyzed the populations of PC subspecies in the enterocyte plasma membrane using LC-MS/MS and found that Lpcat3 deficiency decreased the amount of polyunsaturated PCs (16:0/18:2, 18:1/18:2, 18:1/18:3, and 18:0/20:4) in the plasma membrane (Table 1). These changes could impact lipid uptake by the apical membrane. We also measured lyso-PCs but did not find significant changes (data not shown). We measured PC subspecies in the liver and kidney homogenates of the deficient mice and controls, and we did not find significant changes (Table 1).
We next used Western blotting to measure levels of NPC1L1, CD36, ABCA1, and ABCG8 in enterocyte homogenates; the level of each of these proteins was significantly decreased in Lpcat3-deficient primary enterocytes compared with controls (Fig. 4, A and B). We also measured MTP and FATP4 levels and did not find any significant differences in MTP. Interestingly, however, Lpcat3 deficiency increased FATP4 levels as a compensatory mechanism.
FIGURE 4.

Western blotting and immunostaining. A and B, Western blot fluorograms (A) and quantitation (B) of NPC1L1, ABCA1, ABCG8, CD36, FATP4, and MTP in enterocyte homogenates from Lpcat3 KO and control small intestine. GAPDH was used as a loading control. Values are means ± S.D., n = 5, *, p < 0.05. C, immunostaining for small intestine NPC1L1 and Villin. The data are representative of three independent experiments. D, primary enterocytes were isolated and treated with cycloheximide (20 μg/ml), and cells were harvested at 0, 1.5, and 3 h after treatment. Western blot was performed on cell homogenates. The signal was scanned and decay curve was drawn. Values are means ± S.D., n = 3.
To rationalize the defects in lipid absorption, we performed immunofluorescence staining for the localization of NPC1L1 in the small intestine. NPC1L1 co-localized with the plasma membrane marker villin in the small intestine of control mice (Fig. 4C). This co-localization was greatly reduced, however, in the small intestine of Lpcat3-deficient mice, especially at the tip of the villus. We also isolated primary enterocytes from both control and Lpcat3 KO mice and treated the cells with cycloheximide to block protein biosynthesis. Then we did Western blot for NPC1L1 at different time points. We did not find significant changes in the NPC1L1 protein decay curve within 3 h, suggesting Lpcat3 deficiency might not influence NPC1L1 protein stability (Fig. 4D).
We next immuno-stained stem cells in the crypt, using leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5) antibody (18), and we did not find significant changes (Fig. 5). We also measured SREBP-1c and PPARα levels, as well as their downstream target mRNA levels, and we did not find significant changes (data not shown).
FIGURE 5.

Stem cell staining. Leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5) antibody was used to immuno-stain stem cells in the crypt of the small intestine. The staining was indicated by red arrows. The data are representative of three independent experiments.
Enterocytes secrete cholesterol as a part of apoB lipoproteins into lymph and as a part of HDL into the circulation (14, 19). The latter is influenced by the ABCA1 level (19). Because we found that both NPC1L1 and ABCA1 levels were significantly reduced in enterocytes (Fig. 4, A and B), we next assessed the two aspects of cholesterol absorption, uptake and secretion. Primary enterocytes were incubated with [14C]cholesterol for 1 h, after which Lpcat3 KO enterocytes were found to contain significantly less [14C]cholesterol compared with controls (25%, p < 0.05; Fig. 6A), indicating a defect in cholesterol uptake. The radiolabeled cells were then chased in the presence of oleic acid for 2 h. Lpcat3 KO enterocyte medium contained much less [14C]cholesterol compared with control medium (45%, p < 0.01; Fig. 6B). The conditioned medium was then subjected to density gradient ultracentrifugation to determine the effect of Lpcat3 ablation on cholesterol secretion with chylomicrons and HDL. Cholesterol secreted by control enterocytes was distributed in two separate fractions that corresponded to chylomicrons (fractions 1 and 2) and HDL (fractions 8–10) (Fig. 6C). Similar analyses with Lpcat3 KO enterocytes revealed that cholesterol secretion as part of chylomicron and HDL was significantly reduced (81 and 80% respectively, p < 0.01, Fig. 6C). These studies indicated that cholesterol uptake and secretion (through the chylomicron and HDL pathways) were diminished in the Lpcat3-deficient animals.
