

Abstract
The KCNJ10 gene encoding Kir4.1 contains numerous SNPs whose molecular effects remain unknown. We investigated the functional consequences of uncharacterized SNPs (Q212R, L166Q, and G83V) on homomeric (Kir4.1) and heteromeric (Kir4.1-Kir5.1) channel function. We compared these with previously characterized EAST/SeSAME mutants (G77R and A167V) in kidney-derived tsA201 cells and in glial cell-derived C6 glioma cells. The membrane potentials of tsA201 cells expressing G77R and G83V were significantly depolarized as compared with WTKir4.1, whereas cells expressing Q212R, L166Q, and A167V were less affected. Furthermore, macroscopic currents from cells expressing WTKir4.1 and Q212R channels did not differ, whereas currents from cells expressing L166Q, G83V, G77R, and A167V were reduced. Unexpectedly, L166Q current responses were rescued when co-expressed with Kir5.1. In addition, we observed notable differences in channel activity between C6 glioma cells and tsA201 cells expressing L166Q and A167V, suggesting that there are underlying differences between cell lines in terms of Kir4.1 protein synthesis, stability, or expression at the surface. Finally, we determined spermine (SPM) sensitivity of these uncharacterized SNPs and found that Q212R-containing channels displayed reduced block by 1 μm SPM. At 100 μm SPM, the block was equal to or greater than WT, suggesting that the greater driving force of SPM allowed achievement of steady state. In contrast, L166Q-Kir5.1 channels achieved a higher block than WT, suggesting a more stable interaction of SPM in the deep pore cavity. Overall, our data suggest that G83V, L166Q, and Q212R residues play a pivotal role in controlling Kir4.1 channel function.
Keywords: epilepsy, kinetics, molecular cell biology, patch clamp, potassium channel, potassium transport, single nucleotide polymorphism (SNP), Kir4.1, spermine, KCNJ10
Introduction
Inwardly rectifying potassium (Kir)3 channels are involved in maintenance of negative resting membrane potential (1–3), potassium buffering (1, 4), extracellular glutamate clearance (1, 4, 5), myelination (6), and cell volume regulation (3, 7). Kir4.1 channels are tetramers formed by Kir4.1 (KCNJ10) subunits. Kir4.1-containing channels are expressed in glial cells (8–14) including in astrocytic end feet surrounding blood vessels (9, 15–17) and surrounding neuronal synapses (9, 17) where they are involved in maintenance of extracellular [K+]o and glutamate homeostasis (4, 11, 18). These functions are critical as inability to control [K+]o and glutamate alters neuronal excitability and may lead to seizures and neuronal death (4, 18–20). Similarly in the retina, Kir4.1-dependent Kir channels are involved in homeostasis of extracellular potassium produced by neuronal activity in a process called potassium siphoning (9, 14, 21, 22).
Kir4.1 subunits are also prominently expressed in the distal convoluted tubules in the kidneys (23) where they are involved in K+ recycling (24) and in the ear, specifically in the stria vascularis, where they are responsible for producing the endocochlear potential (7). Complete absence or loss-of-function mutations in these channel subunits cause EAST/SeSAME syndrome characterized by seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (25, 26). In temporal lobe epilepsy (27), Kir4.1 subunit variants have been implicated in perturbation of neuronal excitability and increasing the propensity of seizures due to inappropriate K+ clearance (28, 29).
Interestingly, there are over 120 coding region single nucleotide polymorphisms (SNPs) in the KCNJ10 gene reported in publicly accessible genome databases, and the electrophysiological consequences of these variants have not been examined thoroughly. Kir4.1 can form homotetrameric channels but can also heteromultimerize with Kir5.1 (KCNJ16) subunits (30). Heteromeric Kir4.1-Kir5.1 channels exhibit distinct biophysical properties including larger single channel conductance together with greater pH sensitivity (23, 31, 32), weaker inward rectification, and different expression patterns. Heteromeric Kir4.1-Kir5.1 channels are expressed in the brain stem (33), neocortex, glomeruli of the olfactory bulb (30), retina (34) and kidney cortex, especially in the basolateral membrane of the cortical collecting ducts where they are thought to be responsible for K+ recycling (23). In retinal Müller glial cells, there appears to be a subcellular localization of these channels with homomeric channels being localized in the end feet and heteromeric channels being localized in the somata and distal processes of these cells (34).
In the present study, we investigated the functional consequences of previously uncharacterized KCNJ10 variants, Q212R (rs36040296), L166Q (rs1130182), and G83V (rs17853258) that are predicted by Polymorphism Phenotyping (PolyPhen) analysis to be “probably damaging” (35) but have not been functionally examined. Furthermore, we examined and compared the functional consequences of previously uncharacterized SNPs with EAST/SeSAME-causing mutants A167V and G77R using tsA201 cells and a glial cell-derived glioma cell line. Using a heterologous expression system with whole-cell and excised patch voltage clamp techniques, we evaluated the impact of these variants on homomeric Kir4.1 and heteromeric Kir4.1-Kir5.1 channel activity.
Experimental Procedures
Expression of Wild-type and Mutant Kir4.1 Channels in tsA201 Cells
Rat Kir4.1 cDNA fused with enhanced green fluorescent protein (EGFP) on its N-terminal end was inserted into pcDNA3.1 vector (Invitrogen). This Kir4.1-EGFP was used as a template into which Q212R, L166Q, and G83V variants were introduced using a QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, Ca). All SNPs and mutations were confirmed by DNA sequencing (Genewiz, Inc., South Plainfield, NJ). The EAST/SeSAME-causing mutations A167V and G77R were the same as used previously (36). KCNJ10 mutants were transformed into Escherichia coli XLS Blue supercompetent cells (Stratagene) to obtain DNA for further experiments. tsA201 cells (a kind gift from Dr. William Green, University of Chicago) and rat C6 glioma cells (number CCL-107, American Type Culture Collection, Manassas, VA) were plated in dishes on poly-d-lysine-coated glass coverslips (15-mm diameter) and cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum, 20 mm HEPES, and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin) for tsA201 cells and HEPES-free with 100 units/ml penicillin (Invitrogen) for C6 glioma cells. In both cases, pH was adjusted to 7.4. Cells were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37 °C, the medium was replaced every 3rd day, and cells were passaged twice a week. Cells to be used for whole-cell patch clamp experiments were cultivated to 40% confluence before transfection to avoid cell to cell coupling. For inside-out patch clamp recording, cells were cultivated to 70% confluence before transfection to increase the chances of finding transfected cells. The appropriate cDNAs were transfected into the cells using FuGENE 6 transfection reagent (Roche Applied Science). Normally in tsA201 cells, 600 ng of DNA was diluted in 100 μl of serum-free DMEM, further mixed with 2 μl of FuGENE 6 for each 500 μl of DMEM, and incubated on ice for 45 min before application. To increase channel expression in C6 glioma cells, 700 ng of DNA was used. Cells were incubated in the presence of the transfection mixture for 24–36 h before experiments.
