

Abstract
The bacterial catabolism of lignin and its breakdown products is of interest for applications in industrial processing of ligno-biomass. The gallate degradation pathway of Pseudomonas putida KT2440 requires a 4-carboxy-2-hydroxymuconate (CHM) hydratase (GalB), which has a 12% sequence identity to a previously identified CHM hydratase (LigJ) from Sphingomonas sp. SYK-6. The structure of GalB was determined and found to be a member of the PIG-L N-acetylglucosamine deacetylase family; GalB is structurally distinct from the amidohydrolase fold of LigJ. LigJ has the same stereospecificity as GalB, providing an example of convergent evolution for catalytic conversion of a common metabolite in bacterial aromatic degradation pathways. Purified GalB contains a bound Zn2+ cofactor; however the enzyme is capable of using Fe2+ and Co2+ with similar efficiency. The general base aspartate in the PIG-L deacetylases is an alanine in GalB; replacement of the alanine with aspartate decreased the GalB catalytic efficiency for CHM by 9.5 × 104-fold, and the variant enzyme did not have any detectable hydrolase activity. Kinetic analyses and pH dependence studies of the wild type and variant enzymes suggested roles for Glu-48 and His-164 in the catalytic mechanism. A comparison with the PIG-L deacetylases led to a proposed mechanism for GalB wherein Glu-48 positions and activates the metal-ligated water for the hydration reaction and His-164 acts as a catalytic acid.
Keywords: bacterial metabolism, enzyme mechanism, lignin degradation, metalloenzyme, Pseudomonas, LigJ, PIG-L deacetylase, gallate, hydratase, protocatechuate
Introduction
Lignin, a complex biopolymer found in the plant cell wall, represents a large carbon reservoir in the environment. The polymer is built from nonrepeating aromatic units connected via many different linkages. Bacterial species from the genera Pseudomonas, Comamonas, and Sphingomonas utilize the aromatic metabolites produced from lignin depolymerization, and their catabolic capabilities can therefore be harnessed for industrial processing of biomass to produce valuable products such as biofuels (reviewed in Ref. 1).
Lignin is composed mainly of guaiacyl and syringyl units, which are connected through ether and biphenyl linkages (2). In strains of Pseudomonas, Comamonas, and Sphingomonas, diverse guaiacyl and biphenyl lignin derivatives are metabolized to vanillate (Fig. 1). Vanillate is subsequently demethylated into protocatechuate (PCA),2 which in turn is metabolized through the PCA 4,5-cleavage pathway (3–6). Syringyl lignin derivatives, however, are transformed into gallate, which is then metabolized through the gallate pathway (7). Both the PCA 4,5-cleavage and gallate pathways lead to the production of 4-oxalomesaconate (OMA) or its tautomer, 4-carboxy-2-hydroxy-muconate (CHM), with both tautomers being in equilibrium at physiological pH (8–10). An OMA tautomerase (galD) has been identified in a gallate utilization gene cluster of Pseudomonas putida KT2440, with homologous gene copies found in the canonical PCA 4,5-cleavage pathway, which enzymatically transforms OMA to CHM (9). In both the gallate and PCA 4,5-cleavage pathways CHM is transformed by a hydratase to produce 4-carboxy-4-hydroxy-2-oxoadipate (CHA), which is subsequently cleaved by an aldolase to produce the TCA cycle metabolites pyruvate and oxaloacetate. The aldolase from these pathways is commonly referred to as the 4-hydroxy-4-methyl-2-oxoglutarate (HMG)/CHA aldolase, as the first characterizations of the enzyme identified it as using HMG as a physiological substrate, although to date no enzymatic pathway to HMG production has been fully explicated (11, 12). Thus both the PCA 4,5-cleavage and gallate pathways have the same last three chemical transformations using an OMA tautomerase, CHM hydratase, and HMG/CHA aldolase. The OMA tautomerases and HMG/CHA aldolases found in the pathways share >50% sequence identities. However, the hydratases from the pathways, LigJ and GalB, share only a 12% sequence identity and are unlikely to be evolutionarily related.
FIGURE 1.

Metabolism of guaiacyl, syringyl, and biphenyl lignin metabolites by Pseudomonas and Sphingomonas. A, guaiacyl metabolites are metabolized to vanillate (VAN) that is O-demethylated by LigM to PCA, which is subsequently transformed through the protocatechuate 4,5-cleavage pathway. Sequential conversion of PCA to pyruvate (PYR) and oxaloacetate (OAA) is catalyzed by the following: LigAB, a 4,5-dioxygenase; 4-carboxy-2-hydroxymuconate semialdehyde (CHMS) is converted to the hemiacetyl form non-enzymatically; LigC, a CHMS dehydrogenase; LigI, a PDC lactonase that produces either OMA or CHM; LigJ, a CHM hydratase; and LigK, an HMG/CHA aldolase. Syringyl metabolites are metabolized to gallate (GA), which is transformed sequentially to pyruvate and oxaloacetate by: GalA, a gallate dioxygenase; GalD, a OMA tautomerase; GalB, a CHM hydratase; and GalC, a HMG/CHA aldolase. B, the lignin biphenyl DDVA is transformed to vanillate for utilization in the PCA 4,5-cleavage pathway by: LigX; a DDVA O-demethylase; LigZ, a 2,2′,3-trihydroxy-3′-methoxy-5,5′-dicarboxybiphenyl (HO-DDVA) dioxygenase; LigY, a HO-DDVA hydrolase, which produces both 5-carboxyvanillate (5CVA) and CHPD; and LigW/LigW2, a 5-carboxyvanillate O-demethylase. Here, the enzymatic conversion of CHPD to HMG by either GalB or LigJ hydratase is proposed. HMG is a known substrate for the HMG/CHA aldolase (LigK or GalC), which would produce 2 mol of pyruvate, connecting the CHPD metabolite to central cellular metabolism.
Both LigJ and GalB are divalent metal ion-dependent enzymes that have a preference for Zn2+ (9, 10). A structure of LigJ from Rhodopseudomonas palustris (PDB code 2GWG) has been determined and it is a member of the amidohydrolase II family (PF04909) comprising a (α/β)8 triose-phosphate isomerase (TIM) barrel that is structurally distinct from other characterized divalent metal-dependent hydratases (13–15). The regions in the substrate binding pocket of LigJ are not modeled in the final structure, and accordingly, the catalytic mechanism of this enzyme has not been elucidated. Like LigJ, GalB has no sequence similarity to the previously characterized divalent metal-dependent hydratases. The initial characterization of the gallate pathway identified (2E,4E)-CHM as the substrate for GalB, whereas (2Z,4E)-CHM is the proposed substrate for LigJ (8, 9). The proposed differences in substrate specificity between the two enzymes would give a biological rationale for the two possibly distinct CHM hydratases. However, no structural or mechanistic investigations have been reported in the literature to elucidate the mechanism of GalB and its relationship to LigJ CHM hydratases.
Herein we report the first structure and in-depth kinetic characterization of the GalB CHM hydratase from Pseudomonas putida KT2440. GalB, unlike LigJ, adopts a α/β Rossmann-like fold that resembles the PIG-L deacetylase family of hydrolytic enzymes. A comparative analysis of PmdE from Comamonas sp. strain E6 (herein referred to as LigJCsE6) and GalB indicates that both enzymes in fact utilize the same CHM isomer leading to the same (−)-CHA enantiomer product. LigJCsE6 shares 63 and 80% sequence identity to LigJ from Sphingomonas sp. SYK-6 and R. palustris, respectively. We also show that both GalB and LigJCsE6 catalyze the reversible hydration of 4-carboxy-2-hydroxypenta-2,4-dienoate (CHPD; a CHM analog lacking the C5 carboxylate) to HMG and propose a previously undescribed route for HMG production from the metabolism of the lignin metabolite 5,5′-dehydrodivanillate (DDVA). Together, our findings shed light on the analogous enzymes GalB and LigJ, which have evolved convergently to perform the same chemical reaction.
Experimental Procedures
Chemicals
l-Lactate dehydrogenase (rabbit muscle), l-malate dehydrogenase (porcine heart), and DOWEX® 1X8-200 ion-exchange resin were from Sigma-Aldrich. Restriction enzymes and Pfu polymerase were from Invitrogen or New England Biolabs. All other chemicals were analytical grade and were obtained from either Sigma-Aldrich or Fisher Scientific.
DNA Manipulations
pET29a plasmids harboring galA, galB, and galD from P. putida KT2440 were a generous gift from E. Díaz, Biological Research Center-CSIC, Madrid, Spain (9). GalB variants A16D, R21A, E48A, R67A, H127A, Y123A, H164A, and R216A were produced by site-specific mutagenesis using the QuikChange method (16). A GalB variant, GalBNt, containing a truncation of four residues from the N-terminal end of the gene product, was created with primers to amplify the region of interest with flanking NdeI and HindIII restriction sites. The amplified galBNt was digested and ligated into pT7-7 (17). Primer sequences utilized for mutagenesis and the creation of the GalBNt construct are available upon request. For native overexpression of GalB in P. putida KT2442, galB was subcloned from pET-29a into pVLT-31 using the XbaI and HindIII sites (18). The gene encoding PmdE (ligJCsE6) from Comamonas sp. strain E6 (GenBankTM accession number BAI50711.1) was codon-optimized for expression in Escherichia coli and was synthesized by Biobasic Canada Inc. (Markham, Ontario, Canada) (4). The sequence was designed with flanking NdeI and HindIII restriction sites, and ligJCsE6 was subcloned into pET28a for expression with an N-terminal hexahistidine tag. All plasmids were transformed into E. coli DH5α for propagation with gene sequences, and mutations were confirmed by DNA sequencing at the Guelph Molecular Supercenter (University of Guelph).
Gene Expressions
With the exception of the native expression of galB in Pseudomonas, all of the genes utilized in this study were recombinantly overexpressed in E. coli BL21(λDE3). E. coli cells harboring plasmids encoding galA, galD, ligJCsE6, galB, or galB enzyme variants were propagated in 1 liter of lysogeny broth (LB) containing the appropriate antibiotics for their respective plasmid selection. Cells were grown at 37 °C until an optical density of 0.5 at 600 nm wavelength was reached. For native expression of galB, P. putida KT2442 cells harboring pVLT31 encoding galB were grown in LB medium supplemented with 15 μg/ml tetracycline at 30 °C until an optical density of 0.5 at 600 nm was reached. In each case, protein expression was induced by the addition of 0.75 mm isopropyl β-d-thiogalactopyranoside with E. coli and P. putida cultures incubated overnight at 15 and 30 °C, respectively, before both were harvested by centrifugation at 5000 × g for 10 min.
Protein Purifications
Buffers containing 20 mm sodium-HEPES, pH 7.5, were used throughout each purification procedure unless indicated otherwise. Each cell pellet was resuspended in buffer and disrupted by French press at 1200 p.s.i. Cell debris was removed by centrifugation (17,500 × g for 15 min) and supernatants filtered through a 0.45-μm filter.
The purification of GalA, GalB, and GalB variants was carried out by chromatography on an ÄKTA Explorer 100 (GE Healthcare Life Sciences). All column resins were from GE Healthcare Life Sciences, and column chromatography utilized a SourceTM 15Q anion-exchange column (2 × 13 cm), phenyl-SepharoseTM hydrophobic interaction column (1 × 18.5 cm), and a HiLoad 26/60 Superdex 200 gel filtration column (2.6 × 60 cm).
For purification of GalB and variants, crude extract was loaded onto the anion-exchange column, and the column was washed with 2 column volumes of buffer followed by a linear gradient of NaCl from 0.0 to 0.3 m over 12 column volumes. GalB was eluted with ∼0.15 M NaCl. Active fractions were pooled and concentrated to ∼10 ml by ultrafiltration using a YM10 filter (Millipore, Nepean, Ontario, Canada). Ammonium sulfate was added to the concentrated protein extract to a final concentration of 1.0 m, and the extract was loaded onto the hydrophobic interaction column, which was pre-equilibrated with 1.0 m ammonium sulfate. A 12-column volume linear gradient from 1.0 to 0.0 m ammonium sulfate was applied, and GalB was eluted in ∼0.5 m ammonium sulfate. Active fractions were pooled, concentrated to ∼2 ml by ultrafiltration, and loaded onto the gel filtration column. Protein was eluted using an isocratic flow of buffer containing 0.15 m NaCl. The purified enzyme was dialyzed in the buffer to remove salt, concentrated to ∼40 mg protein/ml by ultrafiltration, and stored at −80 °C.