FIGURE 6.
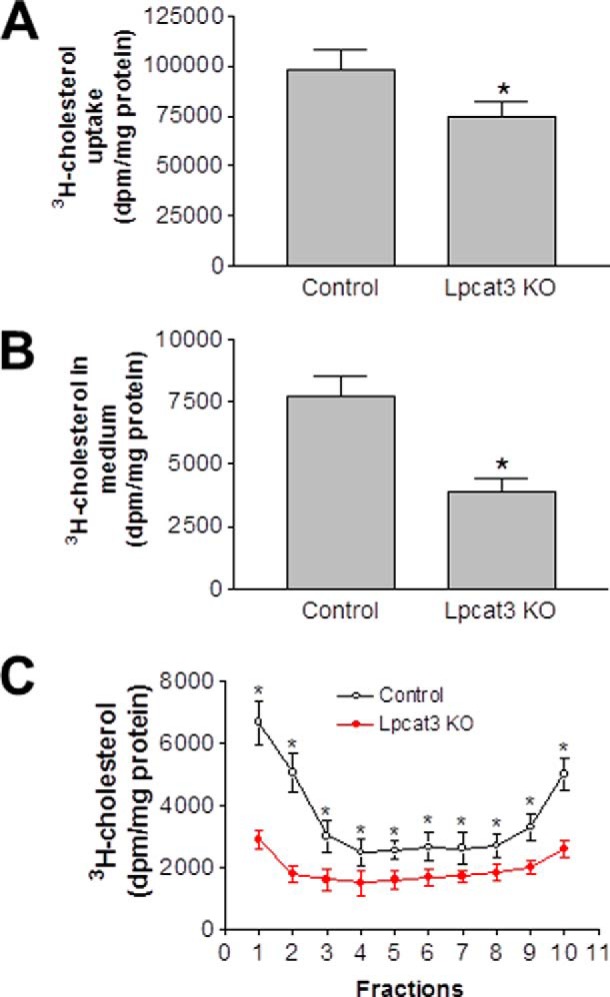
Decreased cholesterol transport across Lpcat3 KO enterocytes. Lpcat3-Flox/Villin-Cre-ERT2 and control female mice were treated with tamoxifen. At day 9 post-treatment, primary enterocytes were isolated and incubated with 0.05 μCi/ml of [14C]cholesterol as well as unlabeled cholesterol (0.5 mg/ml) for 1 h and washed, and then the radioactivity was counted (A). Cells were also incubated with medium supplemented with oleic acid-containing micelles. After 2 h, the medium was collected and radioactivity counted (B). The conditioned medium was subjected to density gradient ultracentrifugation to determine the distribution of secreted cholesterol in different lipoproteins. Fractions were collected from the top and used to measure cholesterol in triplicate (C). Fractions 1 and 2 represent large and small chylomicrons, respectively. Fractions 8–10 represent intestinal HDL. Values are means ± S.D., n = 3, *, p < 0.01.
It has been reported that Lpcat3 protects against ER stress in the liver (8). To determine whether this also applies to the small intestine, we measured mRNA levels for three well known ER stress markers, namely PERK, IRE-1α, and BIP. PERK and IRE-1α levels were significantly reduced in Lpcat3 KO enterocytes compared with controls, whereas the BIP level was not significantly altered (Fig. 7).
FIGURE 7.
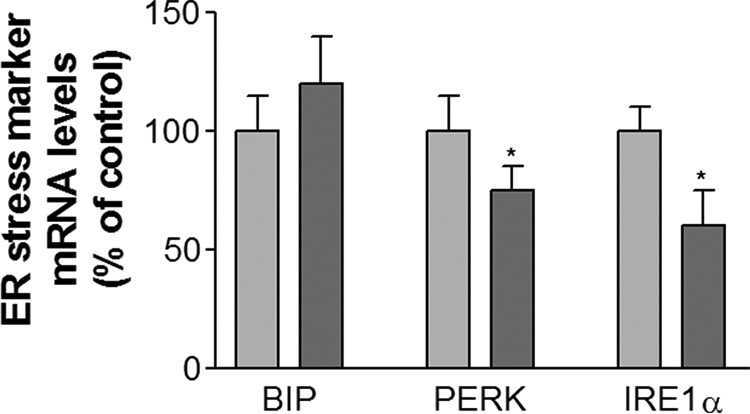
Small intestine lipid and ER stress marker measurements. Lpcat3-Flox/Villin-Cre-ERT2 and control female mice were treated with tamoxifen. At day 9, small intestines were used to measure ER stress markers. Values are means ± S.D. n = 5. *, p < 0.05.