For the expression of heteromeric Kir4.1-Kir5.1 channels or Kir4.1 variant/Kir5.1 channels, cells were transfected with each cDNA at a ratio of 1:1 while maintaining unchanged the overall amount of 600 ng of DNA for tsA201 cells and 700 ng for C6 glioma cells. Patch clamp experiments were conducted at room temperature on cells that were positive for EGFP-Kir4.1 expression as assessed by fluorescence under ultraviolet light. Cells exposed to FuGENE 6 with no cDNA or cDNA for Kir5.1 alone served as controls for homomeric and heteromeric expression, respectively. Multiple transfections were performed in parallel.
Electrophysiology
Membrane potential (Vm) was measured immediately after the establishment of whole-cell configuration. In most experiments, membrane currents were measured with the single electrode whole-cell patch clamp technique. Two Narishige hydraulic micromanipulators (MMW-203, Narishige, Japan) were used for 1) voltage clamp recording and 2) positioning a perfusion micropipette with a 30–50-μm tip diameter positioned at a very close distance from the tip of the patch pipette to allow for rapid application of test solutions. For whole-cell experiments, electrodes with resistances of 4–6 megaohms were pulled from borosilicate glass capillaries (1B150F-4, World Precision Instruments, Inc., Sarasota, FL) in two steps using a Sutter P-97 puller (Sutter Instruments Corp., Novato, CA). Pipettes were filled with intracellular solution containing 120 mm potassium gluconate, 10 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm EGTA, 10 mm HEPES, and 0.3 mm spermine tetrahydrochloride with pH adjusted to 7.2 with KOH/HCl. After achieving whole-cell mode, access resistance was 10–15 megaohms, compensated by at least 75%. The extracellular solution contained 145 mm NaCl, 2.5 mm CaCl2, 2 mm MgCl2, and 10 mm HEPES; KCl was varied from 3 to 10 mm (substituted by NaCl to adjust osmolarity). High frequencies (>1 kHz) were cut off using an Axopatch-200B and a CV-203BU head stage or Axon multiclamp-700B amplifier and a CV-7B head stage. Signals were digitized at 5 kHz through a DigiData 1322A or 1440 interface (Axon Instruments, Sunnyvale, CA). The pClamp 9 (Axon Instruments) software package was used for data acquisition and analysis.
For whole-cell experiments, a pulse protocol was applied in which Vm was held at resting membrane potential and then stepped to test potentials between −100 and +100 mV for 140 ms before returning to the holding potential. To examine sensitivity to spermine (SPM), experiments using the inside-out patch clamp configuration were implemented by applying a step protocol from −100 to +100 mV in 10-mV increments. Electrodes with resistances of 2–3 megaohms were pulled from borosilicate glass capillaries (1B150F-4, World Precision Instruments, Inc.) in four steps using a Sutter P-97 puller. For SPM sensitivity experiments, the solutions used in pipettes (extracellular) and bath (cytoplasmic) were symmetrical, containing 138 mm KCl, 2 mm KOH, 1 mm EGTA, 1 mm EDTA, and 10 mm HEPES; pH was adjusted using KOH/HCl. Spermine tetrahydrochloride was purchased from (Sigma-Aldrich).
Data Analysis
Data were analyzed using Molecular Devices Clampfit 10.4 (Sunnyvale, CA), Origin 6.1 (Northampton, MA), and Microsoft Excel programs. The results are presented as the mean ± S.E.
A descriptive analysis of the variables was done (frequencies, percentages, and central tendency measures as well as variability measures). Normality criteria were evaluated to select the correct parametric or non-parametric test using the Shapiro-Wilk estimator. In normality tests, Ho determines Gaussian distribution, whereas Ha delimits non-Gaussian distribution. The test determined that in most cases a parametric approach should be used (p > 0.05). In some cases, if n is small and to avoid a possible type II error scenario, theory establishes that non-parametric tests are the best choice. A type II error occurs when small sample size blurs statistical significance. Differences among means were evaluated using the k independent sample ANOVA test with Levene's homogeneity of variances test. Pairwise comparisons were made using the Tukey adjustment. Differences among medians were evaluated using the non-parametric k independent sample Kruskal-Wallis test with Bonferroni correction (pairs to be tested), m = p/k, where m is the adjusted p value is 0.05 and k is the number of pairs to be post-tested. The statistical software used was SPSS 23.0.0.1 (Chicago, IL). The overall significance level (α) was set to 0.05. Error bars represent ±S.E. The analyses used for each series of experiments are described in the corresponding figure legends.
Boltzmann Equation Fit
Microsoft Solver, a function of Microsoft Excel, was used to fit the data by a least square algorithm. Currents in the presence of the polyamine SPM were expressed relative to the current in the absence of SPM (Irel). Irel-voltage relationships were fit by a Boltzmann function plus offset as described previously (37).
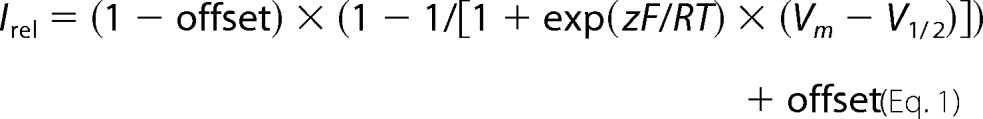 |
where R, T, and F have their usual meanings; z is the effective valence of block; and Vm and V1/2 are the membrane voltage and the voltage at half-maximal block, respectively. Data are presented as mean ± S.E.
Results
In these studies, we evaluated the impact of three novel KCNJ10 SNPs (Q212R, L166Q, and G83V) and two previously characterized EAST/SeSAME mutants (A167V and G77R) on the channel-dependent electrical activity. We assessed this in tsA201 and C6 glioma cells expressing Kir4.1 channels using whole-cell patch clamp recordings.