For purification of GalA, crude extract was loaded onto the anion-exchange column, and the column was washed with 2 column volumes of buffer containing 0.1 m NaCl followed by a linear gradient of NaCl from 0.1 to 0.45 m over 12 column volumes. GalA was eluted with ∼0.25 m NaCl. Active fractions were pooled and concentrated to ∼2 ml by ultrafiltration. GalA did not bind sufficiently to the hydrophobic resin, and the concentrated sample was loaded onto the gel filtration column with the protein eluted with an isocratic flow of buffer containing 0.15 m NaCl. The purified enzyme was dialyzed into 20 mm MES buffer, pH 6.5, containing 50 μm (NH4)2Fe(SO4)2 with 250 μm DTT and concentrated to ∼20 mg protein/ml by ultrafiltration. The protein solution was flushed with N2 and degassed before being flash-frozen in liquid N2. The frozen protein was stored at −80 °C.
For purification of GalD and LigJCsE6, the crude extract was incubated with Ni2+- nitrilotriacetic acid (NTA) affinity resin containing 10 mm imidazole at 4 °C for 16 h with constant mixing. The mixture was then passed through a gravity column, retaining the Ni2+-NTA beads. The Ni2+-NTA resin was washed with 20 ml of buffer containing 15 mm imidazole, and protein was eluted with buffer containing 250 mm imidazole. The polyhistidine tag on LigJCsE6 was removed by first changing the buffer to 50 mm sodium phosphate, pH 8.0, containing 100 mm NaCl and 10% glycerol by ultrafiltration using a YM10 filter. The His6-LigJCsE6 was diluted to ∼10 mg/ml and incubated with 2 units of thrombin for 16 h at 15 °C. The thrombin was removed by incubation with p-aminobenzimide-agarose for 1 h at 15 °C, and the beads were subsequently removed by filtration through a 0.2-μm filter. The cleaved His6 tags, and any tagged protein remaining after the thrombin incubation, were removed by incubation with fresh Ni2+-NTA for 1 h at 15 °C and subsequently passed through a 0.2-μm filter to remove Ni2+-NTA beads. GalD contains an internal thrombin cleavage site at residue 239 of the 361-residue protein, and thus removal of the His6 tag was not attempted. The purified protein was concentrated by ultrafiltration and stored at −80 °C in 20 mm HEPES, pH 7.5.
The molecular mass of the GalA, GalB, GalD, and LigJCsE6 subunits, as determined by SDS-PAGE, was ∼45, ∼25, ∼40, and ∼40 kDa (data not shown), which is consistent with the predicted molecular mass values of 47.6, 27.5, 37.6, and 38.2 kDa, respectively, from previous reports (9, 10, 19, 20).
Determination of Protein Concentration, Purity, and Molecular Mass
Protein concentrations were determined by the Bradford assay using bovine serum albumin as the standard (21). SDS-PAGE was performed and stained with Coomassie Blue according to established procedures (22). The molecular weight of the GalB holoenzyme was determined by gel filtration using a HiLoad 26/60 Superdex 200 prep column (2.6 × 60 cm).
Crystallization and Structure Determination of GalB
Wild type GalB was screened using a Qiagen Classic suite and yielded crystals in which diffraction did not exceed 4.0 Å resolution. The secondary structural prediction using Phyre 2.0 indicated that the N-terminal four residues of the protein are likely to be unstructured and may contribute to disorder in the crystallization (23). An enzyme variant lacking the four N-terminal residues (GalBNt) was screened, and the conditions were optimized. The crystallization condition contained 15% glycerol in the reservoir solution with 2 μl of the reservoir solution mixed with 2 μl of 80 mg/ml purified GalBNt. Cubic crystals grew within a month at 4 °C and were diffracted to ∼3.0 Å resolution. Crystals left for ∼10 months to natively dehydrate at 4 °C were soaked in 50% glycerol for 1 min before being flash-frozen in liquid nitrogen. More than 20 of these crystals were screened with few of them diffracting beyond 2.5 Å resolution and only one diffracting to 2.1 Å resolution. The best diffracting crystal was utilized for both the native and anomalous data collections.
Data were collected at the Canadian Light Source. A fluorescence scan revealed a strong zinc signal and only minimal signals for other elements (data not shown). The zinc, iron, and cobalt K-edges were scanned; however, only the zinc K-edge showed scattering factors consistent with the presence of the metal in the crystal. A data set was collected to 2.0 Å at a wavelength of 0.97949 Å, on the Canadian Light Source 08ID-1 beamline. An attempt to solve the structure by molecular replacement using PIG-L deacetylase structures as search models was unsuccessful. Thus, an anomalous data set for phasing was collected up to 2.2 Å on the same crystal at the wavelength 1.2817 Å (zinc K-edge), on the Canadian Light Source 08B1-1 beamline. The crystal was of the cubic space group P4132, with cell dimensions a = b = c = 201.66 Å. Diffraction data were processed in XDS, and the structure was solved in Phenix (24, 25). The crystal had significant radiation damage by the end of the native data set collection, but Autosol was able to find two zinc sites in the anomalous data set (26). An initial model was built and refined from the anomalous data in Phenix using Autobuild and Refine (27, 28). The native structure was determined by molecular replacement with Phaser using one protomer of the structure from the anomalous data as the search model (29). The structure was rebuilt in Coot, and further refinement was completed in Phenix Refine (28, 30). NCS restraints were not used during refinement, but TLS atomic displacement parameters (with the protein subdivided into five residue groups) were utilized in the final stages of refinement (31). Table 1 lists data collection and final model refinement statistics. The representations of the structure of GalB presented in Figs. 2–4 were prepared with PyMol v1.4.1 (Schrödinger, LLC).
TABLE 1.
GalBNt data collection and refinement statistics
Data shown in parentheses are for the highest resolution shell.
| Native | Zn-K edge | |
|---|---|---|
| Data collection | ||
| Space group | P41 32 | P41 32 |
| Unit cell: a = b = c (Å) | 201.66 | 201.66 |
| Wavelength (Å) | 0.97949 | 1.2817 |
| Resolution (Å) | 48.83-2.10 (2.18-2.10) | 9.84-2.20 (2.28-2.20) |
| No. observed reflections | 638,492 (63,458) | 1,539,423 (92,226) |
| No. unique reflections | 81,109 (7,977) | 135,263 (10,008) |
| Redundancy | 7.9 | 21.5 |
| Data completeness (%) | 99.8 (99.9) | 99.9 (99.9) |
| <I/σI> | 17.9 (2.34) | 22.20 (2.17) |
| Rmerge (%) | 8.31 (99.5) | 8.8 (103) |
| Rpim (%) | 3.1 (37.5) | 1.9 (24.3) |
| CC(1/2) (%) | 99.9 (79.0) | 99.9 (75.5) |
| Wilson average atomic displacement parameters (Å2) | 35.5 | 37.5 |
| Model refinement | ||
| Rwork/Rfree (%) | 15.01/17.09 | |
| Root-mean-square deviation | ||
| Bond lengths (Å) | 0.007 | |
| Bond angles (°) | 1.01 | |
| No. of atoms | ||
| Protein | 3,921 | |
| Water | 512 | |
| Glycerol | 96 | |
| Zinc | 2 | |
| Ramachandran statistics (%) | ||
| Favored | 98 | |
| Outliers | 0 | |
| Atomic displacement parameters (Å2) | ||
| Overall | 37.70 | |
| Protein | 35.90 | |
| Solvent | 47.20 | |
| Ligands | 60.30 | |
FIGURE 2.

Overall structure of the GalB CHM hydratase. A, the GalB protomer colored blue to red from the N to the C terminus. The bound zinc ion is shown as a black sphere, and the secondary structural elements are indicated. Note that the α1 is kinked at a diglycine (Gly-23 and Gly-24) separating α1a (residues 16–22) and α1b (residues 25–32). B, the GalB hexamer with one protomer colored as described in A, with the surface of the rest of the oligomer shown with each protomer differently colored. The interfaces of the protomer within the oligomer are shown: C, 3-fold interface; D, 2-fold β8-β6′ interface; E, 2-fold (α8/α6)2 packing. The interface representations show only the most significant interacting residues.
FIGURE 3.

The GalB metal binding site. A, the zinc ion and ligating water are shown as spheres colored black and red, respectively. The binding site is shown with electron density from a 2mFo − DFc omit map (green), which was derived by setting occupancies of the metal and all atoms in a 5 Å radius to zero (contoured at 1σ). The density for zinc from the anomalous difference map (magenta) is shown (contoured at 15σ). B, representation of the metal binding site showing coordinate distances in angstroms (Å). The distance between Glu-48 and the water (Wat) is shown in gray, as the positioning of the Glu-48, as described under “Results,” is unlikely to fulfill a coordinate with the water. C, comparison of the zinc binding site in GalB and the PIG-L family of N-deacetylases. GalB is shown in white with residues shown as sticks overlaid with the putative or confirmed PIG-L deacetylases: BC1534 from Bacillus cereus (PDB code 2IXD) in green, TT1542 from Thermus thermophilus HB8 (PDB code 1UAN) in cyan, N,N′-diacetylchitobiose deacetylase from Pyrococcus furiosus (PDB code 3WL4) in magenta, LnmX from Streptomyces atroolivaceus (PDB code 5BMO) in yellow, Dbv21 from Actinoplanes teichomyceticus (PDB code 3DFI) in pink, and MshB from Mycobacterium tuberculosis (PDB code 1Q74) in blue. The GalB-bound zinc ion is shown as a black sphere, and the residues are numbered by the GalB sequence. Note that the PIG-L N-deacetylases contain a general base aspartate that is found as an alanine (Ala-16) in GalB and that the PIG-L N-deacetylases lack an acidic residue equivalent to GalB Glu-48.
FIGURE 4.

The GalB and LigJ active sites. A, the active site pocket of GalB. One GalB protomer is shown in white and the other in beige. The metal-ligated water is shown as a red sphere, and significant residues in the binding pocket are shown as sticks. B, model of (2E,4E)-CHM in the GalB active site. The (2E,4E)-CHM is shown in green, and all of the coordinates shown are within 2.8 to 4.2 Å. The side chains of Arg-67, His-164, and His-198 are rotated from their original locations, which are shown as wire and indicated with arrows. The (2E,4E)-CHM is positioned with the C4 in line with the metal-ligated water, which would be activated by the Glu-48. The His-164 and the protonated Glu-48 would act as general acids protonating C5 and C3, respectively. C, zinc binding site of LigJ from R. palustris. The LigJ protomer is shown in cyan with contribution from a second protomer shown in green. The LigJ zinc ion is shown as a black sphere. Two regions in LigJ within proximity to the active site were not modeled in the structure. The missing region between Ser-180 and Thr-190 is indicated by an orange dashed line and the missing region between Ala-72 and Gly-80 is indicated by magenta dashed lines. The dashed lines are used only to highlight the approximate location of the missing regions.
Preparation of 4-Carboxy-2-hydroxymuconate
CHM was synthesized enzymatically from gallic acid using the gallate dioxygenase GalA and the OMA tautomerase GalD. 5-ml reactions were set up in 750 mm sodium phosphate buffer, pH 7.0, containing 150 mm gallic acid. 300 μg of both GalA and GalD was added every 5 min for 50 min with the solution being mixed constantly. After the final addition, the reaction was further incubated with mixing for an additional 30 min. Reactions took place in a closed cell, and throughout the course of the reaction the solution was kept oxygenated by bubbling oxygen through the solution. At the completion of the reaction the enzymes were removed by ultrafiltration through a YM10 filter. The solution was treated with Chelex, sodium form, for 10 min, which was subsequently removed by filtration through a 0.2-μm filter. By the end of the reaction >95% of the gallic acid was catabolized and >65% of the product was CHM, as determined by kinetic assays. CHM was purified by HPLC on an ÄKTA Explorer 100 (GE Healthcare Life Sciences) with 500-μl aliquots of the reaction applied to an Aminex fast acid ion-exchange column, HPX-87H (100 × 7.8 mm) and was separated with an isocratic elution using 100 mm H2SO4. Compounds were detected at 215 nm; using this method CHM, OMA, and gallic acid were resolved to retention volumes of 6.5, 8.0, and 22.5 ml, respectively. When purified CHM was incubated with GalB, converting the CHM to CHA, both the CHA and CHM co-eluted from the column using the isocratic flow of 100 mm H2SO4. Changing the procedure to an isocratic flow of 500 mm H2SO4 facilitated the separation of CHA from CHM, with the compounds having retention volumes of 6.7 and 9.8 ml, respectively.