Lpcat3 Deficiency in the Liver Decreases Plasma Triglyceride but Not Other Lipid Levels
We also injected (intraperitoneally) AAV-Cre (1 × 1012) into the Lpcat3-Flox mice to prepare liver-specific Lpcat3-deficient mice. AAV-LacZ was used as controls. We measured liver and small intestine Lpcat3 mRNA levels in AAV-LacZ and AAV-Cre-treated mice (after 1 month) using real time PCR, and we found the liver-specific Lpcat3 depletion (Fig. 8A). Moreover, we found that AAV-Cre-treated Lpcat3-Flox male animals significantly decreased liver total Lpcat activity (67%, p < 0.001) and plasma triglyceride (40%, p < 0.05) but not other lipid and apolipoprotein levels (Fig. 8, B–D). Also, we did not find any significant changes in liver triglyceride, cholesterol, and phospholipid levels between the two groups (Fig. 8E). We stained the livers and did not find morphological difference between liver Lpcat3-deficient and control mice (Fig. 8F). These results were consistent with a previous report under chow diet (9).
FIGURE 8.

Liver Lpcat3-deficient mice. Lpcat3-Flox male mice were treated with AAV-LacZ (control) and AAV-Cre. At day 30 post-treatment, the liver was excised, and the following were measured. A, Lpcat3 mRNA; B, total Lpcat activity; C, plasma triglyceride, (TG), total cholesterol (Chol), and phospholipid (PL) levels; D, apolipoprotein levels. E, liver lipid levels. Values are means ± S.D., n = 5, *, p < 0.05. F, liver sections were stained with hematoxylin and eosin. The data are representative of three independent experiments.
Discussion
PC is the major phospholipid of mammalian cell membranes and also is abundant in circulating lipoproteins (20, 21). Although it is not completely understood why so many different PC species exist in nature, it is likely that the relative levels of these different PC species in membranes determine membrane fluidity and thus help cells adapt to environmental changes or other physiological needs. With the cloning of Lpcat family members in recent years, it has become clear that these enzymes are responsible for PC remodeling and the generation of polyunsaturated PCs in various tissues. Lpcat3 is the major Lpcat isoform in the small intestine and liver (6). It has been shown that ablation of Lpcat3 in the small intestine at early stages of life is neonatally lethal and diminishes the incorporation of arachidonic acid into PCs within the cell membrane, leading to the impaired triglyceride mobilization and lipoprotein production (9, 10). We previously reported that the neonatal lethality of global Lpcat3 KO mice could be rescued. However, the KO mice had shorter and wider small intestinal villi and a longer and larger small intestine (11). Thus, the impact of Lpcat3 deficiency on small intestine-related lipoprotein metabolism in adult animals remains unknown.
In this study, we used an inducible intestine-specific Lpcat3-deficient and a liver-specific Lpcat3-deficient mouse model to investigate the role of PC remodeling on plasma lipoprotein metabolism. Our major findings are as follows. 1) Small intestine-specific Lpcat3 deficient in adult mice have dramatically reduced all plasma lipid and lipoprotein levels, whereas liver deficiency only reduces plasma triglyceride levels at a lower extent compared with intestinal deficiency (40% versus 81%) (Figs. 2D and 8B). A previous report indicated that albumin-Cre-mediated liver Lpcat3 deficiency only caused about 25% reduction of plasma triglyceride levels with no changes of other lipids (9). Thus, manipulation Lpcat3 activity in the small intestine has a dominant effect on plasma lipid metabolism. 2) Under a chow diet-fed condition, Lpcat3 deficiency in adult mice do not cause lipid accumulation in the small intestine and liver. 3) The small intestine deficiency results in a significant reduction of NPC1L1, CD36, ABCA1, and ABCG8 in the enterocyte plasma membrane, thus reducing lipid uptake and reducing cholesterol secretion through both apoB-dependent and apoB-independent pathways. It is conceivable that inhibition of small intestine Lpcat3 might constitute a novel approach for treating hyperlipidemia.