We first determined that the cell lines chosen for these studies did not express appreciable functional Kir channels. We used untransfected tsA201 cells (a human embryonic kidney-derived cell line) and C6 glioma cells (a rat glial cell-derived glioma cell line) and induced currents by using a step protocol from −100 to 100 mV from the holding potential (Vh). We measured whole-cell currents in the absence and presence of 100 μm barium (a selective Kir channel blocker). In untransfected tsA201 cells (Fig. 1A), the average inward currents measured at −120 mV were −0.15 ± 0.035 nA (n = 3). Similar whole-cell currents of −0.14 ± 0.03 nA (n = 3) were seen in the presence of 100 μm barium, giving negligible barium-sensitive currents of −0.01 ± 0.002 nA (Fig. 1, A3).
FIGURE 1.

Whole-cell currents measured from tsA201 (A1–A4 and B1–B4) and C6 glioma cells (C1–C4 and D1–D4) in response to a voltage step protocol from −100 to +100 mV in the presence and absence of 100 μm barium. Cells were clamped at Vh, which was equal to the resting Vm (Vh = Vm). Vh values were −28.6 ± 1.8 and −74.0 ± 0.6 mV for non-transfected (A1) and Kir4.1-transfected (B1) tsA201 cells and −38.9 ± 0.8 and −83.2 ± 1.7 mV for non-transfected (C1) and Kir4.1-transfected (D1) C6 glioma cells. Representative current traces within the −120 to +20-mV range from non-transfected tsA201 cells (A), Kir4.1-transfected tsA201 cells (B), non-transfected C6 glioma cells (C), and Kir4.1-transfected C6 glioma cells (D) are shown. Whole-cell currents were recorded from tsA201 cells (A1 and B1) and C6 glioma cells (C1 and D1) in response to a voltage step protocol from −100 to +100 mV from the holding potential. Note that 1) Vh is more hyperpolarized in Kir4.1-transfected cells and 2) where there is no error bar shown it is smaller than the size of the symbol. Whole-cell currents were recorded from tsA201 cells (A2 and B2) and C6 glioma cells (C2 and D2) in response to a voltage step protocol in the presence of 100 μm Ba2+ (a blocker of Kir channels). Barium-sensitive currents from tsA201 cells (A3 and B3) and C6 glioma cells (C3 and D3) are shown. The graph shows the subtraction of currents obtained in the presence of barium from total whole-cell currents (Control). Barium-sensitive currents reflect the contribution of Kir channels to the whole-cell currents, and in untransfected cells, there is virtually no barium-sensitive current. Summaries of inward current measured at −120 mV from tsA201 cells (A4 and B4) and C6 glioma cells (C4 and D4) are also shown. Data (A4–D4) were analyzed using central tendency measures (means), dispersion measures (standard deviations), and Kruskal-Wallis k independent group test with a Bonferroni correction (p < 0.05). Results (A4–D4) are expressed as mean ± S.E., and significant differences from control are shown (*). Error bars represent ±S.E.
It has previously been shown that Kir4.1 channels in C6 glioma cells are mislocalized to the nucleus and not functional (38, 39). To verify that these cells were not natively expressing functional Kir4.1 channels, we determined whole-cell currents for untransfected cells in response to a step protocol in the absence and presence of 100 μm barium as for tsA201 cells above. Inward currents obtained at −120 mV in the absence and presence of 100 μm barium were −0.20 ± 0.01 (n = 3) and −0.18 ± 0.02 nA (n = 3), respectively, giving a barium-sensitive current of −0.02 ± 0.01 nA (n = 3) (Fig. 1, C1–C3).
For comparison, we expressed WTKir4.1 in tsA201 cells (Fig. 1B) and in C6 Glioma cells (Fig. 1D) to demonstrate 1) that these cell lines were amenable to transfection and 2) that Kir4.1 currents could be blocked by 100 μm barium. The average whole-cell currents elicited at −120 mV from a step protocol were −1.71 ± 0.14 and −1.58 ± 0.24 nA for tsA201 (Fig. 1, B1) and C6 glioma cells (Fig. 1, D1), respectively. The currents in the same cells measured in the presence of 100 μm barium were −0.15 ± 0.08 and −0.15 ± 0.06 nA, respectively, indicating that the majority of the current was blocked by barium.
After confirming that both tsA201 and C6 glioma cells were suitable candidates for our experiments, we then proceeded to measure the resting membrane potential of cells expressing uncharacterized SNPs and EAST/SeSAME-causing mutations. Membrane potentials were recorded immediately after cell penetration and are summarized in Fig. 2A for transfected tsA201 cells and in Fig. 2C for transfected C6 glioma cells. Cell membrane potentials were considerably more hyperpolarized (∼−75 and ∼−82 mV) in cells expressing homomeric WT channels compared with mock-transfected cells (∼−30 and ∼−35 mV) in tsA201 and C6 glioma cells, respectively. Cells expressing G83V or G77R mutant had membrane potentials similar to untransfected cells. In contrast, cells expressing Q212R, L166Q, or A167V were significantly hyperpolarized compared with untransfected cells, but most of them were less hyperpolarized compared with WTKir4.1-transfected cells (Fig. 2, A and C). A notable difference was found between tsA201 and C6 glioma cells expressing the L166Q variant. The membrane potential of the L166Q-expressing tsA201 cells was slightly less hyperpolarized than those expressing WTKir4.1. In contrast, the membrane potential of C6 glioma cells expressing L166Q was substantially depolarized compares with WTKir4.1.
FIGURE 2.

Membrane potential of tsA201 and C6 glioma cells expressing WT, different SNPs, and EAST/SeSAME mutant Kir4.1 channels. A, average membrane potential of tsA201 cells expressing homomeric Kir4.1 channels. B, average membrane potential of tsA201 cells expressing heteromeric Kir4.1-Kir5.1 channels. C, average membrane potential of C6 glioma cells expressing homomeric Kir4.1 channels. D, average membrane potential of C6 glioma cells expressing heteromeric Kir4.1-Kir5.1 channels. Data were analyzed using central tendency measures (medians), dispersion measures (standard deviations), and a k independent group one-way ANOVA test with Tukey-Kramer post hoc tests (p < 0.05). Statistical differences from Mock or Kir5.1 control (*) or from WTKir4.1- or Kir4.1-Kir5.1-transfected cells (¤) are shown. Error bars represent ±S.E.
One distinguishing characteristic of Kir4.1 subunits is that natively they can be found co-assembled with Kir5.1 subunits, forming functional channels with biophysical properties that are different from those of homomeric channels (40–43) Therefore, we also assessed the membrane potential of cells co-expressing Kir4.1 variants with Kir5.1 (Fig. 2B). Membrane potentials were similar between cells expressing heteromeric WTKir4.1-Kir5.1, Q212R-Kir5.1, L166Q-Kir5.1, and A167V-Kir5.1. In both cell lines, cells expressing G83V-Kir5.1 or G77R-Kir5.1 were again not different from cells transfected with Kir5.1 alone, which does not form functional homomeric channels (43).
