

Abstract
Background/Aims:
Hepatitis B virus (HBV) continues to be one of the most important viral pathogens in humans. Surface (S) protein is the major HBV antigen that mediates virus attachment and entry and determines the virus subtype. Mutations in S gene, particularly in the “a” determinant, can influence virus detection by ELISA and may generate escape mutants. Since no records have documented the S gene mutations in HBV strains circulating in Saudi Arabia, the current study was designed to study sequence variation of S gene in strains circulating in Saudi Arabia and its correlation with clinical and risk factors.
Patients and Methods:
A total of 123 HBV-infected patients were recruited for this study. Clinical and biochemical parameters, serological markers, and viral load were determined in all patients. The entire S gene sequence of samples with viral load exceeding 2000 IU/mL was retrieved and exploited in sequence and phylogenetic analysis.
Patients and Methods:
A total of 123 HBV-infected patients were recruited for this study. Clinical and biochemical parameters, serological markers, and viral load were determined in all patients. The entire S gene sequence of samples with viral load exceeding 2000 IU/mL was retrieved and exploited in sequence and phylogenetic analysis.
Results:
A total of 48 mutations (21 unique) were recorded in viral strains in Saudi Arabia, among which 24 (11 unique) changed their respective amino acids. Two amino acid changes were recorded in “a” determinant, including F130L and S135F with no evidence of the vaccine escape mutant G145R in any of the samples. No specific relationship was recognized between the mutation/amino acid change record of HBsAg in strains in Saudi Arabia and clinical or laboratory data. Phylogenetic analysis categorized HBV viral strains in Saudi Arabia as members of subgenotypes D1 and D3.
Conclusion:
The present report is the first that describes mutation analysis of HBsAg in strains in Saudi Arabia on both nucleotide and amino acid levels. Different substitutions, particularly in major hydrophilic region, may have a potential influence on disease diagnosis, vaccination strategy, and antiviral chemotherapy.
Keywords: Chronic hepatitis, hepatitis B virus, liver cirrhosis, phylogeny, sequence analysis, surface gene
Hepatitis B virus (HBV) infection is a serious public health problem worldwide. It is estimated that 400 million people are living with chronic hepatitis B. High prevalence of HBV infection in Saudi Arabia is well reported. In the late 1980s, approximately 7% of apparently healthy children were shown to be positive for hepatitis B surface antigen (HBsAg; i.e. HBV carriers).[1] Since the introduction of HBV vaccination in October of 1989, the prevalence of HBV in Saudi children has sharply declined to less than 0.2%.[2] In Saudi adult population, the prevalence of HBsAg is variable based on age, region, and other factors but is generally around 3%.[3] Because of the error-prone nature of the reverse transcriptase enzyme, HBV displays remarkable genetic diversity compared with other DNA viruses.[4] Eight major genotypes (A–H) and several subgenotypes have been identified worldwide.[5] The distribution of HBV genotypes varies in different geographical regions, whereas genotype D is the most prevalent in Saudi Arabia.[6,7]
HBV genome is made up of a condensed coding region that includes four overlapping genes termed X, C, P, and S in order. C gene codes for the core protein (HBcAg), Pgene for the viral polymerase, and S gene for the surface antigen. The function of the protein coded by X gene is not fully understood.[8] Among these genes, S gene is composed of a long open reading frame that contains three in-frame “start” (ATG) codons dividing the gene into three discrete fragments; pre-S1, pre-S2, and S. Because of the multiple start codons, polypeptides of three different sizes are produced, including Large (pre-S1 + pre-S2 + S), medium (pre-S2 + S), and small (S).[9] S protein is a key viral antigen that binds to cell receptors and mediates virus entry. It also encompasses an antigenic structure located between amino acids 99 and 169, which is termed “Major Hydrophilic Region (MHR).” The amino acid composition of this region principally decides the HBV subtype.[10] Furthermore, a distinct region locates within MHR, between residues 124 and 147, and named “a” determinant, constituting the major target for neutralizing antibodies.[11]
Since serological detection of HBsAg is the foremost approach used for diagnosis of HBV infection, mutations within MHR, particularly in “a” determinant, potentially affect the antigenicity of HBsAg and might lead to false-negative results.[4] The antigenic alterations may also help the virus to escape from the host immune system.[12] In the short frame of “a” determinant (24 amino acids),[4] the amino acid changes are diverse in different geographical regions, even in the same country.[13,14,15,16,17] It seems that almost all of the individual amino acid residues in “a” determinant have the potential to change. However, in a Spanish study, the genome fragment that encodes the amino acids 112–212 of HBsAg was amplified and sequenced in 272 sera sample collected from randomly selected HBV chronic carriers. The authors identified 17 amino acid residues that are well conserved in all cases.[18]
Currently, there is no data that describes the mutations and amino acid changes in S gene/protein of HBV strains circulating among Saudi population. The present study was designed to record the prevalent and unique mutations and amino acid changes in HBsAg of strains in Saudi Arabia and investigate their potential relationship with different clinical parameters and risk factors such as liver function tests, serological markers, viral load, and disease stage.