It has been reported that ablation of Lpcat3 reduces arachidonoyl-PCs in the embryonic and postnatal small intestine (9, 10). We found that Lpcat3 deficiency in the small intestine significantly decreased polyunsaturated PCs, including 16:0/18:2, 18:1/18:2, 18:1/18:3, and 18:0/20:4 (Table 1), and this modification could disrupt the structural integrity and fluidity of the plasma membrane. We also found that intestinal Lpcat3 deficiency significantly decreased all circulating lipids and lipoproteins (Fig. 2, B–F). These phenotypes are the manifestation of the lipid absorption defects in the Lpcat3-deficient small intestine (Fig. 3, A–D) because liver Lpcat3 expression was not influenced (Fig. 1C).
Dietary lipid absorption occurs in the lumen of the small intestine and on the apical surface of enterocytes. NPC1L1 and ABCG5/8 are the two major factors mediating net cholesterol uptake. The former mediates cholesterol uptake, and the latter mediates excretion of excessive cholesterol into the intestinal lumen (22, 23). CD36 also participates in cholesterol uptake at the brush border of enterocytes (24, 25). Three proteins, namely CD36 (25), plasma membrane-associated fatty acid-binding protein (26), and FATP4 (27), are involved in free fatty acid uptake by enterocytes. The former two proteins are located on the apical surface of enterocytes, and the latter is located on the ER membrane (24). It is conceivable that ablation of Lpcat3 may reduce the incorporation of polyunsaturated PCs in the enterocyte plasma membrane with a consequent increase in membrane rigidity, which may perturb endocytosis and protein recruitment to the plasma membrane and ultimately lead to defective lipid uptake and secretion. Indeed, intestinal cholesterol absorption is mediated by endocytosis of NPC1L1 (28), and change in the composition of polyunsaturated PCs affects the endocytosis process (29, 30).
Although the Lpcat3-deficient small intestine did not show obvious morphological alterations (Fig. 1F), the levels of both NPC1L1 and CD36 were significantly reduced in Lpcat3-deficient primary enterocytes compared with control mice (Fig. 4, A and B). Immunofluorescence staining revealed that NPC1L1 abundance was significantly reduced in Lpcat3-deficient villi compared with the control (Fig. 4C). These results represent a plausible explanation for the observed reduction in cholesterol and triglyceride absorption. The reduction of ABCG8 abundance (Fig. 4, A and B) could explain the compensation for defective cholesterol uptake in Lpcat3-deficient enterocytes. It has been shown that global deletion of Lpcat3 at an early stage of development results in the accumulation of lipid droplets in enterocytes (9, 10). However, we did not find any difference in lipid levels in the small intestine of Lpcat3-deficient and control mice (Fig. 2G). Thus, the effects of Lpcat3 deletion differ depending on whether the gene is deleted in utero or in adult tissues. This is not an uncommon phenomenon; indeed, depleting liver kinase B1 at different stages of life results in completely different phenotypes (31, 32).
Lpcat3 deficiency in the small intestine significantly decreased the concentration of plasma apoA-I (Fig. 2E). It has been demonstrated that the small intestine is one of the sources of apoA-I in blood (33, 34). Plasma apoA-I levels in healthy humans increases after ingestion of a fatty meal (35). Moreover, apoA-I can be transferred from chylomicrons to HDL (36). Thus, in our Lpcat3 KO mice, the observed significant reduction in plasma apoA-I level is the major outcome of defective absorption of cholesterol, triglycerides, and phospholipids (Fig. 3, A–D).