In whole-cell voltage clamp, similar current amplitudes were detected in tsA201 cells expressing homomeric WT and Q212R channels. In contrast, currents recorded from cells expressing homomeric L166Q and A167V were significantly lower, and the currents recorded from G83V- and G77R-expressing cells were negligible and similar to control (Fig. 3A). Similar current amplitudes were recorded from tsA201 cells expressing heteromeric WTKir4.1-Kir5.1, Q212R-Kir5.1, or L166Q-Kir5.1 channels. Cells expressing A167V-Kir5.1 channels had appreciable but reduced current, whereas cells expressing G83V-Kir5.1 and G77R-Kir5.1 channels were again not significantly different from control cells expressing the Kir5.1 subunit alone (Fig. 3B). It is interesting to note that, although whole-cell currents obtained from cells expressing homomeric L166Q channels were about 50% reduced as compared with WT, currents from cells co-expressing either Kir4.1-Kir5.1 or L166Q-Kir5.1 were similar, i.e. suggesting rescue of channel function. In contrast, the whole-cell currents in cells expressing A167V-Kir5.1 heteromeric channels remain reduced; i.e. channel function was not rescued.
FIGURE 3.

Whole-cell currents measured in tsA201 cells using a voltage step protocol. A step protocol with a 10-mV step increment from −100 to 100 mV from the holding potential (Vh = Vm) was applied. A, representative current traces from cells expressing homomeric Kir4.1 channels. B, representative current traces from cells expressing heteromeric Kir4.1-Kir5.1 channels. C, summary of the results within the range of −120 to 30 mV. D, summary of the results within the range of −120 to 30 mV. Error bars represent ±S.E.
There were notable differences in channel activity between C6 glioma cells and tsA201 cells expressing L166Q and A167V. When expressed in C6 glioma cells, inward currents from homomeric A167V were similar to WTKir4.1 and not reduced as in tsA201 cells (Figs. 3, A and C, and 4, A and C). Furthermore, L166Q-expressing glioma cells showed negligible currents very similar to G83V-, G77R-, and mock-transfected cells (Fig. 4A). Consistent with their expression in tsA201 cells, currents from L166Q-Kir5.1- transfected C6 glioma cells were partially rescued when compared with homomeric L166Q-transfected cells (Fig. 4B).
FIGURE 4.

Whole-cell currents measured in C6 glioma cells using a voltage step protocol. A step protocol with a 10-mV step increment from −100 to 100 mV from the holding potential (Vh = Vm) was applied. A, representative current traces from cells expressing homomeric Kir4.1 channels. B, representative current traces from cells expressing heteromeric Kir4.1-Kir5.1 channels. C, summary of the results within the range of −140 to 20 mV. D, summary of the results within the range of −140 to 20 mV. Error bars represent ±S.E.
One of the major functions of Kir4.1 channels is to take up extracellular potassium. In the brain, Kir4.1 channels in astrocytes have a prominent role in maintaining extracellular potassium homeostasis. In the kidney, potassium uptake by these channels is a key component of potassium recycling. To assess the effect of these variants in the KCNJ10 gene on the ability to take up potassium, each cell was clamped at their resting membrane potential, thus zero current, and then we measured currents induced by K+ steps (from 3 to 10 mm) at resting potential (44). K+-induced currents from cells expressing homomeric WTKir4.1 and Q212R channels were not significantly different, whereas currents from cells expressing L166Q, G83V, and G77R were significantly reduced (Fig. 5, A and C). Consistent with the findings of the whole-cell studies (Figs. 3 and 4), currents elicited by a step in [K+]o from 3 to 10 mm from tsA201 cells expressing A167V were reduced from WTKir4.1, whereas C6 glioma cells expressing A167V were no different from WTKir4.1. Channel function was rescued when L166Q was co-expressed with Kir5.1 in either tsA201 cells or C6 glioma cells, whereas there was no rescue of channel function for heteromeric A167V-Kir5.1 when expressed in tsA201 cells.
FIGURE 5.

Summary of the inward currents measured in response to a step from 3 to 10 mm extracellular K+ for tsA201 cells expressing homomeric Kir4. 1 channels (A), tsA201 cells expressing heteromeric Kir4.1-Kir5.1 channels (B), C6 glioma cells expressing homomeric Kir4.1 channels (C), and C6 glioma cells heteromeric Kir4.1-Kir5.1 channels (D). Data were analyzed using central tendency measures (medians), dispersion measures (standard deviations), and a k independent group one-way ANOVA test with Tukey-Kramer post hoc tests (p < 0.05) for tsA201 cells. For C6 glioma cells, data were analyzed by frequencies and a Kruskal-Wallis k independent group test. Results are expressed as mean ± S.E., and significant differences (p < 0.05) from mock- or Kir5.1 control- (*) or from WTKir4.1- or WTKir4.1-Kir5.1-transfected cells (¤) are shown. Vh = Vm. Error bars represent ±S.E.
One of the defining features of Kir channels is decreased conductance in the outward direction relative to the inward direction (inward rectification). Inward rectification of Kir4.1-containing channels is caused by voltage-dependent block of the channel pore by intracellular Mg2+ and polyamines such as SPM (45). We measured the SPM dependence of WT and mutated Kir4.1 and Kir4.1-Kir5.1 channels expressed in tsA201 cells. Figs. 6, A1 and B1, and 7, A1–C1, show representative excised patch recordings in the presence of varying [SPM]. Figs. 6, A2 and B2, and 7, A2–C2, show steady state current-voltage relationships obtained from such recordings, whereas Figs. 6, A3 and B3, and 7, A3–C3, summarize the Irel-voltage relationships utilized to obtain the fitted parameters summarized in Table 1.
FIGURE 6.

Intracellular SPM sensitivity experiments on tsA201 cells expressing homomeric Kir4.1 and Q212R mutant channels. Representative inside-out patch clamp recordings of currents recorded in response to voltage steps from −100 and +100 mV in the presence of 0, 1, 10, and 100 μm SPM or in extracellular solution at pH 5.5 (A1 and B1) are shown. A2 and B2, steady state current-voltage relationships from A1 and B1. A3 and B3, mean Irel-voltage relationships from WTKir4.1 (n = 7) and Q212R (n = 6) experiments as in A1 and B1.