PATIENTS AND METHODS
Patients
Patients were sequentially recruited from the hepatology clinics at King Khalid University Hospital in Riyadh, Saudi Arabia, during the period from June 2012 to January 2013. Patients were included if they were HBsAg positive regardless of the liver disease stage. Patients were excluded if they had co-infection with any other virus. Clinical data including age, gender, duration of infection, clinical symptoms, and treatment regime were documented for all patients. The study design and experimental protocols were approved by the Institutional Review Board (IRB) of the College of Medicine at King Saud University (code E-12-711) and informed consent was obtained from all patients.
Liver function tests and serological markers
Biochemical liver testing including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), bilirubin (BIL), and albumin (ALB) were assessed in all clinical samples using Dimension RxL Max Clinical Chemistry System, DADE Behring (Deerfield, IL, USA). Serological markers (HBsAg, HBeAg, and anti-HBeAg) were evaluated in patients' samples using ARCHITECT system assays (Abbott laboratories, Wiesbaden, Germany) according to the manufacturer's instructions.
Quantitation of HBV DNA
The level of HBV DNA was determined in all samples using COBAS® Ampliprep/COBAS® TaqMan® HBV Test, version 2.0 (Roche, Mannheim, Germany), which is based on two major processes: I) preparation of specimen for isolation of HBV DNA; II) simultaneous polymerase chain reaction (PCR) amplification of target DNA sequence and detection of cleaved dual-labeled target-specific oligonucleotide probe. For sample preparation, plasma was separated from blood samples by centrifugation at 1500 rpm for 10 min and transferred to 1.5 mL screw-capped tubes. Approximately 1.05 mL of each plasma sample was placed into the S-input tube provided by COBAS® Ampliprep and DNA extraction was performed according to the manufacturer's instructions. For amplification/detection, processed specimens were added to the amplification mixture in k-tubes and transferred to COBAS® TaqMan® analyzer, in which the HBV DNA copy number was automatically determined as international unit (IU)/mL. The association between HBV viral load and clinical risk factors such as gender, disease category, and response to treatment was calculated and analyzed.