Does Lpcat3 deficiency affect lipid secretion? Based on studies of the small intestine in neonatal mice, two groups of researchers proposed models for the assembly of Lpcat3-related apoB-containing lipoproteins (9, 10). However, cholesterol secretion in cultured primary enterocytes is mediated by both apoB-dependent and -independent pathways (14). In this study, we found that Lpcat3 deficiency in primary enterocytes reduced not only cholesterol uptake (Fig. 6A) but also secretion (Fig. 6B). Moreover, the deficiency reduced cholesterol secretion through both the chylomicron and HDL pathways (Fig. 6C). This reflects the fact that Lpcat3 deficiency, at least in part, reduces polyunsaturated PCs within the enterocyte plasma membrane (Table 1). This modification influences basal as well as apical membrane PC composition. Indeed, we found a significant reduction in the level of ABCA1 (Fig. 4, A and B), which is located on the basal membrane of enterocytes and is a major player in the apoB-independent pathway (14, 37).
The results obtained from our liver Lpcat3-deficient mice (Fig. 8, A–D) echoed what has been reported previously (9), showing a reduction in total liver Lpcat activity and plasma triglyceride but no other lipid levels. Importantly, we also confirmed that Lpcat3 deficiency in the liver does not cause significant lipid accumulation in the liver under chow diet conditions (Fig. 8E) (9). Why then does liver deficiency have a smaller effect than that of the small intestine in terms of plasma lipid metabolism? One explanation could be due to the compensation of other Lpcats. Indeed, we previously reported that global Lpcat3 deficiency causes about 90% reduction of total Lpcat activity in the small intestine, whereas the deficiency only causes about a 70% reduction of total activity in the liver (11). We confirmed this phenomenon in this study (Fig. 1E and 8B).
Acute knockdown of liver Lpcat3 in genetically obese mice exacerbates lipid-induced ER stress (8). However, the same group recently reported that Lpcat3 deficiency in the liver does not affect the expression of ER stress markers (9). We also recently reported that global Lpcat3 deficiency has no impact on ER stress (11). In this study, we again measured the expression of ER stress markers in the Lpcat3-deficient small intestine and found that the deficiency attenuated (rather than enhanced) the expression of PERK and IRE-1α (Fig. 7).
In this study, the Lpcat3 depletion was achieved by tamoxifen treatment in adult Lpcat3-Flox/Villin-Cre-ERT2 mice over a period of 9 days so that the effect was acute, similar to drug intervention. Given the fact that small intestine Lpcat3 deficiency has 1) a much bigger impact on plasma lipid levels than that of liver deficiency, 2) no effect on lipid retention in the tissue, and 3) a marginal effect on ER stress, selective inhibition of Lpcat3 activity in the small intestine could be a novel approach for treating hyperlipidemia.
Author Contributions
I. K. and Z. L. designed and performed experiments, analyzed data, interpreted results, discussed implications, and critically evaluated the manuscript. H. H. B. and M. S. K. used measured PC subspecies using LC/MS/MS and reviewed the manuscript. G. G. provided AAV-LacZ and AAV-Cre and reviewed the manuscript. X. C. J. conceived the study, supervised the project, interpreted results, discussed implications, and wrote the manuscript.
Acknowledgments
We are grateful to Drs. Jahangir Iqbal and Mahmood Hussain for their technical support and Villin-Cre-ERT2 transgenic mice.
This work was supported by Veterans Affairs Merit Grant 000900-01 (to X. C. J.), National Institutes of Health Grant R56HL121409 (to X. C. J.), National Natural Scientific Foundation (NSF) of China Grant 31371190 (to X. C. J.), and Scientist Development Grant 10SDG4040054 from the American Heart Association (to Z. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PC
- phosphatidylcholine
- Lpcat
- lysophosphatidylcholine acyltransferase
- AAV
- adenovirus-associated virus
- ER
- endoplasmic reticulum
- MTP
- microsomal triglyceride transfer protein
- PERK
- protein kinase RNA-like endoplasmic reticulum.