Preparation of CHPD
CHPD was synthesized enzymatically with GalB. 5-ml reactions comprised 50 mm (R/S)-HMG, 50 μm CoCl2, and 100 μg of GalB in 100 mm HEPES, pH 7.0. Reactions were incubated at 15 °C overnight with constant mixing. Reactions were stopped by removing the enzyme by ultrafiltration through a YM10 filter. The reaction was subsequently treated with Chelex, sodium form, for 1 h to remove metals, with the beads removed by filtration through a 0.2-μm filter. 500-μl samples of the reaction were separated by HPLC via an Aminex fast acid ion-exchange column using 100 mm H2SO4. HMG and CHPD were resolved to retention volumes of 6.4 and 9.0 ml, respectively.
GalB Metal Analysis
To assess the metal utilized by the enzyme in vivo, GalB expressed in and purified from P. putida KT2442 was analyzed by inductively coupled plasma mass spectrometry (ICP-MS). The enzyme was purified as described previously, and the purified protein was washed with Chelex (sodium form)-treated buffer several times in an ultrafiltration stir cell using a YM10 filter to remove exogenous metals. The final protein sample was concentrated to 5 ml in buffer before being diluted to 125 ml in HPLC-grade water (Fisher Scientific) and sent for ICP-MS analysis at ALS Global (Waterloo, Ontario, Canada). The final concentration of enzyme analyzed by ICP-MS was 24.1 μm. The detection limit for iron (9.0 μm) with the ICP-MS methodology utilized was too low relative to the stoichiometric amounts of enzyme supplied to draw conclusions about its presence. Thus the iron content of the GalB-purified proteins was assessed using a ferrozine assay (32, 33).
Protein Metal Ion Substitution
All buffers were treated with Chelex, sodium form, for 20 min to remove exogenous metals, with the beads removed by filtration through a 0.45-μm filter. Metal-free apoenzymes were prepared by treating 50 mg of purified enzyme in 50 ml of 100 mm sodium-HEPES, pH 7.0, with 1 mm EDTA for 12 h at 4 °C with constant mixing. Using ultrafiltration with YM10 filter, the EDTA was removed from the enzyme solution by successive washes with buffer, and the final apoprotein was concentrated to ∼10 mg/ml and stored at 4 °C. Apoenzyme was preincubated with 50 μm metal chlorides for a minimum of 1 h at 15 °C prior to use in the kinetic assays. For incubation with Fe2+, a 200-μl solution comprising 0.92 μg/ml apoenzyme was transferred to a Baker Ruskinn Bugbox anaerobic chamber filled with nitrogen and degassed for 5 min. The Fe2+ enzyme solutions were made up in the anaerobic chamber to 50 μm (NH4)2Fe(SO4)2 from a freshly degassed stock of 5 mm (NH4)2Fe(SO4)2, prepared in 50 mm H2SO4, and incubated on ice for 1 h. Less than 3% of the specific activity was lost after uncapping and exposing the Fe2+ anaerobically incubated enzyme sample to air for 1 h.
Zinc and Cobalt Dissociation Constant Determination
Apoprotein of either the wild type or variants of GalB was incubated with varying concentrations of either ZnSO4 or CoCl2 in a total volume of 600 μl of 100 mm HEPES, pH 7.0, and was kept at 15 °C for 1 h with constant mixing. Binding reactions were then filtered by ultrafiltration using an Amicon Microcon YM10 to remove enzyme from the solution. Samples of the enzyme-free solutions were mixed with 250 μm freshly prepared 4-(2-pyridylazo)resorcinol and made up to 1 ml with Chelex (sodium form)-treated buffer. The amount of free Co2+ or Zn2+ present in the reaction was determined in at least duplicate spectrophotometrically at 505 nm for Co2+ and 497 nm for Zn2+ using extinction coefficients described previously (34). The concentration of enzyme-bound Zn2+ or Co2+ was calculated using Equation 1, and dissociation constants were determined by nonlinear regression in GraphPad Prism with Equation 2, where [M2+] is concentration of metal and C is the saturated concentration of the metal.
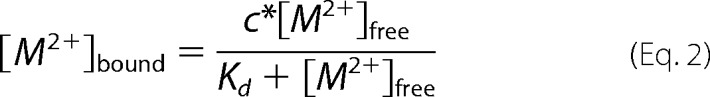 |
Enzyme Assays
All of the kinetic assays were performed at least in duplicate at 25 °C using a Varian Cary 3 spectrophotometer equipped with a thermostatted cuvette holder. Assays for CHM hydration and CHA dehydration were completed similarly to that described previously (9). Briefly, the hydration of CHM to CHA, or the dehydration of CHA to CHM, was monitored by detecting the olefinic signal of CHM at 265 nm. The hydration of CHPD to HMG, or the dehydration of HMG to CHPD, was similarly monitored at 260 nm, where UV scans indicate the maximum absorbance of the CHPD olefinic signal. Standard assays were completed in 100 mm HEPES, pH 7.5, and the extinction coefficients of 9451 m−1cm−1 utilized for CHM and 8221 m−1cm−1 for CHPD were determined experimentally. All assays were carried out with apoGalB preincubated for at least 1 h with the stated metal at 50 μm prior to the assay. Metal ion specificity assays were completed in 100 mm HEPES, pH 7.0, with 50 μm CHM and 50 μm metal chloride, EDTA, or with enzyme prior to metal substitution.
The efficiency of the removal of bound metals by EDTA was assessed using enzyme activity assays. ApoGalB (1.5 μm) that had been preincubated with 50 μm ZnCl2 was incubated with either 250 or 1000 μm EDTA, and the specific activity of the enzyme toward 50 μm CHM under standard conditions was measured at successive time points. The loss of activity in the enzyme solutions was fit to a one-phase exponential decay, Equation 3, where SA is the specific activity, SA0 is the initial specific activity, k is the rate constant, and t is time.
The pH dependence of CHM utilization kinetics was determined with pH varying between pH 5.5 and 9.5 in a three-component constant ionic strength buffer containing 0.1 m Tris, 0.05 m acetic acid, and 0.05 m MES. Enzyme was preincubated with 50 μm ZnCl2 or 50 μm CoCl2 in the stated pH buffer for a minimum of 30 min before assays were completed. The extinction coefficient for CHM at each pH point was determined experimentally. All data were fitted by nonlinear regression in GraphPad Prism. The pH profile of kcat shows a single ionization event and is fitted to Equation 4. The pH profile of pKm is bell-shaped with slopes of unity and is fitted to Equation 5. The pH profile of kcat/Km is bell-shaped, with the ascending limb of the curve having a slope of ≈2 and a descending slope of unity, and is fitted to Equation 6.
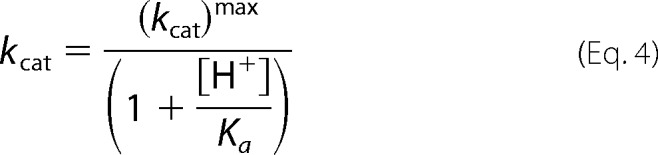 |
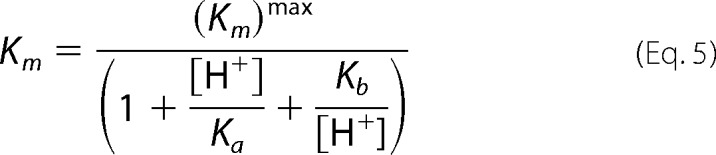 |
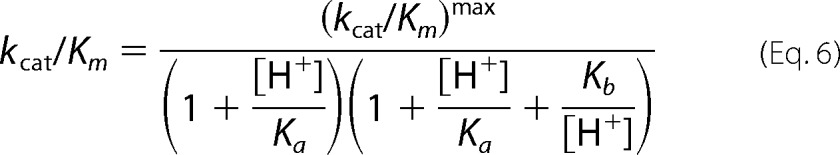 |
Polarimetry assays were completed in a Rudolph IV polarimeter using a 100-mm cuvette. Reactions of 7 ml contained 15 mm racemic CHA, 30 mm racemic HMG, or 5 mm CHM in 100 mm HEPES, pH 7.5, and 0.62, 8.7, or 0.10 μm GalB preincubated with 50 μm CoCl2, respectively. Reactions were initiated with the addition of enzyme, and the solution was passed through a 0.2-μm filter prior to data acquisition.
Results
Expression and Purification of GalB
The gene for GalB from P. putida KT2440 was overexpressed both natively in P. putida KT2442 and recombinantly in E. coli BL21(λDE3) cells with typical yields of 15 and 22 mg of purified protein/liter of bacterial culture, respectively. The subunit molecular mass of the enzyme as determined by SDS-PAGE is 27 kDa, consistent with the predicted molecular mass (27,461 Da). GalB variants and the protein containing an N-terminal truncation were overexpressed and purified from E. coli BL21(λDE3) similar to the wild type protein.
Overall Structure of GalB
An initial substructure of GalBNt was determined by zinc single-wavelength anomalous diffraction (SAD) phasing to 2.2 Å resolution, which was then subsequently utilized as a model for molecular replacement of the native data set (2.1 Å) (Table 1). There are two protomers in the asymmetric unit, which can be superimposed with a root-mean-square deviation of 0.087 Å, indicating only minor differences. Each protomer has an overall fold similar to the PIG-L family of N-deacetylases, described in more detail below, consisting of an α-β-α sandwich with the N-terminal portion forming a five-stranded Rossmann fold (residues 1–160) leading to a mixed α/β C-terminal region (residues 161–234) and a β-strand (β8, residues 237–240) (Fig. 2A). The N-terminal Rossmann fold is composed of a β-sheet (β1–β5) wrapped by α4 and α5 on one face and α1 and α3 on the other. The C-terminal portion consists of two anti-parallel β-strands (β6–β7) interspersed with a helical hairpin (α7 and α8). The β8 from an adjacent monomer packs parallel to β6 making a continuous β-sheet of β7-β6-β8′ with the β7 anti-parallel to the rest. The protomer is composed of the Rossmann fold, the C-terminal region, and contributions by α8′ and β8′ from adjacent units. The bound zinc ion is found at the bottom of a ∼22 Å-deep, solvent-accessible pocket that has a total volume of ∼1060 Å3 as determined by DogSiteScorer (35).
Oligomeric State
The interface between the two protomers in the asymmetric unit constitutes 125 Å2 (∼1%) of each molecules surface area, as determined by PISA, and is likely not a physiologically relevant interaction (36). Instead, each protomer acts as the asymmetric fraction of a distinct hexamer, point group D3 (Fig. 2B). The hexamer is assembled by interactions along three distinct interfaces: dimerization through packing of β8 on β6′ of an adjacent protomer along a 2-fold axis, which buries ∼13% of the total surface area (Fig. 2C); dimerization through packing of α8/α6with α6′/α8′ along a 2-fold axis, which buries ∼9% of the total surface area (Fig. 2D); and packing of α5 and the loop between β5 and α5 against α2′ and the loop region between α3′ and β3′ related by a 3-fold axis, which buries ∼6% of the total surface area (Fig. 2E). Together with additional interactions, the hexameric oligomerization buries ∼50% of the total surface area on the protomer, indicating that the hexamer is the biological unit. Analysis of the GalB protein by gel filtration yields a mass of 156.2 kDa, consistent with a hexameric organization (164.8 kDa; data not shown).
GalB Metal Binding Site
Scans of the GalB crystal revealed fluorescence consistent with zinc being the only transitional metal present in the crystal. The GalB zinc ion is coordinated in a skewed trigonal bipyramidal fashion by His-14 Nδ, Asp-17 Oδ1, Glu-48 Oϵ1, His-127 Nϵ, and a water molecule (Fig. 3).