FIGURE 7.

Intracellular SPM sensitivity experiments on tsA201 cells expressing heteromeric Kir4.1-Kir5.1, Q212R-Kir5.1, and L166Q-Kir5.1 mutant channels. A1, B1, and C1, representative inside-out patch clamp recordings of currents recorded in response to voltage steps from −100 and +100 mV in the presence of 0, 1, 10, and 100 μm SPM or in extracellular solution at pH 5.5. A2, B2, and C2, steady state current-voltage relationships from A1, B1, and C1. A3, B3, and C3, mean Irel-voltage relationships from heteromeric Kir4.1-Kir5.1 (n = 9), Q212R-Kir5.1 (n = 6) and L166Q-Kir5.1 (n = 7) experiments as in A1, B1, and C1.
TABLE 1.
V1/2 obtained from fitting currents of cells expressing homomeric Kir4.1 and heteromeric Kir4.1-Kir5.1 channels
Statistical probability was calculated using Student's t test for homomeric channels and one-way ANOVA followed by Tukey-Kramer post hoc test for heteromeric channels. Data are expressed as mean ± S.E.
|
V1/2 (mV) |
|||
|---|---|---|---|
| 1 μm SPM | 10 μm SPM | 100 μm SPM | |
| Kir4.1 | 12.3 ± 0.8 (n = 9) | −9.9 ± 0.8 (n = 9) | −25.1 ± 1.0 (n = 9) |
| Q212R | 76.6 ± 2.6 (n = 5)a | −4.0 ± 1.6 (n = 6)a | −24.0 ± 0.8 (n = 6) |
| Kir4.1-Kir5.1 | 38.7 ± 2.2 (n = 9) | 8.2 ± 1.0 (n = 8) | −6.7 ± 1.2 (n = 7) |
| Q212R-Kir5.1 | 92.5 ± 7.4 (n = 6)a | −3.7 ± 1.1 (n = 6)a | −22.1 ± 1.3 (n = 6)a |
| L166Q-Kir5.1 | 45.3 ± 0.6 (n = 6) | 24.0 ± 1.3 (n = 4)a | 5.1 ± 1.0 (n = 3)a |
a Significant difference from WT (either Kir4.1 or Kir4.1-Kir5.1) control (p < 0.05).
When excised patches expressing Kir4.1 are exposed to SPM-, Mg+-, and Na+-free solution, they lose their rectification properties, and the outward current becomes linear (37). This occurs within ∼1–3 min of excision after which time we began the SPM dose-response experiments. Kir4.1 channels are also strongly blocked by acidic intracellular pH (46). To confirm that the conductance measured was due to Kir channels, we consistently exposed our inside-out patches to SPM-free, pH 5.5 solution, and the mostly negligible remaining current was subtracted from the total current obtained for each concentration of SPM before plotting the steady state current-voltage relationships.
Irel-voltage relationships in SPM were fit by a Boltzmann function plus offset, and V1/2 values are summarized in Table 1 for WTKir4.1 and Q212R and in Table 2 for Kir4.1-Kir5.1, Q212R-Kir5.1, and L166Q-Kir5.1 at 1, 10, and 100 μm SPM. G83V-expressing cells were not examined because there was no appreciable current when this variant was expressed alone or together with Kir5.1. Similarly, the reduced whole-cell current observed for the L166Q variant when expressed alone made the curve fitting unreliable; therefore, we only provide the calculated parameters for heteromeric L166Q-Kir5.1 channels. In addition to the V1/2, the offset values were calculated and are summarized in Table 2. Because Q212R-containing channels exhibited a considerable slowing of the blocking rate in response to 1 μm SPM (Figs. 6, B2, and 7, B2), steady state block was difficult to achieve for this channel. Therefore, the offset for 1 μm SPM that was used for curve fitting of Q212R and Q212R-Kir5.1 channel parameters was estimated based on the offset obtained for 10 μm SPM. Most variants displayed incomplete block at high voltage as reported for WTKir4.1 channels due to SPM permeation through the channel pore (Table 2 and Refs. 37 and 47), although L166Q-Kir5.1 currents appear to show more complete block at 100 μm SPM (see “Discussion”).
TABLE 2.
Offset obtained from fitting currents of cells expressing homomeric Kir4.1 and heteromeric Kir4.1-Kir5.1 channels
Statistical probability was calculated using Student's t test for homomeric channels and one-way ANOVA followed by Tukey-Kramer post hoc test for heteromeric channels. Data are expressed as mean ± S.E.
| Offset (Irel) |
|||
|---|---|---|---|
| 1 μm SPM | 10 μm SPM | 100 μm SPM | |
| Kir4.1 | 0.15 ± 0.01 | 0.12 ± 0.01 | 0.10 ± 0.01 |
| Q212R | 0.18 ± 0.02a | 0.14 ± 0.02a | |
| Kir4.1-Kir5.1 | 0.22 ± 0.03 | 0.12 ± 0.02 | 0.05 ± 0.01 |
| Q212R-Kir5.1 | 0.15 ± 0.01 | 0.11 ± 0.01a | |
| L166Q-Kir5.1 | 0.24 ± 0.04 | 0.12 ± 0.03 | 0.00 ± 0.02a |
a Significant difference from WT (either Kir4.1 or Kir4.1-Kir5.1) control (p < 0.05).
Discussion
Here we have analyzed the effects of previously uncharacterized KCNJ10 SNPs and EAST/SeSAME-causing mutations found in humans on channel activity. Each SNP variant was predicted by PolyPhen analysis to be probably damaging. PolyPhen software makes prediction based on physical and evolutionary comparative considerations, and naïve Bayes classifier is used to calculate the deleterious effect of non-synonymous variants based on an eight sequence- and three structure-based predictive feature (35). Mutation L166Q (rs1130182) analyzed by SIFT PolyPhen-2, I-Mutant 2.0, PANTHER, and FASTSNP is predicted to be probably damaging to channel function and protein structure (35).