References
- 1. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schiller J., Zschörnig O., Petković M., Müller M., Arnhold J., and Arnold K. (2001) Lipid analysis of human HDL and LDL by MALDI-TOF mass spectrometry and (31)P-NMR. J. Lipid Res. 42, 1501–1508 [PubMed] [Google Scholar]
- 3. Lands W. E. (2000) Stories about acyl chains. Biochim. Biophys. Acta 1483, 1–14 [DOI] [PubMed] [Google Scholar]
- 4. Lands W. E. (1958) Metabolism of glycerolipids; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 231, 883–888 [PubMed] [Google Scholar]
- 5. Hishikawa D., Shindou H., Kobayashi S., Nakanishi H., Taguchi R., and Shimizu T. (2008) Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. U.S.A. 105, 2830–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao Y., Chen Y. Q., Bonacci T. M., Bredt D. S., Li S., Bensch W. R., Moller D. E., Kowala M., Konrad R. J., and Cao G. (2008) Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J. Biol. Chem. 283, 8258–8265 [DOI] [PubMed] [Google Scholar]
- 7. Li Z., Ding T., Pan X., Li Y., Li R., Sanders P. E., Kuo M. S., Hussain M. M., Cao G., and Jiang X. C. (2012) Lysophosphatidylcholine acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine accumulation promotes very low density lipoprotein production by enhancing microsomal triglyceride transfer protein expression. J. Biol. Chem. 287, 20122–20131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rong X., Albert C. J., Hong C., Duerr M. A., Chamberlain B. T., Tarling E. J., Ito A., Gao J., Wang B., Edwards P. A., Jung M. E., Ford D. A., and Tontonoz P. (2013) LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 18, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rong X., Wang B., Dunham M. M., Hedde P. N., Wong J. S., Gratton E., Young S. G., Ford D. A., and Tontonoz P. (2015) Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. eLife 10.7554/eLife.06557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hashidate-Yoshida T., Harayama T., Hishikawa D., Morimoto R., Hamano F., Tokuoka S. M., Eto M., Tamura-Nakano M., Yanobu-Takanashi R., Mukumoto Y., Kiyonari H., Okamura T., Kita Y., Shindou H., and Shimizu T. (2015) Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. eLife 4, e06328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Z., Jiang H., Ding T., Lou C., Bui H. H., Kuo M. S., and Jiang X. C. (2015) Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology 149, 1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hayashi Y., Mori Y., Janssen O. E., Sunthornthepvarakul T., Weiss R. E., Takeda K., Weinberg M., Seo H., Bell G. I., and Refetoff S. (1993) Human thyroxine-binding globulin gene: complete sequence and transcriptional regulation. Mol. Endocrinol. 7, 1049–1060 [DOI] [PubMed] [Google Scholar]
- 13. Kitajima K., Marchadier D. H., Miller G. C., Gao G. P., Wilson J. M., and Rader D. J. (2006) Complete prevention of atherosclerosis in apoE-deficient mice by hepatic human apoE gene transfer with adeno-associated virus serotypes 7 and 8. Arterioscler. Thromb. Vasc. Biol. 26, 1852–1857 [DOI] [PubMed] [Google Scholar]
- 14. Iqbal J., and Hussain M. M. (2005) Evidence for multiple complementary pathways for efficient cholesterol absorption in mice. J. Lipid Res. 46, 1491–1501 [DOI] [PubMed] [Google Scholar]
- 15. Liu R., Iqbal J., Yeang C., Wang D. Q., Hussain M. M., and Jiang X. C. (2007) Phospholipid transfer protein-deficient mice absorb less cholesterol. Arterioscler. Thromb. Vasc. Biol. 27, 2014–2021 [DOI] [PubMed] [Google Scholar]
- 16. Jiang X. C., Qin S., Qiao C., Kawano K., Lin M., Skold A., Xiao X., and Tall A. R. (2001) Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat. Med. 7, 847–852 [DOI] [PubMed] [Google Scholar]
- 17. Li Z., Zhang H., Liu J., Liang C. P., Li Y., Li Y., Teitelman G., Beyer T., Bui H. H., Peake D. A., Zhang Y., Sanders P. E., Kuo M. S., Park T. S., Cao G., and Jiang X. C. (2011) Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol. Cell. Biol. 31, 4205–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barker N., and Clevers H. (2010) Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology 138, 1681–1696 [DOI] [PubMed] [Google Scholar]