Sequencing of HBV surface gene
HBV DNA was extracted from serum/plasma samples using QIAamp DNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The complete sequence of HBV S gene was retrieved by amplification of two overlapping fragments that span the entire gene as described by Zhang et al.[19] The primer sets included the following: HBV-S1-F (5′-GCGTCGCAGAAGATCTCAAT-3′; positions 593-613 of HBV genome) and HBV-S1-R (5′-TTGAGAGAAGTCCACCACGAG-3′; positions 1667-1688) for the first fragment, and HBV-S2-F (5′-CTGCTGGTGGCTCCAGTT-3′; positions 1451-1469) and HBV-S2-R (5′-GCCTTGTAAGTTGGCGAG AA-3′; positions 2510-2530) for the second fragment. PCR was conducted in a total volume of 50 μL including 10 μL of DNA extract, 25 μL GoTaq® Long PCR Master Mix (Promega, Madison, WI), 1.5 mM Magnesium Chloride (Qiagen), 0.2 μM of each primer and nuclease free water. Amplification was carried out in GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA) using the following protocol: Initial denaturation at 95°C for 2 min, 35 cycles at 94°C for 30 sec, 58°C for 30 s and 65°C for 1 min, and final extension at 72°C for 10 min. PCR amplicons were separated in 1% agarose gel and purified using Illustra™ Gel Band Purification Kit (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's instructions. The purified products were sequenced on both strands with relevant sequence-specific primers at GATC Biotech (Cologne, Germany). DNA sequences were edited and assembled using Bioedit program, version 7.0 (Ibis Biosciences, Carlsbad, CA). Full-length S gene sequences were deposited in GenBank under the accession numbers KF018213 to KF018217.
Sequence and phylogenetic analysis
Multiple sequence alignment was conducted between S gene sequence of viral strains in Saudi Arabia and 30 different international strains that represent all recorded genotypes and subgenotypes, and available in GenBank. Alignments were performed using Clustal W algorithm of MegAlign program, Lasergene software, version 3.18 (DNAStar, Madison, WI) for divergence analysis, recognition of mutation sites and prediction of amino acid changes. Phylogenetic tree was constructed using the neighbor-joining method of Molecular Evolutionary Genetics Analyses (MEGA) software, version 5.1. The accuracy of tree topology was evaluated by bootstrapping of 1000 replicates. Only values exceeding 50% were indicated at the branch nodes. Unique Saudi HBV surface gene sequences were only included in tree construction.
Clinical classification of patients
Patients were classified according to standard clinical criteria based on international guidelines as follows:
(1) Inactive HBV carriers defined as HBsAg-positive and hepatitis B e antigen (HBeAg)-negative with persistently normal alanine aminotransferase (ALT) (less than or equal to laboratory defined upper limit of normal [ULN]) at enrollment and HBV DNA persistently <20,000 IU/mL (105 copies/mL). (2) Active HBV chronic hepatitis, either HBeAg-negative or HBeAg-positive, with elevated ALT levels (>ULN) and HBV DNA ≥20,000 IU/mL. (3) HBV cirrhosis, which was diagnosed based on liver biopsy or the presence of (a) at least two of the following: Platelet count <90,000/L, radiological (ultrasonography [US] or computed tomography [CT]) evidence of cirrhosis, or esophageal varices (demonstrated by endoscopy), and (b) at least two signs of liver dysfunction; albumin level <30 g/L, INR ≥1.5, or bilirubin level >35 µmol/L. All participants had abdominal ultrasound (US) screening. The US was performed and interpreted by trained radiographers according to a standardized protocol, and the records reviewed by the investigators. Cirrhosis was inferred based on the appearance of the liver surface, liver parenchymal texture, portal vein size, splenic size, and presence of ascites and varicose veins in the portal and perisplenic area.
RESULTS
One hundred and twenty-three patients were recruited in this study, 114 (92.7%) with chronic hepatitis and 9 (7.3%) with cirrhosis. Inactive carriers were not included due to the low viral DNA load. Eighty-four patients (68.3%) were males and 39 (31.7%) were females. The average age of patients was 40.7 ± 1.32 and 41.8 ± 1.99 years in males and females, respectively. Testing samples for serological markers has identified 100% and 28.5% of samples positive for HBsAg and HBeAg, respectively. As expected, all HBeAg-positive samples were negative for anti-HBeAg antibodies, whereas 66.7% of HBeAg-negative samples have generated distinct antibody response.