More importantly, each of these residues is located in a known functionally critical region of the Kir channel structure (Fig. 8). G83V is located in the first transmembrane domain of the Kir4.1 subunit, immediately behind the pore helix and selectivity filter. A switch from glycine to valine changes the hydropathy index from −0.4 to 4.2, which may affect protein insertion into the lipid bilayer but may render the channels non-functional in the membrane, potentially via effects on ion permeation or on gating, perhaps by interfering with the channel bending and straightening in TM1 as proposed for channel opening (36, 48). We found that cells expressing the G83V-Kir4.1 variant showed negligible macroscopic whole-cell recordings, and no current was elicited upon elevation of extracellular [K+] in the bath solution. This variant also failed to alter the membrane potential of the cells in which it was transfected. This variant behaved similarly to the EAST/SeSAME mutant G77R. We would predict a mild EAST/SeSAME-like syndrome in heterozygous carriers of the G83V variant (36) and a complete loss of function of Kir4.1 and Kir4.1-Kir5.1 channels in individuals who are homozygous for this variation, which would be expected to result in deficits similar to those seen in Kir4.1 knock-out animals (21) including small size, higher rate of mortality, motor coordination deficits, and ultimately death in early childhood. Kir4.1(−/−) mice survive to about 3 weeks of age (21).
FIGURE 8.

Proposed model of the Kir4.1 channel protein and location of Gln-212, Leu-166, and Gly-83 amino acid changes.
Cells expressing the Q212R variant of the Kir4.1 channel showed membrane potentials and overall conductance levels that were similar to WT, although mean membrane potential was slightly depolarized as compared with WTKir4.1 when expressed in tsA201 cells. This may have reflected the changes in SPM-dependent rectification that were present for the Q212R variant. This variant displayed an apparent reduction in SPM blocking affinity with SPM block at 1 μm displaying a shift to more positive values of membrane voltage and voltage of half-maximal block than the WT counterpart in both homomeric and heteromeric (Q212-Kir5.1) conformations (Table 1). At such low SPM concentrations, SPM block was markedly slowed (taking minutes to achieve steady state), and hence the blocking potency may be underestimated experimentally. We would suggest that this residue, which lines the cytoplasmic pore of the channel (Fig. 8), could be affecting the entry of SPM into the Kir4.1 inner cavity because extensive mutagenesis has shown that the introduction of positive charge to (or removal of negative charge from) the wall of the Kir cytoplasmic domain can provide a major energetic barrier to SPM entry (49, 50). This effect was no longer observed at 10 and 100 μm SPM probably because the driving force and concentration of SPM were considerably greater, making it possible to achieve steady state.
Adding to this interpretation, position 224 in Kir2.1 has been shown to contribute to rectification (51). In non-excitable cells such as glial cells where the voltage variation is not high, we suggest that this effect in kinetics of SPM block at 1 μm may not have any negative effect; i.e. it is a benign mutation. Although Kir4.1 is not expressed in most neurons, theoretically a comparable shift in the kinetics of SPM block in a Kir channel expressed in neurons could affect the neuronal firing pattern because the channel would not achieve block by intracellular SPM due to constant high voltage variations.
Cells expressing the L166Q variant (whether in homomeric conformation or as heteromeric channels together with Kir5.1) had a hyperpolarized membrane potential only slightly depolarized when compared with that of the WTKir4.1-expressing kidney-derived tsA201cells. tsA201 cells expressing the homomeric form of this variant displayed significant whole-cell current and were responsive to elevated extracellular K+ concentrations, but the magnitude of the current in both cases was substantially less than in wild type. Fully functional channel capabilities were obtained when L166Q was co-expressed together with the Kir5.1 subunit. In this situation, total whole-cell currents and response to elevated extracellular K+ were rescued. Homomeric L166Q-containing channels displayed decreased channel function when expressed in C6 glioma cells as compared with tsA201 cells, although there was significant recovery of function when heteromeric L166Q-Kir5.1 channels were expressed in glioma cells. Taken together, these data suggest that the Kir5.1 subunit hides the negative effect of the leucine substitution, thereby re-establishing channel function and/or expression.
In experiments studying SPM block of these channel variants, we could only assess the effect of SPM on heteromeric L166Q-Kir5.1 channels as the currents obtained from cells expressing homomeric L166Q channels were too small. However, heteromeric L166Q-Kir5.1 channels had V1/2 values similar to those of Kir4.1-Kir5.1 channels for 1 μm SPM. With increasing SPM concentrations and most notably with 100 μm SPM, heteromeric L166Q-Kir5.1 channels achieved a higher block, which could be explained if this residue helps to keep SPM in a stable conformation in the deep pore cavity and (somehow) reduces SPM permeation through the L166Q Kir channel. The L166Q variant is located in the second transmembrane domain at the “bundle crossing” of these domains in the so-called “hot spot” near positions where reported mutations are known to be causative of EAST/SeSAME syndrome, e.g. A167V mutation (52). Other mutants near this position have been shown to reduce protein surface expression and function (36). Mutations in this region of other inward rectifier channels are known to have drastic effects on channel gating, and thus we would suggest that this effect is most likely due to the modification from a highly hydrophobic leucine to the hydrophilic amino acid glutamine.
Surprisingly, expression of L166Q channels in glial cell-derived C6 glioma cells resulted in a different profile with the membrane potential substantially depolarized as compared with C6 glioma cells expressing WTKir4.1. Furthermore, the whole-cell currents and response to elevated extracellular K+ concentrations of C6 glioma cells expressing L166Q were similar to mock-transfected controls. This possibly could be due to reduced channel expression in the membrane because C6 glioma cells mistraffic native Kir4.1 channels to the nucleus (39). Even when expressed in C6 glioma cells, L166Q channel function was partially restored when co-expressed with Kir5.1.
Interestingly, when expressed in Xenopus oocytes (53) or three kidney-derived cell lines (monkey Cosm6 cells (36), human HEK293 (54), and human tsA201 cells), the EAST/SeSAME-causing mutation A167V displayed reduced inward current compared with its WTKir4.1 counterpart, but when this mutant was expressed in glial cell-derived C6 glioma cells, there was no loss of channel function. These data suggest that there are very important but often neglected underlying differences between cell lines in terms of Kir4.1 protein synthesis, stability, or expression at the surface.
Overall, our data suggest that these coding variants of KCNJ10 that have been identified in human subjects are likely to affect channel function and may cause pathological features in carrier individuals. We show that different mechanisms are likely to be responsible. The G83V variation renders the channel to be non-functional. Such an effect could lead to hypokalemia, metabolic alkalosis, deafness, neuronal hyperexcitability, and neuronal death especially in the homozygous state. In contrast, the L166Q variant reduced function, particularly when expressed in glia-derived cells, and this partial function may be insufficient to fulfill the CNS needs to buffer extracellular K+ under pathological conditions such as an ischemic stroke. Finally, the Q212R mutant did not reduce overall conductance in kidney cells but did demonstrate subtle effects on SPM sensitivity.