- 19. Iqbal J., Parks J. S., and Hussain M. M. (2013) Lipid absorption defects in intestine-specific microsomal triglyceride transfer protein and ATP-binding cassette transporter A1-deficient mice. J. Biol. Chem. 288, 30432–30444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vance D. E. (2002) Phospholipid Biosynthesis in Eukaryotes, 4th Ed., Elsevier Science B. V. [Google Scholar]
- 21. Quehenberger O., Armando A. M., Brown A. H., Milne S. B., Myers D. S., Merrill A. H., Bandyopadhyay S., Jones K. N., Kelly S., Shaner R. L., Sullards C. M., Wang E., Murphy R. C., Barkley R. M., Leiker T. J., et al. (2010) Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altmann S. W., Davis H. R. Jr., Zhu L. J., Yao X., Hoos L. M., Tetzloff G., Iyer S. P., Maguire M., Golovko A., Zeng M., Wang L., Murgolo N., and Graziano M. P. (2004) Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303, 1201–1204 [DOI] [PubMed] [Google Scholar]
- 23. Yu L., York J., von Bergmann K., Lutjohann D., Cohen J. C., and Hobbs H. H. (2003) Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J. Biol. Chem. 278, 15565–15570 [DOI] [PubMed] [Google Scholar]
- 24. Pan X., and Hussain M. M. (2012) Gut triglyceride production. Biochim. Biophys. Acta 1821, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nassir F., Wilson B., Han X., Gross R. W., and Abumrad N. A. (2007) CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J. Biol. Chem. 282, 19493–19501 [DOI] [PubMed] [Google Scholar]
- 26. Stremmel W., Lotz G., Strohmeyer G., and Berk P. D. (1985) Identification, isolation, and partial characterization of a fatty acid binding protein from rat jejunal microvillous membranes. J. Clin. Invest. 75, 1068–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stahl A., Hirsch D. J., Gimeno R. E., Punreddy S., Ge P., Watson N., Patel S., Kotler M., Raimondi A., Tartaglia L. A., and Lodish H. F. (1999) Identification of the major intestinal fatty acid transport protein. Mol. Cell 4, 299–308 [DOI] [PubMed] [Google Scholar]
- 28. Li P. S., Fu Z. Y., Zhang Y. Y., Zhang J. H., Xu C. Q., Ma Y. T., Li B. L., and Song B. L. (2014) The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L1. Nat. Med. 20, 80–86 [DOI] [PubMed] [Google Scholar]
- 29. Koeberle A., Shindou H., Koeberle S. C., Laufer S. A., Shimizu T., and Werz O. (2013) Arachidonoyl-phosphatidylcholine oscillates during the cell cycle and counteracts proliferation by suppressing Akt membrane binding. Proc. Natl. Acad. Sci. U.S.A. 110, 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinot M., Vanni S., Pagnotta S., Lacas-Gervais S., Payet L. A., Ferreira T., Gautier R., Goud B., Antonny B., and Barelli H. (2014) Lipid cell biology. Polyunsaturated phospholipids facilitate membrane deformation and fission by endocytic proteins. Science 345, 693–697 [DOI] [PubMed] [Google Scholar]
- 31. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., and Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woods A., Heslegrave A. J., Muckett P. J., Levene A. P., Clements M., Mobberley M., Ryder T. A., Abu-Hayyeh S., Williamson C., Goldin R. D., Ashworth A., Withers D. J., and Carling D. (2011) LKB1 is required for hepatic bile acid transport and canalicular membrane integrity in mice. Biochem. J. 434, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glickman R. M., and Kirsch K. (1973) Lymph chylomicron formation during the inhibition of protein synthesis. Studies of chylomicron apoproteins. J. Clin. Invest. 52, 2910–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glickman R. M., and Green P. H. (1977) The intestine as a source of apolipoprotein A1. Proc. Natl. Acad. Sci. U.S.A. 74, 2569–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Glickman R. M., Green P. H., Lees R. S., and Tall A. (1978) Apoprotein A-I synthesis in normal intestinal mucosa and in Tangier disease. N. Engl. J. Med. 299, 1424–1427 [DOI] [PubMed] [Google Scholar]
- 36. Tall A. R., Green P. H., Glickman R. M., and Riley J. W. (1979) Metabolic fate of chylomicron phospholipids and apoproteins in the rat. J. Clin. Invest. 64, 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hussain M. M. (2014) Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 25, 200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]