Three conformers of the Glu-48 residue are seen in the two chains. The predominant conformer, with occupancy of ∼60%, is found in both chains with the Oϵ1 of Glu-48 coordinating the zinc ion. The alternate conformation of the residue is slightly different between the two chains. In chain B the residue is flipped, having the Oϵ2 coordinating the zinc ion and the Oϵ1 positioned by interaction with Asn-124 Nδ and the Tyr-90 hydroxyl (Fig. 3A). In chain A the carboxylate rotated away from the metal ion, with the Oϵ1 interacting with Asn-124 Nδ and the hydroxyl of Tyr-90, whereas the Oϵ2 projects out of the metal binding site (Fig. 3C). The average atomic displacement parameter for the Glu-48 is ∼34.5 Å2, which is similar to the atomic displacement parameter of the other metal binding residues, indicating that the residue is well ordered.
The coordinate distance of the water to the metal (2.3 Å in chain A and 2.8 Å in chain B) is longer than that the protein ligands (∼2.1 Å) to the metal (Fig. 3B). The metal ligating water is also positioned by interaction with the Oδ2 of Asp-17 (2.8 Å). The water is in close proximity to the Glu-48 Oϵ1 (2.1Å), but the water is not in line with the atom for ideal interaction, suggesting that the Glu-48 does not interact directly with the water. The average atomic displacement parameter for the metal ligating water is 51.7 Å2, indicating that the water is possibly present at less than full occupancy and its presence may be modified by the different conformers of the Glu-48.
Outside of the metal binding residues, the pocket is lined with hydrophilic residues with several residues (such as Arg-21, Arg-67, His-164, and Arg-216′ from the α8′ on the adjacent protomer) providing electropositive character to the pocket and likely facilitating the binding of the tricarboxylic acid substrate, CHM (Fig. 4A).
Structurally Related Proteins
A search of structural homologs to GalB using DALI revealed members of the PIG-L family of N-deacetylases as the closest structural homologs (37). There are currently eight structures of unique PIG-L family members in the protein database, and GalB aligns with these proteins with root-mean-square deviation values of <3.0 Å resulting in Z-scores of >20 and coverage of >70% of the deacetylase sequence. Members of this family, such as the N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase from Mycobacterium tuberculosis (MshB), N,N′-diacetylchitobiose deacetylase from Pyrococcus species, and N-acetyl-glucosaminylpseudoaglycone deacetylase from Actinoplanesteichomyceticus, are divalent metal-dependent hydrolytic enzymes with preference for zinc or iron (II) ions (38–40). Oligomerization differs across the PIG-L family of proteins with variability in the both GalB N- and C-terminal portions, resulting in proteins predicted or observed to exist as hexamers, trimers, or monomers (40–43). Common among all of the PIG-L deacetylases and GalB is the single α/β domain with a Rossmann-like fold containing a solvent-accessible pocket. With the exception of the Glu-48 residue, zinc binding in these structural homologs is facilitated by a His-X2-Asp-Xn-His motif similarly found in GalB (for GalB: His-14-X2-Asp-17-Xn-His-127) (Fig. 3C). MshB is the only PIG-L family member for which the structure has been determined that contains a glutamate in the primary sequence aligned to GalB Glu-48. However, the equivalent MshB residue is found on a loop region which is rotated away from the bound zinc ion and active site (43, 44). The bound zinc ion in GalB is exposed on one face to a solvent-accessible pocket, which is similar to the PIG-L family and is the substrate binding pocket in these enzymes. Outside of the metal binding residues, only the GalB residues Arg-67 and Gln-196 are conserved in the binding pocket among all PIG-L N-deacetylases.
In MshB (and other PIG-L deacetylases), the active site residues His-144, Asp-15, and Tyr-142 are implicated as the general acid, general base, and oxyanion transition state stabilizer, respectively (45). The equivalent residues in GalB are Asn-124, Ala-16, and Tyr-123, respectively (Fig. 4A), implying that the general base and acid roles of the PIG-L deacetylases at least are not recapitulated by these residues. The hydration of CHM to CHA is likely to proceed through deprotonation of the CHM enol, producing an enolate that is analogous to the oxyanion transition state in the PIG-L deacetylases. The MshB oxyanion stabilizing the Tyr-142 residue is found in a glycine-rich loop (Gly-140/Gly-141/Tyr-142/Gly-143), which provides conformational flexibility to allow the positioning of Tyr-142 with its side chain projecting into the active site pocket. The GalB Tyr-123, however, is not found in as flexible a region (Asp-121/Pro-122/Tyr-123/Asn-124), and the phenol side chain is observed rotated away from the active site pocket. The GalB Tyr-123 hydroxyl is stabilized by interaction with the Nϵ and Oϵ of Gln-217 with the tyrosine π electrons interacting with the Nϵ of Gln-165. Although some movement is likely to occur upon substrate binding, the lack of conformational freedom and interactions made by the GalB Tyr-142 suggests that it is unlikely to rotate into the active site pocket and fulfill the enolate stabilization role. Thus, although conserving the protein fold and metal ion binding, GalB lacks key residues that in PIG-L enzymes facilitate hydrolase activity, likely reflecting differences in the chemical reaction of the enzymes.
Metal and Kinetic Analysis of GalB
To ascertain the native metal ion utilized by GalB in vivo, protein purified from the native host, P. putida KT2442, was analyzed. When overexpressed and purified, using three chromatographic steps, the specific activity of the native protein was 1.02 μmol min−1μg−1. The specific activity of the native protein increased 4-fold when 50 μm exogenous Co2+ was added, indicating that either the native enzyme was not fully saturated with metal during overexpression or else the metal was lost during purification. ICP-MS analyses on the native purified protein, without the addition of any exogenous metals, revealed the presence of zinc, cobalt, and copper in relative stoichiometric quantities of 23, 0.1, and 0.1%, respectively. Because of the high background, amounts of iron could not be estimated by ICP-MS. Instead, Fe was determined by the ferrozine assay to be below 0.6%. Thus zinc is the predominant metal ion found in the enzyme through the aerobic purification methods utilized here.
The GalB N-terminal truncation variant used for crystallization maintained the same activity toward CHM as the wild type enzyme, indicating that the observed structure represents the fully active zinc bound enzyme. A metal cofactor is required for enzyme activity, and the half-life of Zn2+-saturated GalB enzyme activity when treated with either 250 μm or 1 mm EDTA was 32.3 or 17.1 min, respectively. After treatment with 1 mm EDTA for 12 h, the protein lacked detectable enzyme activity above the error of the assay conditions. CHM hydratase activity could be restored by incubating the apoenzyme with divalent metals (Table 2). The enzyme showed maximal activity with Fe2+ and Co2+. The presence of all other metal ions, including Zn2+, resulted in less than 30% of the activity observed with Fe2+.
TABLE 2.
Relative activity of GalB-catalyzed CHM hydration with various metal ions
Maximal activity was observed with Fe2+ (11.9 μmol min−1μg−1) taken as 100%. The native activity is that of the purified enzyme before treatment with EDTA. An increase in activity for the natively purified enzyme was observed when it was incubated with exogenous 50 μm CoCl2 for 10 min, indicating that the enzyme was not fully saturated with metal.
| Metal ion | Relative activity |
|---|---|
| % | |
| Fe2+ | 100 ± 4.6 |
| Co2+ | 79 ± 4.0 |
| Native + Co2+ | 34 ± 1.5 |
| Mn2+ | 30 ± 1.9 |
| Zn2+ | 17 ± 0.66 |
| Fe3+ | 14 ± 2.2 |
| Ni2+ | 13 ± 0.33 |
| Ca2+ | 11 ± 1.0 |
| Mg2+ | 9.8 ± 0.66 |
| Native | 8.6 ± 0.95 |
| Cd2+ | 7.4 ± 0.66 |
| Apo | 4.0 ± 0.33 |
| Cu2+ | 0.19 ± 0.10 |
| EDTA | 0.030 ± 0.0033 |
The metal binding ligands observed in GalB are the same as those observed in the PIG-L deacetylase family, except that Glu-48 in GalB represents a potential extra ligand; however this residue shows alternative orientations, implying that its interactions with the metal ion are weaker than the other three ligating residues (Fig. 3). Both Zn2+ and Co2+ bind strongly to GalB with dissociation constants (Kd) of 0.63 ± 0.05 and 0.056 ± 0.009 μm, respectively (Table 3). The H127A variant showed a 28- and 1625-fold increase in Kd for Zn2+ and Co2+, respectively. The E48A variant, however, showed only a 5- and 26-fold increase in Kd for Zn2+ and Co2+, respectively. The effects of the Glu-48 substitution were less pronounced than that of the substitution of His-127, a conserved metal ligand in the PIG-L family, therefore suggesting a minimal role for Glu-48 in metal binding.
TABLE 3.
Dissociation constants (Kd) of the wild type and variants of GalB
The GalB metal dissociation constants were determined by titration using a PAR assay as described under “Experimental Procedures.” NS indicates that the protein could not be saturated with metal up to 500 μm.
| Protein | Zn2+ | Co2+ |
|---|---|---|
| μm | μm | |
| Wild type | 0.63 ± 0.048 | 0.056 ± 0.0085 |
| A16D | 0.75 ± 0.095 | 0.071 ± 0.0043 |
| E48A | 3.2 ± 0.29 | 1.5 ± 0.19 |
| H127A | 17.6 ± 1.4 | 90 ± 10 |
| H14A/D17A | NS | NS |
In the GalB CHM hydration steady state kinetics, the choice of metal ion (among Fe2+, Co2+, and Zn2+) affected the specificity constant (kcat/Km) by less than 3-fold (Table 4). However, the reaction rates with the metals at high substrate concentrations diverged markedly, with both kcat and Km being ∼7-fold higher for Fe2+ or Co2+ versus Zn2+; the highest catalytic activity was therefore observed with Fe2+, the but the increase in kcat was offset by a parallel increase in Km. GalB specificity was >200-fold lower for CHPD, indicating an important role for the CHM C5 carboxylate group in substrate binding. The enzymatic reaction is reversible, with the dehydration of CHA and HMG being catalyzed with kinetic parameters similar to the hydration reaction. Polarimetric analysis of the GalB-catalyzed hydration of CHM indicates that only the (−)-enantiomer of CHA was produced (Fig. 5). Similarly, the enzyme was found to dehydrate only the (−)-enantiomers of both CHA and HMG in the reverse reaction, leaving the positive enantiomers, when exposed to racemic mixtures of the substrates.
TABLE 4.
Steady state kinetic parameters of the GalB reversible reactions in the presence of 50 μm (NH4)2Fe(SO4)2, CoCl2, or ZnCl2
The A16D-catalyzed reaction was not saturable (NS) up to 2 mm CHM, and the kcat/Km was determined by the linear regression of velocity over substrate concentration.
| Enzyme | Metal | Substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| μm | s−1 | m−1·s−1 | |||
| Wild type | Fe2+ | CHM | 11.8 ± 1.4 | 14.6 ± 0.54 | 1.23 × 106 |
| Wild type | Co2+ | CHM | 5.28 ± 0.37 | 12.9 ± 0.21 | 2.44 × 106 |
| Wild type | Zn2+ | CHM | 2.56 ± 0.27 | 2.08 ± 0.043 | 8.12 × 105 |
| A16D | Zn2+ | CHM | NS | 8.51 | |
| R21A | Zn2+ | CHM | 860 ± 50 | 0.0347 ± 0.00088 | 4.03 × 101 |
| E48A | Zn2+ | CHM | 24.3 ± 2.9 | 0.00274 ± 0.00013 | 1.13 × 102 |
| R67A | Zn2+ | CHM | 72.5 ± 5.1 | 0.0927 ± 0.0019 | 1.28 × 103 |
| Y123A | Zn2+ | CHM | 5.28 ± 0.42 | 1.78 ± 0.037 | 3.38 × 105 |
| H164A | Zn2+ | CHM | 1.78 ± 0.13 | 0.0372 ± 0.00042 | 2.09 × 104 |
| R216A | Zn2+ | CHM | 894 ± 93 | 0.0146 ± 0.00067 | 1.63 × 101 |
| Wild type | Zn2+ | CHPD | 22.6 ± 1.7 | 0.0898 ± 0.0016 | 3.97 × 103 |
| R21A | Zn2+ | CHPD | 89.6 ± 12 | 0.000454 ± 0.000023 | 5.06 |
| R67A | Zn2+ | CHPD | 444 ± 49 | 0.00279 ± 0.00013 | 6.29 |
| R216A | Zn2+ | CHPD | 81.1 ± 4.7 | 0.107 ± 0.023 | 1.32 × 103 |
| Wild type | Zn2+ | CHA | 6.80 ± 0.48 | 0.538 ± 0.0088 | 7.91 × 104 |
| Wild type | Zn2+ | HMG | 54.5 ± 5.6 | 0.0197 ± 0.00051 | 3.61 × 102 |
FIGURE 5.