The majority of patients had low viral load (mostly below 104 IU/mL). The average HBV viral load was significantly higher in males (8.5 × 106 IU/mL) than in females (1.9 × 104 IU/mL); P = 0.028. The average viral load was relatively similar in patients with chronic hepatitis (1.8 × 107 IU/mL) and those with cirrhosis (1.3 × 107 IU/mL; P = 0.831). On the contrary, liver enzymes were significantly higher in cirrhotic than in chronic hepatitis patients; ALT (78.4 ± 11.89 vs. 49.92 ± 2.09 IU/L, P = 0.044; upper normal level 56 IU/L), AST (53.5 ± 14.8 vs 23.4 ± 1.1 IU/L, P = 0.076; upper normal level 40 IU/L), and ALP (137 ± 44.2 vs. 110.6 ± 4.2 IU/L, P = 0.085; upper normal level 120 IU/L). There were no significant differences in the level of bilirubin and albumin in both the groups.
Of the collected samples, only 20 (16.3%) samples possessed viral load that exceeded 2000 IU/mL. This load was identified as the cutoff value for generation of sequencing fragments in conventional PCR (data not shown). Among these samples, sequence analysis identified five unique S gene sequences represented by samples Riyadh 96/2012 and 108/2013 for chronic hepatitis and samples Riyadh 8/2012, 112/2013 and 121/2013 for cirrhosis. The genetic diversity among viral strains in Saudi Arabia was very limited with an overall homology of 97.1%–100% and 96.4%–100% for the level of nucleotides and amino acids, respectively.
Multiple sequence alignment with 30 strains, that were recovered on different geographical and temporal bases and represent the entire genotypic and subgenotypic range of HBV, identified 48 mutation sites (among which 21 are unique) in viral strains in Saudi Arabia. All strains that have been sequenced showed amino acid substitutions (no = 24) in S protein [Table 1]. These changes include an overall of 11 unique sites that locate in the three different fragments of S proteins; D16E, T40P, F56L, Q75H, L77R, Q107K, and W111Sc in pre-S1, S135F in pre-S2, and F183S, C239Y, and Y369H in S. No specific relationship was identified between the mutations/amino acid changes of HBsAg in viral strains in Saudi Arabia and different clinical data (eg, age, gender, disease category, response to treatment) or laboratory parameters (eg, serological markers, liver function indices, viral load). Phylogenetic analysis has enabled the plotting of viral strains in Saudi Arabia as members of genotype D. Riyadh strains 8/2012, 108/2013, 112/2013, and 121S/2013 belong to subgenotype D1, whereas Riyadh 96/2012 strain belong to subgenotype D3 [Figure 1].
Table 1.
A collective record of the mutations and amino acid changes in HBV surface gene/protein of viral strains in Saudi Arabia
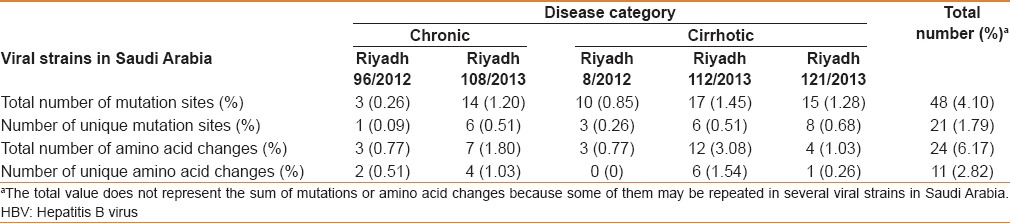
Figure 1.

Phylogenetic tree of representative HBV strains: S gene sequence of 5 Saudi and 30 international HBV strains was aligned using Megalign program of Lasergene software (DNASTAR) and tree was constructed via the neighborhood-joining method of MEGA 5.1 program. Confidence of the tree was verified by bootstrapping for 1000 times and only percentages above 50% were denoted at the branch nodes. Genotypes and subgenotypes are indicated at the right side of the tree. HBV strains were classified into 8 genotypes A–H, whereas genotype D in further divided into 5 subgenotypes D1–D5. All viral strains in Saudi Arabia are categorized within genotype D in both subgenotypes D1 and D3
In the context of “a” determinant, four mutation spots have been described in viral strains in Saudi Arabia including a single previously recorded site (T388C in Riyadh 112/2013 strain) and three unique sites (C381A, C404T and A414G in Riyadh 121/2013, Riyadh 108/2013 and Riyadh 121/2013 in order). Among these mutations, only two were capable to change their respective amino acids in S protein. The first; T388C, changed the amino acid at position 130 from phenylalanine (F) to leucine (L), which is a common amino acid change recorded previously in many countries such as China and Philippines. The second; C404T, induced a unique amino acid change at position 135 from serine (S) to phenylalanine (F). No record for the well-established “vaccine escape” mutant G145R was observed in any of the sequenced samples.