Author Contributions
M. P. M.-G. performed the experiments. M. P. M.-G., Y. V. K., G. M.-M., and M. J. E. analyzed the data. M. P. M.-G. and W. V.-C. made the mutant cDNA. C. G. N., A. Z.-S., and L. A. C. helped in the interpretation of the obtained results. S. N. S. and M. J. E. designed the experiments. M. P. M.-G. wrote the first version of the paper, and this was edited to the final submitted version by C. G. N., S. N. S., and M. J. E. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Sunjoo Lee for developing the three-dimensional channel model and Paola López Pieraldi and Natalia Skachkova for superior technical assistance.
This work was supported by National Institutes of Health Grants SC1GM088019, R01NS065201, G12MD007583, and R25GM110513 and Department of Education Title V Promoting Postbaccalaureate Opportunities for Hispanic Americans Grant P031M105050. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Kir
- inwardly rectifying potassium
- ANOVA
- analysis of variance
- EGFP
- enhanced green fluorescent protein
- Vm
- membrane potential
- V1/2
- voltage at half-maximal block
- PolyPhen
- Polymorphism Phenotyping.
References
- 1. Kucheryavykh Y. V., Kucheryavykh L. Y., Nichols C. G., Maldonado H. M., Baksi K., Reichenbach A., Skatchkov S. N., and Eaton M. J. (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281 [DOI] [PubMed] [Google Scholar]
- 2. Nichols C. G., and Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191 [DOI] [PubMed] [Google Scholar]
- 3. Bichet D., Haass F. A., and Jan L. Y. (2003) Merging functional studies with structures of inward-rectifier K+ channels. Nat. Rev. Neurosci. 4, 957–967 [DOI] [PubMed] [Google Scholar]
- 4. Djukic B., Casper K. B., Philpot B. D., Chin L. S., and McCarthy K. D. (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inyushin M., Kucheryavykh L. Y., Kucheryavykh Y. V., Nichols C. G., Buono R. J., Ferraro T. N., Skatchkov S. N., and Eaton M. J. (2010) Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia 51, 1707–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neusch C., Rozengurt N., Jacobs R. E., Lester H. A., and Kofuji P. (2001) Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 21, 5429–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen J., and Zhao H. B. (2014) The role of an inwardly rectifying K+ channel (Kir4.1) in the inner ear and hearing loss. Neuroscience 265, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishii M., Horio Y., Tada Y., Hibino H., Inanobe A., Ito M., Yamada M., Gotow T., Uchiyama Y., and Kurachi Y. (1997) Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: their regulation by insulin and laminin signals. J. Neurosci. 17, 7725–7735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kofuji P., Biedermann B., Siddharthan V., Raap M., Iandiev I., Milenkovic I., Thomzig A., Veh R. W., Bringmann A., and Reichenbach A. (2002) Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia 39, 292–303 [DOI] [PubMed] [Google Scholar]
- 10. Li L., Head V., and Timpe L. C. (2001) Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia 33, 57–71 [DOI] [PubMed] [Google Scholar]
- 11. Neusch C., Papadopoulos N., Müller M., Maletzki I., Winter S. M., Hirrlinger J., Handschuh M., Bähr M., Richter D. W., Kirchhoff F., and Hülsmann S. (2006) Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J. Neurophysiol. 95, 1843–1852 [DOI] [PubMed] [Google Scholar]
- 12. Olsen M. L., Higashimori H., Campbell S. L., Hablitz J. J., and Sontheimer H. (2006) Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia 53, 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poopalasundaram S., Knott C., Shamotienko O. G., Foran P. G., Dolly J. O., Ghiani C. A., Gallo V., and Wilkin G. P. (2000) Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia 30, 362–372 [DOI] [PubMed] [Google Scholar]
- 14. Wu J., Xu H., Shen W., and Jiang C. (2004) Expression and coexpression of CO2-sensitive Kir channels in brainstem neurons of rats. J. Membr. Biol. 197, 179–191 [DOI] [PubMed] [Google Scholar]
- 15. Heuser K., Eid T., Lauritzen F., Thoren A. E., Vindedal G. F., Taubøll E., Gjerstad L., Spencer D. D., Ottersen O. P., Nagelhus E. A., and de Lanerolle N. C. (2012) Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 71, 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hibino H., Fujita A., Iwai K., Yamada M., and Kurachi Y. (2004) Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 279, 44065–44073 [DOI] [PubMed] [Google Scholar]
- 17. Higashi K., Fujita A., Inanobe A., Tanemoto M., Doi K., Kubo T., and Kurachi Y. (2001) An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931 [DOI] [PubMed] [Google Scholar]
- 18. Olsen M. L., Khakh B. S., Skatchkov S. N., Zhou M., Lee C. J., and Rouach N. (2015) New insights on astrocyte ion channels: critical for homeostasis and neuron-glia signaling. J. Neurosci. 35, 13827–13835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Ambrosio R., Wenzel J., Schwartzkroin P. A., McKhann G. M. 2nd, and Janigro D. (1998) Functional specialization and topographic segregation of hippocampal astrocytes. J. Neurosci. 18, 4425–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bristol L. A., and Rothstein J. D. (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 39, 676–679 [DOI] [PubMed] [Google Scholar]
- 21. Kofuji P., Ceelen P., Zahs K. R., Surbeck L. W., Lester H. A., and Newman E. A. (2000) Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci. 20, 5733–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newman E. A., Frambach D. A., and Odette L. L. (1984) Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225, 1174–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tucker S. J., Imbrici P., Salvatore L., D'Adamo M. C., and Pessia M. (2000) pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J. Biol. Chem. 275, 16404–16407 [DOI] [PubMed] [Google Scholar]