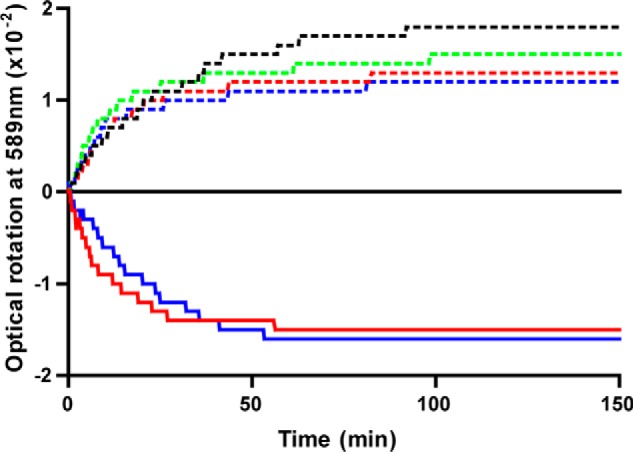
Analysis of the GalB and LigJCsE6 enzyme-catalyzed reactions by polarimetry. Hydration of CHM to CHA (solid lines) and dehydration of racemic CHA (dashed lines) by GalB (blue) and LigJCsE6 (red). Dehydration reactions of racemic HMG catalyzed by GalB (green) and LigJCsE6 (black) are shown as dashed lines.
CHM and GalB Hydration pH Profiles
The pH dependence of the GalB-catalyzed hydration of CHM was assessed in a three-component buffer. The extinction coefficient of CHM increases sigmoidally with increasing pH, likely because of the deprotonation of the enol group leading to increased electron delocalization and chromogenicity. Fitting the increase in extinction coefficient to an acid titration resulted in a pKa (pKaenol) of 7.3 ± 0.1 for the CHM substrate (Fig. 6A).
FIGURE 6.
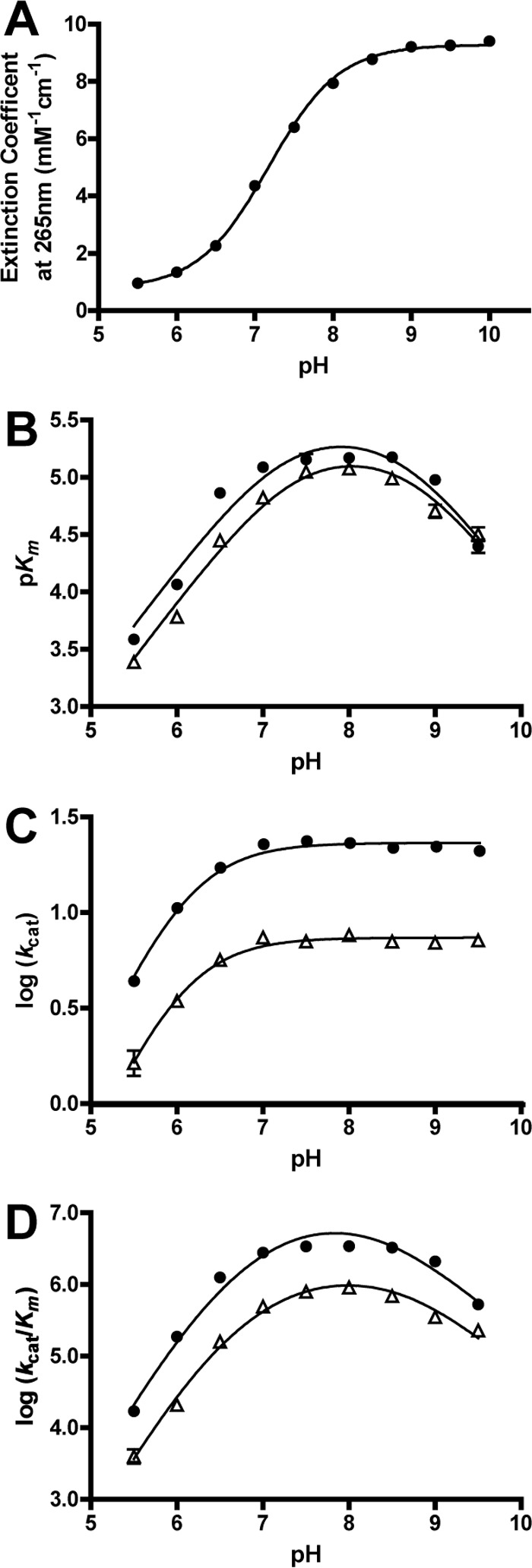
pH profiles of the GalB CHM hydration reaction with Zn2+ or Co2+ cofactors. A, the CHM extinction coefficients determined at each pH value. B–D, the pH dependence of the CHM hydration reaction catalyzed utilizing the cofactors Co2+ (●) and Zn2+ (▵). The profiles of log(kcat) (B), pKm (C), and log(kcat/Km) (D) are shown.
The pKasubstrate values for the other ionizable groups of the CHM (the C2, C4, and C5 carboxylates) were determined by titration. CHM was isolated by chromatography in 100 mm H2SO4 and could not be further purified, hampering the definition of pKa values that were close to the second pKa of sulfuric acid (∼2.0). Titration of the CHM in H2SO4 indicated two ionizable groups with pKasubstrate values around 3.5 and two values of about 6 and 7 (data not shown). Considering the pKaenol value of 7.3 determined for CHM using absorption spectroscopy as described above, the pKasubstrate value for the other ionizable group fitted to the titration curve was estimated to be ∼6.3.
Kinetic parameters for the CHM hydration reaction could not be determined for pH values below 5.5, and those above 9.5 could not be determined due to high Km values for CHM. The dependence of pH on the Zn2+ and Co2+ enzyme-catalyzed reactions was assessed, and the profiles for each were similar (Table 5). In both cases, the pH profile of log(kcat) shows a single ionization event in the enzyme-substrate complex, with a pKaES value of 6.1 ± 0.1 and 6.2 ± 0.1 for the Zn2+ and Co2+ reactions, respectively (Fig. 6C).
TABLE 5.
The pH-dependent parameters of GalB and enzyme variant-catalyzed hydration of CHM
pKaE/S2 was determined from the ascending slope of unity from pKm plots, and the value was fixed for the determination of the pKaE/S1 from plots of log (kcat/Km), which have ascending slopes ≈ 2.
| Enzyme | Metal | pKaE/S1 | pKaE/S2 | pKbE | pKaES |
|---|---|---|---|---|---|
| Wild type | Co2+ | 6.4 ± 0.2 | 7.3 ± 0.2 | 8.3 ± 0.2 | 6.2 ± 0.05 |
| Wild type | Zn2+ | 6.2 ± 0.1 | 7.3 ± 0.2 | 8.6 ± 0.2 | 6.1 ± 0.03 |
| H164A | Zn2+ | 6.4 ± 0.1 | 7.2 ± 0.1 | 9.1 ± 0.1 | 6.3 ± 0.07 |
| E48A | Zn2+ | 5.6 ± 0.2 | 7.1 ± 0.2 | 8.3 ± 0.1 | 6.2 ± 0.04 |
The pH profiles of pKm are bell-shaped, with the ascending and descending limbs having slopes of unity (Fig. 6B). The ascending pKa values for both metal cofactor-catalyzed reactions is 7.3 ± 0.2, with the descending pKb values being 8.7 ± 0.2 for Zn2+ and 8.5 ± 0.3 for Co2+. The close proximity of the ascending pKa and pKaenol, as well as the observation of the same ascending pKa in both the Zn2+ and Co2+ dependences, suggests that the pKa in the pKm plot likely represents the free substrate rather than the free enzyme.
The pH profiles of log(kcat/Km) are also bell-shaped but have the ascending limb of the curve with a slope of ≈2 and a descending slope of unity (Fig. 6D). For both metal ions, fitting of the data to Equation 3 gives rise to two pKa values on the ascending limb with each being ∼6.7 and one pKb on the descending limb being ∼8.4. However, fixing one of the ascending pKa values as pKaenol, as observed for pKm, results in pKa values of 6.2 ± 0.2 and 8.6 ± 0.1 for the Zn2+ cofactor profile and 6.4 ± 0.2 and 8.3 ± 0.2 for the Co2+ cofactor profile. For both cofactor-supported reactions, the ascending pKa values are the same, and the descending pKb values are within close proximity to each other, suggesting that conserved groups contribute to these ionization values, which are only moderately affected by the metal cofactor. The two pKasubstrate values of ∼7.3 and ∼6.3 may correspond to the two pKa values observed in the ascending slope of the pH profile of log(kcat/Km). However, the proximity of the lower pKa from the log(kcat/Km) profile (∼6.3) to the pKaES from the pH profile of log(kcat) (∼6.2) suggests that the value represents an ionizable group on the free enzyme rather than the free substrate.
The lack of an ionizable group on the free substrate above pH 8 suggests that the pKb of the descending bell curves observed in the pH profile of pKm (∼8.7 for Zn2+ and ∼8.5 for Co2+) and log(kcat/Km) (∼8.6 for Zn2+ and ∼8.3 for Co2+) represents an ionizable group on the free enzyme (pKbE).
GalB Mutagenesis Investigation
The PIG-L family of enzymes relies on a conserved aspartate residue to act as general base in their deacetylase reactions. The equivalently positioned residue in GalB is Ala-16; replacement of this residue with aspartate (A16D) led to a significantly increased Km value for CHM, and the enzyme variant could not be saturated with substrate up to 2 mm (Table 4). The catalytic efficiency for the substrate was reduced 105-fold relative to the wild type enzyme, indicating the importance of a small nonpolar residue at this position for hydratase function. Neither the wild type GalB nor the A16D variant exhibited inhibition by glucosamine or N-acetyl glucosamine up to 1 mm in the CHM hydration reaction. In the PIG-L deacetylase MshB, Tyr-142 is proposed to act as an oxyanion stabilizing residue. As described previously, GalB Tyr-123 aligns with Tyr-142 of MshB in the primary sequence, but the GalB residue is differently positioned, with the phenolic side chain rotated away from the active site pocket (38, 44). Accordingly, the GalB Y123A variant resulted in no significant change in CHM utilization relative to the wild type protein (Table 4). Together the results indicate that the catalytic mechanism and substrate binding mode of GalB is distinct from that of the PIG-L N-deacetylases.
The putative active site of GalB encompasses several hydrophilic residues that may support substrate binding and catalysis. Three arginine residues (Arg-21, Arg-67, and Arg-216′) are found within 12 Å of the bound zinc ion and are candidates for ionic interaction with the three carboxylates on CHM (Fig. 4A). Each of the residues was substituted separately with alanine, and their activities were measured with both CHM and CHPD (Table 4). Each variant had decreased specific activities relative to the wild type enzyme for both CHM and CHPD hydration. The GalB R216A variant had a 142-fold decrease in kcat for CHM but a 1.2-fold increase in kcat for CHPD, indicating that Arg-216 may interact with the C5 carboxylate moiety of CHM.
As described previously, the E48A substitution has minimal effect on the metal binding capacity of the enzyme. The E48A substitution, however, resulted in a >7000-fold decrease in the catalytic efficiency of the enzyme for CHM, largely due to a >760-fold decrease in kcat (Table 4). The pH dependence of the E48A-catalyzed hydration of CHM showed no significant change on the pKa of the enzyme-substrate complex or on the descending limb of log kcat/Km (Fig. 7). However, the substitution causes the pKa of the ascending limb of kcat/Km to shift from 6.2 ± 0.2 observed in the wild type to 5.7 ± 0.1 in the enzyme variant (Table 5). Together, the E48A investigations suggest a minimal role for Glu-48 in metal binding but that the residue is important to the enzyme catalytic mechanism by likely supporting proton transfer.
FIGURE 7.