DISCUSSION
Patients infected with HBV have a variety of disease outcomes that principally depend on several factors including age of infection, level of virus replication, and host immune condition. Additionally, the error-prone nature of HBV reverse transcriptase leads to a significant increase in the mutation rate (10 times greater) as compared with other DNA viruses.[20] In particular, S gene mutations can trigger the development of immune escape variants that possess potential challenges to the diagnosis, prevention, and treatment of HBV infections. For instance, the changes in the conformational structure of HBsAg due to specific amino acid alterations may lead to low reactivity and false-negative results in diagnostic assays. Diagnostic failure increases the risk of occult infections and subsequently cause an upsurge in the risk of post-transfusion infection.[21] Avellon and Echevarria [18] have reported that amino acid substitutions in HBV variants are the cause of 12.5% failure in disease diagnosis, 6.6% invalid vaccination, and 9.2% escape from immunoglobulin therapy.
In the present study, a total of 48 nucleotide substitutions were identified in S gene of five Saudi variants [Table 1]. Among these, 24 mutations changed their respective amino acids with six amino acid substitutions located in MHR and two in the “a determinant” motif of HBsAg. Although we did not find an obvious correlation between specific mutation sites and status of HBV infection, we cannot rule out the potential existence of significant mutations that affect the antigenic properties of HBsAg in the circulating viral strains in Saudi Arabia. The patients were enrolled in this study based on HBsAg positivity on ELISA testing and therefore, the HBsAg mutations in our patients were not expected to preclude diagnosis. In addition, the majority of patients' samples (83.7%) were excluded from sequence analysis because of the low virus titer that did not allow amplification of sequencing fragments. In spite of the low number of subjects tested, it is noted that the mean number of mutations is higher in cirrhotic than in chronic patients (14 vs 8.5%). This observation was supported by similar higher levels of ALT, AST, and ALP in cirrhotic patients. Our data logically implies that S gene mutations may provide the way for virus persistence and for the development of more severe liver inflammation that leads to cirrhosis. A further larger sample study is needed to validate these results and conclusion.
The most stable and frequent amino acid change in HBsAg worldwide is G145R. This substitution is known to cause an immune-escape during vaccination and is recorded to be closely associated with genotype D viruses.[22,23] Although our phylogenetic analysis demonstrated that all viral strains in Saudi Arabia are members of genotype D [Figure 1], none of these strains showed the existence of G145R mutation. A comprehensive study that includes a wider range of viral strain isolates in Saudi Arabia, particularly those that escape from standard diagnostic and vaccination measures, will clearly monitor the potential existence of such amino acid substitution in HBsAg of Saudi patients. Conversely, a unique amino acid change F135S was identified in the 'a' determinant of Riyadh 108/2013 strain. Although nearly all the amino acids in this region have the potential to change, a recent Spanish study has pointed out to 17 amino acids including residue 135 to be highly conserved.[18] This observation indicates that the conserved nature of each amino acid in “a” determinant is far from being established and that new investigations embracing a wide array of viral strains circulating in many countries worldwide are essential to accurately identify the conserved amino acids in this region. Another mutation with significant outcome on the protein level involves the change of the amino acid tryptophan into a stop codon at residue 111 within MHR of Riyadh 96/2012 strain. This mutation results in the formation of a truncated S protein lacking the entire “a” determinant, which may preclude the HBsAg detection and induce the development of escape mutants.