- 24. Paulais M., Bloch-Faure M., Picard N., Jacques T., Ramakrishnan S. K., Keck M., Sohet F., Eladari D., Houillier P., Lourdel S., Teulon J., and Tucker S. J. (2011) Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 10361–10366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bockenhauer D., Feather S., Stanescu H. C., Bandulik S., Zdebik A. A., Reichold M., Tobin J., Lieberer E., Sterner C., Landoure G., Arora R., Sirimanna T., Thompson D., Cross J. H., van't Hoff W., Al Masri O., Tullus K., Yeung S., Anikster Y., Klootwijk E., Hubank M., Dillon M. J., Heitzmann D., Arcos-Burgos M., Knepper M. A., Dobbie A., Gahl W. A., Warth R., Sheridan E., and Kleta R. (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scholl U. I., Choi M., Liu T., Ramaekers V. T., Häusler M. G., Grimmer J., Tobe S. W., Farhi A., Nelson-Williams C., and Lifton R. P. (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raphemot R., Lonergan D. F., Nguyen T. T., Utley T., Lewis L. M., Kadakia R., Weaver C. D., Gogliotti R., Hopkins C., Lindsley C. W., and Denton J. S. (2011) Discovery, characterization, and structure-activity relationships of an inhibitor of inward rectifier potassium (Kir) channels with preference for Kir2.3, Kir3.x, and Kir7.1. Front. Pharmacol. 2, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenzen K. P., Heils A., Lorenz S., Hempelmann A., Höfels S., Lohoff F. W., Schmitz B., and Sander T. (2005) Supportive evidence for an allelic association of the human KCNJ10 potassium channel gene with idiopathic generalized epilepsy. Epilepsy Res. 63, 113–118 [DOI] [PubMed] [Google Scholar]
- 29. Buono R. J., Lohoff F. W., Sander T., Sperling M. R., O'Connor M. J., Dlugos D. J., Ryan S. G., Golden G. T., Zhao H., Scattergood T. M., Berrettini W. H., and Ferraro T. N. (2004) Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 58, 175–183 [DOI] [PubMed] [Google Scholar]
- 30. Pessia M., Tucker S. J., Lee K., Bond C. T., and Adelman J. P. (1996) Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987 [PMC free article] [PubMed] [Google Scholar]
- 31. Xu H., Cui N., Yang Z., Qu Z., and Jiang C. (2000) Modulation of kir4.1 and kir5.1 by hypercapnia and intracellular acidosis. J. Physiol. 524, 725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang Z., Xu H., Cui N., Qu Z., Chanchevalap S., Shen W., and Jiang C. (2000) Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1-Kir5.1 channels by CO2 and pH. J. Gen. Physiol. 116, 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pearson W. L., Dourado M., Schreiber M., Salkoff L., and Nichols C. G. (1999) Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver. J. Physiol. 514, 639–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishii M., Fujita A., Iwai K., Kusaka S., Higashi K., Inanobe A., Hibino H., and Kurachi Y. (2003) Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am. J. Physiol. Cell Physiol. 285, C260–C267 [DOI] [PubMed] [Google Scholar]
- 35. Phani N. M., Acharya S., Xavy S., Bhaskaranand N., Bhat M. K., Jain A., Rai P. S., and Satyamoorthy K. (2014) Genetic association of KCNJ10 rs1130183 with seizure susceptibility and computational analysis of deleterious non-synonymous SNPs of KCNJ10 gene. Gene 536, 247–253 [DOI] [PubMed] [Google Scholar]
- 36. Sala-Rabanal M., Kucheryavykh L. Y., Skatchkov S. N., Eaton M. J., and Nichols C. G. (2010) Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J. Biol. Chem. 285, 36040–36048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kucheryavykh Y. V., Pearson W. L., Kurata H. T., Eaton M. J., Skatchkov S. N., and Nichols C. G. (2007) Polyamine permeation and rectification of Kir4.1 channels. Channels 1, 172–178 [DOI] [PubMed] [Google Scholar]
- 38. Higashimori H., and Sontheimer H. (2007) Role of Kir4.1 channels in growth control of glia. Glia 55, 1668–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsen M. L., and Sontheimer H. (2004) Mislocalization of Kir channels in malignant glia. Glia 46, 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Casamassima M., D'Adamo M. C., Pessia M., and Tucker S. J. (2003) Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J. Biol. Chem. 278, 43533–43540 [DOI] [PubMed] [Google Scholar]
- 41. Konstas A. A., Korbmacher C., and Tucker S. J. (2003) Identification of domains that control the heteromeric assembly of Kir5.1/Kir4.0 potassium channels. Am. J. Physiol. Cell Physiol. 284, C910–C917 [DOI] [PubMed] [Google Scholar]
- 42. Pessia M., Imbrici P., D'Adamo M. C., Salvatore L., and Tucker S. J. (2001) Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J. Physiol. 532, 359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanemoto M., Kittaka N., Inanobe A., and Kurachi Y. (2000) In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiol. 525, 587–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skatchkov S. N., Krusek J., Reichenbach A., and Orkand R. K. (1999) Potassium buffering by Müller cells isolated from the center and periphery of the frog retina. Glia 27, 171–180 [PubMed] [Google Scholar]
- 45. Skatchkov S. N., Woodbury-Fariña M. A., and Eaton M. (2014) The role of glia in stress. Psychiatr. Clin. North Am. 37, 653–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Z., and Jiang C. (1999) Opposite effects of pH on open-state probability and single channel conductance of kir4.1 channels. J. Physiol. 520, 921–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kucheryavykh Y. V., Shuba Y. M., Antonov S. M., Inyushin M. Y., Cubano L., Pearson W. L., Kurata H., Reichenbach A., Veh R. W., Nichols C. G., Eaton M. J., and Skatchkov S. N. (2008) Complex rectification of Müller cell Kir currents. Glia 56, 775–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., and MacKinnon R. (2002) The open pore conformation of potassium channels. Nature 417, 523–526 [DOI] [PubMed] [Google Scholar]
- 49. Kurata H. T., Zhu E. A., and Nichols C. G. (2010) Locale and chemistry of spermine binding in the archetypal inward rectifier Kir2.1. J. Gen. Physiol. 135, 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kurata H. T., Cheng W. W., Arrabit C., Slesinger P. A., and Nichols C. G. (2007) The role of the cytoplasmic pore in inward rectification of Kir2.1 channels. J. Gen. Physiol. 130, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu H., Yang Z., Cui N., Chanchevalap S., Valesky W. W., and Jiang C. (2000) A single residue contributes to the difference between Kir4.1 and Kir1.1 channels in pH sensitivity, rectification and single channel conductance. J. Physiol. 528, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pattnaik B. R., Asuma M. P., Spott R., and Pillers D. A. (2012) Genetic defects in the hotspot of inwardly rectifying K+ (Kir) channels and their metabolic consequences: a review. Mol. Genet. Metab. 105, 64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parrock S., Hussain S., Issler N., Differ A. M., Lench N., Guarino S., Oosterveld M. J., Keijzer-Veen M., Brilstra E., van Wieringen H., Konijnenberg A. Y., Amin-Rasip S., Dumitriu S., Klootwijk E., Knoers N., et al. (2013) KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol. 123, 7–14 [DOI] [PubMed] [Google Scholar]
- 54. Tang X., Hang D., Sand A., and Kofuji P. (2010) Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem. Biophys. Res. Commun. 399, 537–541 [DOI] [PMC free article] [PubMed] [Google Scholar]