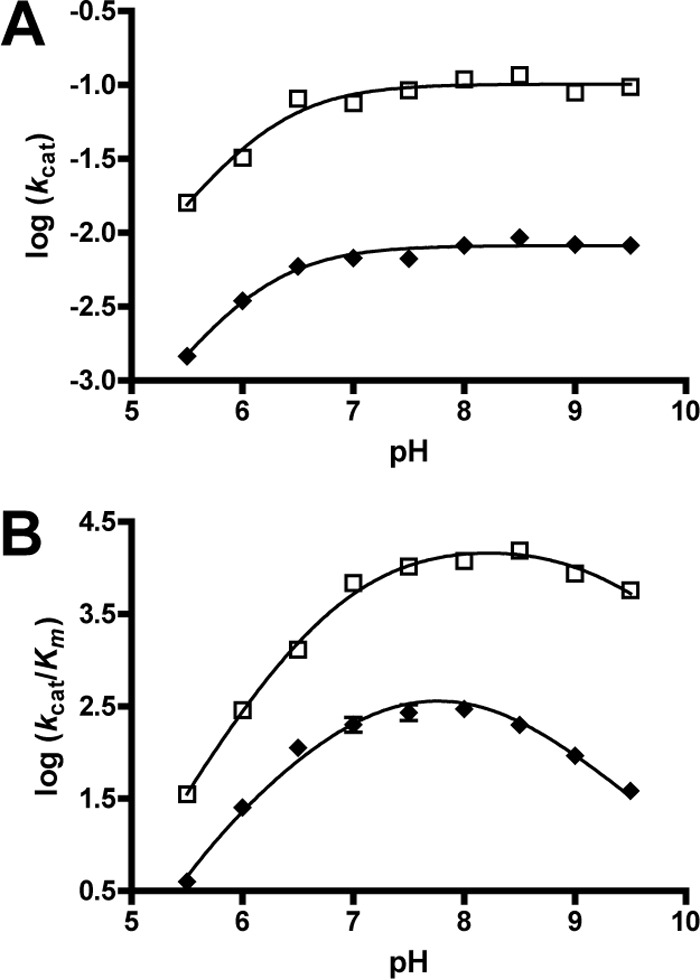
pH profiles of the E48A and H164A GalB variants with Zn2+ cofactor. The pH dependence on log(kcat) (A) and log(kcat/Km) (B) of the CHM hydration reaction catalyzed by the H164A (□) and E48A (♦) enzyme variants is shown.
A histidine residue, His-164, is found in the GalB active site with the Cβ being 8.6 Å from the zinc ion (Fig. 7). Substitution of His-164 with alanine resulted in no significant change in Km for the CHM hydration under standard assay conditions but there was a 56-fold decrease in kcat (Table 4). The pH dependence of the H164A variant-catalyzed hydration of CHM showed no significant change in the pKa of the enzyme-substrate complex or the ascending limb of the kcat/Km profile (Fig. 7). However, the pKa of the descending limb of the kcat/Km profile is shifted up by 0.6 pH unit in the H164A variant to 9.2 ± 0.1, suggesting a role for the residue in proton transfer (Table 5).
Comparative Analysis of GalB and LigJCsE6
Degradation of gallic acid through the gallate pathway can be monitored by coupling the production of oxaloacetate from GalA-GalD-GalB-GalC to NADH oxidation by l-malate dehydrogenase. When GalB is omitted from the coupled reaction, CHM is not hydrated to CHA and the NADH is not oxidized (data not shown). Substitution of LigJCsE6 for GalB in the coupled reaction leads to the oxidation of NADH in a 1:1 stoichiometric ratio to the amount of gallic acid originally present, indicating that LigJCsE6 utilizes the same CHM isomer as GalB. When the tautomerase GalD is omitted from either the GalB- or LigJCsE6-coupled reactions, only a very slow NADH oxidation is observed and the rate is independent of hydratase concentration, demonstrating that only the enol form (CHM) and not the keto form (OMA) of the compound is utilized as the substrate by both hydratases. The slow rate is thus attributable to the rate-limiting spontaneous tautomerization of the OMA to CHM in the absence of GalD. Polarimetric analysis of the LigJCsE6-catalyzed reactions confirms that the enzyme has the same substrate stereo requirement as the GalB enzyme, producing and utilizing only the (−)-CHA and (−)-HMG enantiomers (Fig. 5).
Comparison of GalB and LigJ Active Sites
Although they exhibit different folds, GalB and LigJ share some similarities in their active site organization. In the LigJ structure from R. palustris (PDB code 2GWG) the bound zinc ion is ligated in a tetrahedral fashion by residues His-6, His-8, His-178, and Glu-248 (Fig. 4C). Analogous to the GalB Glu-48 residue, the LigJ metal ligating Glu-248 is found to coordinate the zinc ion on the solvent-accessible face of the metal ion that separates the metal from the remainder of the active site pocket. The GalB Glu-48 is critical to enzyme function and may be functionally convergent on Glu-248 in LigJ. There are two missing regions not modeled in the final R. palustris structure, which map close to the active site. The cysteine residues of the LigJ from Pseudomonas ochraceae NGJ1, including Cys-183 and Cys-186 from the Thr-181–Gly-191 missing region of the R. palustris enzyme, are not involved in CHM catalysis but may be involved with substrate binding (10, 46). Similar to GalB, the LigJ active site pocket is lined with several hydrophilic residues, which could support substrate binding and catalysis. However, to date, no investigations of the LigJ active site residues have been completed, and the role of these residues remains unknown.
Operon Organizations
A MultiGeneBlast search was utilized to identify species and gene clusters containing homologs to the gal and lig operons. Clusters for both gal and lig genes were found across proteo (α, β, and γ)- and actinobacteria (Fig. 8). In general, gene clusters containing a galB homolog were less prevalent in β-proteobacteria, whereas those with a ligJ were found throughout all bacterial clades. The galB gene was not found to replace ligJ in any of the canonical Lig pathway gene clusters. In contrast, galD homologs are frequently found in lig operons and is found in the same gene cluster as the canonical protocatechuate 4,5-cleavage pathway of Sphingobium sp. SYK-6 (9, 47). The presence of a GalD homolog in the Lig pathways suggests that the product of 2-pyrone-4,6-dicarboxylate (PDC) lactonase, LigI, is OMA rather than (2Z,4E)-CHM, which is subsequently tautomerized to (2E,4E)-CHM by the GalD homolog to be used by either GalB or LigJ (Fig. 1). Thus the genetic organization is consistent with our experimental data suggesting that both GalB and LigJ utilize the same isomer of CHM produced by GalD. It is, however, unclear why two evolutionarily distinct hydratases are recruited to perform the same reaction in these related pathways.
FIGURE 8.

Gene clusters containing GalB and LigJ CHM hydratases in α-proteo, β-proteo, γ-proteo, and actinobacteria. Gene clusters comprising known members of aromatic metabolism and either GalB (A) or LigJ (B) were identified using MultiGeneBlast, and a small sample of the “hits” are shown. Genes homologous to the GalB CHM hydratase are shown in cyan, and genes homologous to the LigJ CHM hydratase are shown in purple. Further homologous genes shown are: dioxygenase genes (GalA, LigAB, and LigZ) in red; OMA tautomerase (GalD) in green; HMG/CHA aldolase (GalC and LigK) in yellow; 4-carboxy-2-hydroxymuconate-6-semialdehyde dehydrogenase (LigC) in blue; and PDC lactonase (LigI) in brown.
Discussion
The metabolism of gallic acid by P. putida through the gallate pathway is similar to the metabolism of other aromatic compounds such as hydroxyphenyl acetate (through the Hpa/Hpc pathway) and catechol (through the Xyl 2,3-cleavage pathway). In all of these pathways a dienol or dienolate compound is produced (either as a substrate or transition state intermediate), which is then hydrated to an aldol product to enable C–C cleavage by a subsequent aldolase that connects the products to central cellular metabolism (48–50). GalB is structurally distinct from the previously characterized hydratases, including the CHM hydratase LigJ, and is instead related to the PIG-L family of deacetylases.
In common with the PIG-L deacetylases, GalB has a single α/β domain with a Rossmann-like fold containing a His-X2-Asp-Xn-His divalent metal binding motif found at the bottom of a solvent-accessible pocket. As with members of the PIG-L family, GalB was initially described as a Zn2+-dependent enzyme (9). Kinetic analyses of the PIG-L family protein MshB identified the enzyme activity as maximal when utilizing Fe2+ as a cofactor, and Fe2+ was found to be the predominant ion when the gene is expressed and the product purified under anaerobic conditions (51). When expressed and purified aerobically, however, MshB is found to switch metals, resulting in predominantly Zn2+ found in the purified enzyme. Unlike previous reports on GalB, here we showed that enzymatic activity could be rescued when the apoenzyme was incubated for at least 1 h with the exogenous metal ions (9). GalB exhibits the same trend as MshB in metal ion activity, with the highest turnover catalyzed by cofactors Fe2+> Co2+ ≫ Zn2+ (51). Only small changes in CHM specificity are seen when GalB utilizes Fe2+, Co2+, or Zn2+ as a cofactor, and the increase in kcat for Fe2+ relative to Zn2+-supported reactions is tempered by a simultaneous increase in Km. Zinc is found as the predominant metal ion when GalB is overexpressed in the native strain of Pseudomonas and aerobically purified, with iron and cobalt each found in less than 1% of the total enzyme. The identity of the metal utilized by the enzyme in vivo may not be zinc, however, as the lengthy and aerobic protein isolation strategies utilized in our study may have resulted in a loss of iron, similarly seen in MshB and other iron-dependent enzymes (51). GalB is able to bind both Zn2+ and Co2+ efficiently with submicromolar Kd values. As expected, enzyme variants of the metal ligating residues, except for the Glu-48 residue, resulted in substantial increases in the proteins Kd for metal ion. The Glu-48 residue is not observed to coordinate the metal ion in any of the known PIG-L deacetylases, which contain an equivalent glutamate residue, and its inclusion in GalB is likely to fulfill more of a functional role than that of a metal ligand.
There is some uncertainty as to the isomeric form of CHM utilized by LigJ and GalB (8, 9). The initial characterization of the gal operon identified (2E,4E)-CHM as the product of the GalD-catalyzed reaction, describing the compound as the 2E isomer on the basis of the chemical shift of the C3 olefinic proton (δ 6.73 ppm) being lower than expected if it was the 2Z isomer (δ 7.38 ppm) (9). However, a recent crystal structure of the PDC lactonase LigI, which catalyzes the production of an unknown tautomer of OMA or CHM in the Lig pathway, was solved and found to contain (2Z,4E)-CHM bound to the enzyme. The potential difference in the isomeric forms of CHM produced through the two different pathways was initially hypothesized to be the result of differences in the substrate stereospecificity of the two hydratases, GalB and LigJ, from the distinct pathways. However, here we have shown that both GalB and LigJCsE6 can indeed utilize the same CHM isomer produced from GalD, and this utilization leads to the same (−)-enantiomer of CHA. Similarly, when reactions with either GalB or LigJCsE6 were incubated with racemic CHA, only the (−)-enantiomer was consumed by both of the hydratase enzymes. A galD homolog is commonly found in the protocatechuate 4,5-cleavage pathways, and previous studies have shown that GalB can rescue the growth of a Sphingomonas sp. SYK-6 derivative species (Sphingomonas sp. DLJ), which lacks ligJ when grown on vanillate or syringate, further indicating that the GalB and LigJ enzymes act on the same (2E,4E)-CHM isomer produced physiologically by GalD (9, 10). The assumed CHM product observed in the LigI structure may therefore be an artifact of crystallization arising from utilization of CHM/OMA prepared at pH 10, but when cocrystallized at pH 6.5, OMA underwent nonenzymatic tautomerization to (2Z,4E)-CHM, which may not be the physiological substrate for the enzyme (8).
Previous characterizations of the GalC/LigK gene product have identified both CHA and HMG as substrates for the aldolase enzyme, with the enzyme having stereo preference for the (R)-HMG and (−)-CHA enantiomers (12, 52–54). Physiological routes leading to CHA production, through both the gallate and PCA 4,5-cleavage pathways, have been identified for some time. However, there has yet to be a definition of a physiological route leading to HMG production. Characterizations of DDVA metabolism in Sphingomonas sp. SYK-6 have identified the genes LigXa and LigZ, which transforms DDVA into a meta-cleavage product (4-[2-(5-carboxy-2-hydroxy-3-methoxyphenyl)-2-oxoethylidene]-2-hydroxypent-2-enedioic acid) that is hydrolyzed by LigY into 5-carboxyvanillate and CHPD (5, 55, 56) (Fig. 1). The 5-carboxyvanillate is further metabolized to vanillate and then degraded through the protocatechuate 4,5-cleavge pathway. However, the subsequent metabolism of the CHPD has yet to be defined. Here we have shown that both GalB and LigJCsE6 utilize the (R)-HMG enantiomer from a racemic HMG mixture and that the dehydration reaction is reversible. GalB had a 10-fold increase and a 23-fold decrease in Km and kcat, respectively, for CHPD hydration relative to the CHM substrate. Although the kinetic parameters for CHPD are less ideal relative to the CHM substrate, it is reasonable to assign CHPD hydratase roles to both GalB and LigJ homologs in the LigXa/LigZ/LigY pathway, which gives a route to HMG production for the HMG/CHA aldolase that has yet to be identified.
Attempts to cocrystallize GalB or the E48A variant with either CHM or CHA did not yield new crystallization conditions from the crystallization screens used and yielded only poorly diffracting crystals in the holoenzyme condition. Soaking the crystals with their substrates or products resulted in cracking of the crystals and was not successful. Also, attempts to model the substrates and/or products of GalB into the active site by computational docking methods did not yield productive models that would support catalysis. The kinetic investigations carried out here have led to a proposal for key residues in the GalB active site and have facilitated the generation of a proposed model for substrate binding and catalysis (Fig. 4B). Binding of the substrate is facilitated by the C1 carboxyl positioned by interaction with Nϵ of His-198, the C2 enolate positioned by interaction with Nη1 and Nη2 of Arg-67 and Nϵ of Gln-196, the C4 carboxylate positioned by interaction with Nη1 of Arg-21 and the hydroxyl of Tyr-203, and the C5 carboxylate positioned by interaction with Nη1 of Arg-216. The R216A variant had a ∼50,000-fold decrease in the specificity constant for CHM but only a 3-fold decrease in the specificity constant for CHPD, which is consistent with the residue facilitating (2E,4E)-CHM binding through the C5 carboxylate. In the proposed mechanism the (2E,4E)-CHM enolate serves as the substrate for the enzyme and Glu-48 activates the metal-ligated water for the addition at the C4 with protonation by His-164 at the C5 (Fig. 9). Tautomerization of the enolate leads to abstraction of the proton from Glu-48 to the C3 position, thus completing the reaction cycle.
FIGURE 9.
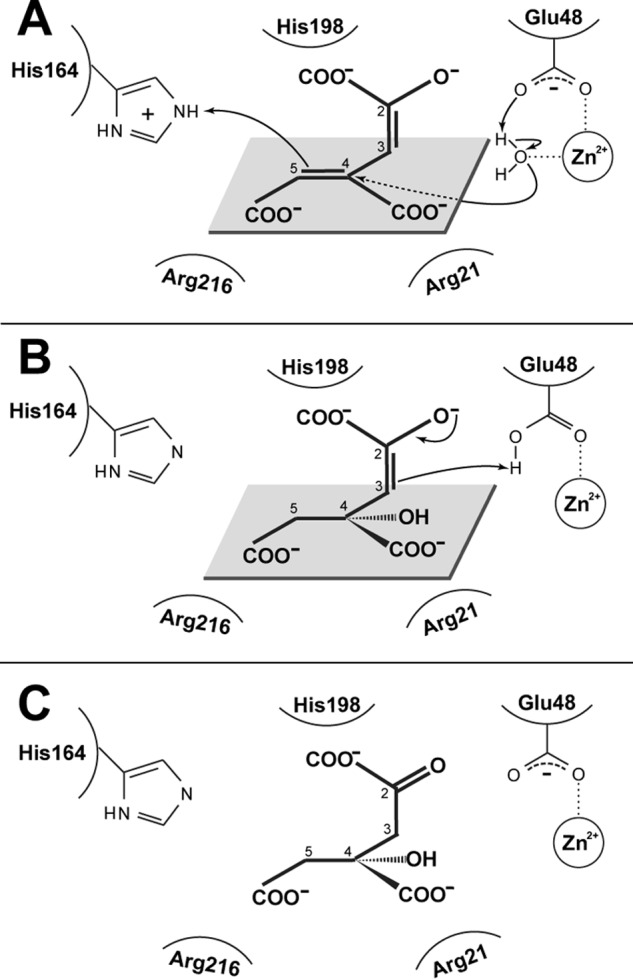
Proposed catalytic mechanism of GalB. A, the (2E,4E)-CHM enolate is positioned in the active site by the interaction of the carboxylate molecules C2, C4, and C5 with His-198, Arg-21, and Arg-216, respectively. The Glu-48 activates the metal-ligated water leading to the addition of the activated water at the C4 with subsequent protonation of the C5 by His-164. B, tautomerization of the enolate and protonation of the C3 by the protonated Glu-48 completes the reaction cycle. C, the CHA product found as the (S)-enantiomer. Note that the GalB is stereospecific for only the (−)-CHA enantiomer and that, although the absolute configuration of the (−)-CHA is not known, only the (S)-CHA product could be modeled consistent the biochemical data presented.
The enol form (CHM), and not the keto form (OMA), acts as the substrate for GalB, indicating that either the enol proton contributes to the catalytic mechanism or the enolate serves as the substrate. The CHM has a pKa of 7.3, which leads to an increase in the extinction coefficient for the substrate with increasing pH. This is consistent with the enol deprotonation leading to an increase in the chromogenicity of the compound, which would not arise from deprotonation of the carboxylate moieties of CHM. The enol pKa is observed in the enzyme pH dependence, with the deprotonation leading to increased GalB hydration activity, indicating that the substrate for the enzyme is in fact the enolate and that the enol proton is not likely to be involved in the catalytic mechanism.
The single ionization in the enzyme-substrate complex (pKa 6.1 ± 0.1) of GalB is analogous to the pKa observed in carbonic anhydrases, where the zinc metal cofactor in the carbonic anhydrases facilitates the decrease in the pKa of water, leading to a hydroxide primed for nucleophilic attack at physiological pH values (reviewed in Ref. 57). In the PIG-L N-deacetylases and some members of the metalloprotease family (such as carboxypeptidase A and thermolysin, which are structurally distinct from the PIG-L N-deacetylases but contain a similar metal ligation and act on similar chemical linkages), a similar Lewis acid mechanism is also proposed (38, 58, 59). In these enzymes an acidic residue (an aspartate in PIG-L and glutamate in the metalloproteases) in close proximity to the metal ligand is proposed to deprotonate the metal-ligated water to act as a nucleophile (45, 60–62). In both cases, after bond scission the protonated acidic residues relinquish the proton either to solvent or to the newly formed terminal atom. Similar to what is observed with the PIG-L N-deacetylases and metalloproteases, the GalB Glu-48 carboxylate ligates the metal ion and is in close proximity to the metal-ligated water. The GalB E48A variant has a 760-fold decrease in kcat for the CHM hydration reaction, and the pKa of the free enzyme in the variant-catalyzed reaction shifts down 0.6 unit (from ∼6.3 in the wild type to ∼5.7 in the variant) supporting the role of Glu-48 in proton transfer and acting as a general base. The abstracted proton from the metal-ligated water could be utilized to protonate the C3 during tautomerization or could be lost to solvent, with the solvent donating the proton to complete the reaction cycle.
His-164 is located in the active site with the imidazole ring facing the pocket and its Nδ making a hydrogen bond to the phenolic hydroxyl of Tyr-203. Rotation of the imidazole group of His-164 such that Nδ projects toward the active site results in Nδ being 6.6 Å from the metal-ligated water (Fig. 4B). The H164A substitution results in a 56-fold decrease in kcat with a relatively negligible effect on the Km of the CHM hydration reaction. The pKb of the free enzyme in the H164A variant increases ∼0.6 unit, from 8.6 in the wild type to 9.2 in the variant, consistent with the proposal that the residue functions as a general acid in the reaction mechanism.
The effect of either Zn2+ or Co2+ as a cofactor on the pH dependence of the enzyme is minimal. The GalB pKa values for both the enzyme-substrate complex and the free enzyme/substrate are unaffected by the metal cofactor utilized. Bell-shaped profiles for pH dependence on kcat/Km for Zn2+ cofactor reactions are observed with the PIG-L deacetylases MshB (pKa values 7.3 and 10.5) and BshB (pKa values 6.5 and 8.5), which are similar to GalB (45, 63). In both MshB and BshB, a minimal or no effect is observed on either pKa when utilizing Co2+ as the cofactor, and in MshB neither Fe2+, Ni2+, nor Mn2+ affected the pKa values (45, 63). In the proposed GalB and PIG-L deacetylase reactions, the metal contributes to the mechanism by positioning and likely lowering the pKa of the catalytic water. However, it appears that in both GalB and the PIG-L deacetylases that the metal does not significantly affect the pKa of the catalyzed reaction, possibly the result of key residues in the enzymes having a more significant effect on the pKa.
GalB is a hydratase from the gallic acid utilization pathway that is structurally distinct from previously characterized hydratase enzymes from other aromatic catabolic gene clusters, including the CHM hydratase LigJ. A comparison of the GalB and LigJ active site organizations indicates similar residues that may facilitate a common mechanism between the convergently evolved enzymes. The structure and kinetic characterization of GalB presented here will help guide future investigations into both the LigJ and GalB CHM hydratases.
Author Contributions
S. M. did most of the experiments, crystallized and solved the GalB structure, and wrote most of the paper. A. S. B. optimized the protocols to synthesize and purify CHM in addition to creating some of the GalB variants. M. S. K. helped with the crystallography and critically appraised the manuscript. S. Y. K. S. coordinated the project and assisted S. M. with writing the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Elyse Roach and Cezar Khursigara for their assistance and expertise with the use of the anaerobic chamber, Prof. David Rose and colleagues from his laboratory at the University of Waterloo for providing an x-ray source and assistance in screening several crystals, and Robert Reed for his assistance with polarimetry assays. The x-ray crystallography data were collected using beamlines 08ID-1 and 08B1–1 at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
This work was supported by Grant 2015-05366 from the National Science and Engineering Research Council of Canada (to S. Y. K. S.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5CGZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PCA
- protocatechuate
- OMA
- 4-oxalomesaconate
- CHM
- 4-carboxy-2-hydroxymuconate
- CHA
- 4-carboxy-4-hydroxy-2-oxoadipate
- CHPD
- 4-carboxy-2-hydroxypenta-2,4-dienoate
- HMG
- 4-hydroxy-4-methyl-2-oxoglutarate
- NTA
- nitrilotriacetic acid
- ICP-MS
- inductively coupled plasma mass spectrometry
- DDVA
- 5,5′-dehydrodivanillate
- PDC
- 2-pyrone-4,6-dicarboxylate
- PDB
- Protein Data Bank.
References
- 1. Ragauskas A. J., Beckham G. T., Biddy M. J., Chandra R., Chen F., Davis M. F., Davison B. H., Dixon R. A., Gilna P., Keller M., Langan P., Naskar A. K., Saddler J. N., Tschaplinski T. J., Tuskan G. A., and Wyman C. E. (2014) Lignin valorization: Improving lignin processing in the biorefinery. Science 344, 1246843. [DOI] [PubMed] [Google Scholar]
- 2. Vanholme R., Demedts B., Morreel K., Ralph J., and Boerjan W. (2010) Lignin biosynthesis and structure. Plant Physiol. 153, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dagley S., Evans W. C., and Ribbons D. W. (1960) New pathways in the oxidative metabolism of aromatic compounds by microorganisms. Nature 188, 560–566 [DOI] [PubMed] [Google Scholar]
- 4. Kamimura N., Aoyama T., Yoshida R., Takahashi K., Kasai D., Abe T., Mase K., Katayama Y., Fukuda M., and Masai E. (2010) Characterization of the protocatechuate 4,5-cleavage pathway operon in Comamonas sp. strain e6 and discovery of a novel pathway gene. Appl. Environ. Microbiol. 76, 8093–8101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng X., Egashira T., Hanashiro K., Masai E., Nishikawa S., Katayama Y., Kimbara K., and Fukuda M. (1998) Cloning of a Sphingomonas paucimobilis SYK-6 gene encoding a novel oxygenase that cleaves lignin-related biphenyl and characterization of the enzyme. Appl. Environ. Microbiol. 64, 2520–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katayama Y., Nishikawa S., Murayama A., Yamasaki M., Morohoshi N., and Haraguchi T. (1988) The metabolism of biphenyl structures in lignin by the soil bacterium (Pseudomonas paucimobilis SYK-6). FEBS Lett. 233, 129–133 [DOI] [PubMed] [Google Scholar]
- 7. Tack B. F., Chapman P. J., and Dagley S. (1972) Metabolism of gallic acid and syringic acid by Pseudomonas putida. J. Biol. Chem. 247, 6438–6443 [PubMed] [Google Scholar]
- 8. Hobbs M. E., Malashkevich V., Williams H. J., Xu C., Sauder J. M., Burley S. K., Almo S. C., and Raushel F. M. (2012) Structure and catalytic mechanism of LigI: insight into the amidohydrolase enzymes of cog3618 and lignin degradation. Biochemistry 51, 3497–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nogales J., Canales A., Jiménez-Barbero J., Serra B., Pingarrón J. M., García J. L., and Díaz E. (2011) Unravelling the gallic acid degradation pathway in bacteria: The gal cluster from Pseudomonas putida. Mol. Microbiol. 79, 359–374 [DOI] [PubMed] [Google Scholar]
- 10. Hara H., Masai E., Katayama Y., and Fukuda M. (2000) The 4-oxalomesaconate hydratase gene, involved in the protocatechuate 4,5-cleavage pathway, is essential to vanillate and syringate degradation in Sphingomonas paucimobilis SYK-6. J. Bacteriol. 182, 6950–6957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shannon L. M., and Marcus A. (1962) γ-Methyl-γ-hydroxy-α-ketoglutaric aldolase. I. Purification and properties. J. Biol. Chem. 237, 3342–3347 [PubMed] [Google Scholar]
- 12. Tack B. F., Chapman P. J., and Dagley S. (1972) Purification and properties of 4-hydroxy-4-methyl-2-oxoglutarate aldolase. J. Biol. Chem. 247, 6444–6449 [PubMed] [Google Scholar]
- 13. Rea D., Fülöp V., Bugg T. D., and Roper D. I. (2007) Structure and mechanism of HpcH: a metal ion dependent class II aldolase from the homoprotocatechuate degradation pathway of Escherichia coli. J. Mol. Biol. 373, 866–876 [DOI] [PubMed] [Google Scholar]
- 14. Horn J. M., Harayama S., and Timmis K. N. (1991) DNA sequence determination of the TOL plasmid (pWWO) xylGFJ genes of Pseudomonas putida: implications for the evolution of aromatic catabolism. Mol. Microbiol. 5, 2459–2474 [DOI] [PubMed] [Google Scholar]
- 15. Prieto M. A., Díaz E., and García J. L. (1996) Molecular characterization of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli W: engineering a mobile aromatic degradative cluster. J. Bacteriol. 178, 111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu H., and Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tabor S., and Richardson C. C. (1985) A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A. 82, 1074–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Lorenzo V., Eltis L., Kessler B., and Timmis K. N. (1993) Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123, 17–24 [DOI] [PubMed] [Google Scholar]
- 19. Nogales J., Canales A., Jiménez-Barbero J., García J. L., and Díaz E. (2005) Molecular characterization of the gallate dioxygenase from Pseudomonas putida KT2440: the prototype of a new subgroup of extradiol dioxygenases. J. Biol. Chem. 280, 35382–35390 [DOI] [PubMed] [Google Scholar]
- 20. Ni B., Zhang Y., Chen D. W., Wang B. J., and Liu S. J. (2013) Assimilation of aromatic compounds by Comamonas testosteroni: characterization and spreadability of protocatechuate 4,5-cleavage pathway in bacteria. Appl. Microbiol. Biotechnol. 97, 6031–6041 [DOI] [PubMed] [Google Scholar]
- 21. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 22. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 23. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 Web portal for protein modeling, prediction, and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., and Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement, and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Winn M. D., Isupov M. N., and Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 32. Riemer J., Hoepken H. H., Czerwinska H., Robinson S. R., and Dringen R. (2004) Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 331, 370–375 [DOI] [PubMed] [Google Scholar]
- 33. Fish W. W. (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364 [DOI] [PubMed] [Google Scholar]
- 34. McCall K. A., and Fierke C. A. (2000) Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal. Biochem. 284, 307–315 [DOI] [PubMed] [Google Scholar]
- 35. Volkamer A., Griewel A., Grombacher T., and Rarey M. (2010) Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Model. 50, 2041–2052 [DOI] [PubMed] [Google Scholar]
- 36. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 37. Holm L., and Rosenström P. (2010) Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maynes J. T., Garen C., Cherney M. M., Newton G., Arad D., Av-Gay Y., Fahey R. C., and James M. N. (2003) The crystal structure of 1-d-myo-inosityl 2-acetamido-2-deoxy-α-d-glucopyranoside deacetylase (MshB) from Mycobacterium tuberculosis reveals a zinc hydrolase with a lactate dehydrogenase fold. J. Biol. Chem. 278, 47166–47170 [DOI] [PubMed] [Google Scholar]
- 39. Zou Y., Brunzelle J. S., and Nair S. K. (2008) Crystal structures of lipoglycopeptide antibiotic deacetylases: Implications for the biosynthesis of A40926 and teicoplanin. Chem. Biol. 15, 533–545 [DOI] [PubMed] [Google Scholar]
- 40. Mine S., Niiyama M., Hashimoto W., Ikegami T., Koma D., Ohmoto T., Fukuda Y., Inoue T., Abe Y., Ueda T., Morita J., Uegaki K., and Nakamura T. (2014) Expression from engineered Escherichia coli chromosome and crystallographic study of archaeal N,N′-diacetylchitobiose deacetylase. FEBS J. 281, 2584–2596 [DOI] [PubMed] [Google Scholar]
- 41. Fadouloglou V. E., Deli A., Glykos N. M., Psylinakis E., Bouriotis V., and Kokkinidis M. (2007) Crystal structure of the BcZBP, a zinc-binding protein from Bacillus cereus. FEBS J. 274, 3044–3054 [DOI] [PubMed] [Google Scholar]
- 42. Chan H. C., Huang Y. T., Lyu S. Y., Huang C. J., Li Y. S., Liu Y. C., Chou C. C., Tsai M. D., and Li T. L. (2011) Regioselective deacetylation based on teicoplanin-complexed Orf2* crystal structures. Mol. Biosyst. 7, 1224–1231 [DOI] [PubMed] [Google Scholar]
- 43. McCarthy A. A., Peterson N. A., Knijff R., and Baker E. N. (2004) Crystal structure of MshB from Mycobacterium tuberculosis, a deacetylase involved in mycothiol biosynthesis. J. Mol. Biol. 335, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 44. Broadley S. G., Gumbart J. C., Weber B. W., Marakalala M. J., Steenkamp D. J., and Sewell B. T. (2012) A new crystal form of MshB from Mycobacterium tuberculosis with glycerol and acetate in the active site suggests the catalytic mechanism. Acta Crystallogr. D Biol. Crystallogr. 68, 1450–1459 [DOI] [PubMed] [Google Scholar]
- 45. Huang X., and Hernick M. (2012) Examination of mechanism of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) reveals unexpected role for dynamic tyrosine. J. Biol. Chem. 287, 10424–10434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li S., Kimura M., Takashima T., Hayashi K., Inoue K., Ishiguro R., Sugisaki H., and Maruyama K. (2007) Role of cysteine residues in 4-oxalomesaconate hydratase from Pseudomonas ochraceae NGJ1. Biosci. Biotechnol. Biochem. 71, 449–457 [DOI] [PubMed] [Google Scholar]
- 47. Masai E., Katayama Y., and Fukuda M. (2007) Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci. Biotechnol. Biochem. 71, 1–15 [DOI] [PubMed] [Google Scholar]
- 48. Izumi A., Rea D., Adachi T., Unzai S., Park S. Y., Roper D. I., and Tame J. R. (2007) Structure and mechanism of HpcG, a hydratase in the homoprotocatechuate degradation pathway of Escherichia coli. J. Mol. Biol. 370, 899–911 [DOI] [PubMed] [Google Scholar]
- 49. Burks E. A., Johnson W. H. Jr., and Whitman C. P. (1998) Stereochemical and isotopic labeling studies of 2-oxo-hept-4-ene-1,7-dioate hydratase: evidence for an enzyme-catalyzed ketonization step in the hydration reaction. J. Am. Chem. Soc. 120, 7665–7675 [Google Scholar]
- 50. Pollard J. R., and Bugg T. D. (1998) Purification, characterization and reaction mechanism of monofunctional 2-hydroxypentadienoic acid hydratase from Escherichia coli. Eur. J. Biochem. 251, 98–106 [DOI] [PubMed] [Google Scholar]
- 51. Huang X., Kocabas E., and Hernick M. (2011) The activity and cofactor preferences of n-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) change depending on environmental conditions. J. Biol. Chem. 286, 20275–20282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maruyama K. (1990) Purification and properties of 4-hydroxy-4-methyl-2-oxoglutarate aldolase from Pseudomonas ochraceae grown on phthalate. J. Biochem. 108, 327–333 [DOI] [PubMed] [Google Scholar]
- 53. Wang W., Mazurkewich S., Kimber M. S., and Seah S. Y. (2010) Structural and kinetic characterization of 4-hydroxy-4-methyl-2-oxoglutarate/4-carboxy-4-hydroxy-2-oxoadipate aldolase, a protocatechuate degradation enzyme evolutionarily convergent with the HpaI and DmpG pyruvate aldolases. J. Biol. Chem. 285, 36608–36615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ritter C. S., Chapman P. J., and Dagley S. (1973) Absolute configuration of a metabolite in the m-fission pathway of protocatechuate. J. Bacteriol. 113, 1064–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yoshikata T., Suzuki K., Kamimura N., Namiki M., Hishiyama S., Araki T., Kasai D., Otsuka Y., Nakamura M., Fukuda M., Katayama Y., and Masai E. (2014) Three-component O-demethylase system essential for catabolism of a lignin-derived biphenyl compound in Sphingobium sp. strain SYK-6. Appl. Environ. Microbiol. 80, 7142–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peng X., Masai E., Katayama Y., and Fukuda M. (1999) Characterization of the meta-cleavage compound hydrolase gene involved in degradation of the lignin-related biphenyl structure by Sphingomonas paucimobilis SYK-6. Appl. Environ. Microbiol. 65, 2789–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Becker H. M., Klier M., and Deitmer J. W. (2014) Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications. Subcell. Biochem. 75, 105–134 [DOI] [PubMed] [Google Scholar]
- 58. Matthews B. W., Jansonius J. N., Colman P. M., Schoenborn B. P., and Dupourque D. (1972) Three-dimensional structure of thermolysin. Nat. New Biol. 238, 37–41 [DOI] [PubMed] [Google Scholar]
- 59. Reeke G. N., Hartsuck J. A., Ludwig M. L., Quiocho F. A., Steitz T. A., and Lipscomb W. N. (1967) The structure of carboxypeptidase A, vi. Some results at 2.0 Å resolution, and the complex with glycyl-tyrosine at 2.8 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 58, 2220–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pelmenschikov V., Blomberg M. R., and Siegbahn P. E. (2002) A theoretical study of the mechanism for peptide hydrolysis by thermolysin. J. Biol. Inorg. Chem. 7, 284–298 [DOI] [PubMed] [Google Scholar]
- 61. Wu S., Zhang C., Xu D., and Guo H. (2010) Catalysis of carboxypeptidase A: promoted-water versus nucleophilic pathways. J. Phys. Chem. B 114, 9259–9267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alvarez-Santos S., González-Lafont A., Lluch J. M., Oliva B., and Avilés F. X. (1994) On the water-prompted mechanism of peptide cleavage by carboxypeptidase A: a theoretical study. Can. J. Chem. 72, 2077–2083 [Google Scholar]
- 63. Fang Z., Roberts A. A., Weidman K., Sharma S. V., Claiborne A., Hamilton C. J., and Dos Santos P. C. (2013) Cross-functionalities of Bacillus deacetylases involved in bacillithiol biosynthesis and bacillithiol-S-conjugate detoxification pathways. Biochem. J. 454, 239–247 [DOI] [PubMed] [Google Scholar]