The global distribution of HBV genotypes seems to have an obvious pattern. Genotype A is the most prevailing in Western Europe, genotypes B, C, I, and J in East Asia, genotype E in Africa, genotype F in America, and genotypes G and H in Europe and Japan, whereas genotype D is widespread globally.[24,25] People in the Middle East region including Saudi Arabia often carry genotype D viruses.[7] All viral strains identified in this study belong to genotype D and more specifically to subgenotypes D1 and D3 [Figure 1]. Despite the fact that the number of viral strains included in the phylogenetic analysis is limited, it may provide at least a preliminary view of the circulating genotypes and subgenotypes. Identification of HBV genotypes, and in some cases subgenotypes, is necessary to determine the clinical consequences of infection and the response to antiviral treatment. For instance, concomitant sustained biochemical remission and clearance of HBV DNA occur at a higher rate in genotype A but not in genotype D infections.[26] On the other hand, genotype A infections mostly lead to chronic hepatitis more than genotype D infections.[27]
It is important to mention that this study is the first that identifies the potential mutation sites in S gene of HBV variants circulating among Saudi population. However, certain unavoidable aspects related to the patients enrolled in the study have restricted surpassed outcomes. These aspects included: (1) The small percentage of patients with cirrhosis, (2) the low virus titer in the majority of clinical samples, which imposed the exclusion of these samples from sequence analysis, (3) the limited number of unique sequences from patients with chronic hepatitis (n = 2) despite the fact that this category of patients represents 92.7% of the study cohort. Indeed, the recruitment of more diverse and spacious spectrum of patients from Riyadh and other districts of Saudi Arabia will provide more evidence that correctly identifies the impact of S gene mutations on clinical outcomes of the disease.
CONCLUSION
The present study has provided important data on the sequence diversity of HBsAg in HBV strains circulating in Saudi Arabia. Different unique nucleotide and amino acid substitutions have been described, particularly in MHR, with potential impact on disease diagnosis and vaccination/treatment outcomes. The correlation between HBsAg mutation record and the clinical/laboratory parameters is not clear enough to justify a chance-base or casual-effect relationship, and thus needs further studying. All viral strains in Saudi Arabia were identified as members of genotype D (subgenotypes D1 and D3).
Footnotes
Source of Support: Nil
Conflict of Interest: The authors declare that they have no conflicts of interest.
REFERENCES
- 1.al-Faleh FZ, Ayoola EA, Arif M, Ramia S, al-Rashed R, al-Jeffry M, et al. Seroepidemiology of hepatitis B virus infection in Saudi Arabian children: A baseline survey for mass vaccination against hepatitis B. J Infect. 1992;24:197–206. doi: 10.1016/0163-4453(92)93006-c. [DOI] [PubMed] [Google Scholar]
- 2.Alfaleh F, Alshehri S, Alansari S, Aljeffri M, Almazrou Y, Shaffi A, et al. Long-term protection of hepatitis B vaccine 18 years after vaccination. J Infect. 2008;57:404–9. doi: 10.1016/j.jinf.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Abdo AA, Sanai FM, Al-Faleh FZ. Epidemiology of viral hepatitis in Saudi Arabia: Are we off the hook? Saudi J Gastroenterol. 2012;18:349–57. doi: 10.4103/1319-3767.103425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pourkarim MR, Sharifi Z, Soleimani A, Amini-Bavil-Olyaee S, Elsadek Fakhr A, Sijmons S, et al. Evolutionary analysis of HBV “S” antigen genetic diversity in Iranian blood donors: A nationwide study. J Med Virol. 2014;86:144–55. doi: 10.1002/jmv.23798. [DOI] [PubMed] [Google Scholar]
- 5.Guirgis BS, Abbas RO, Azzazy HM. Hepatitis B virus genotyping: Current methods and clinical implications. Int J Infect Dis. 2010;14:e941–53. doi: 10.1016/j.ijid.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Khan A, Al Balwi MA, Tanaka Y, Hajeer A, Sanai FM, Al Abdulkarim I, et al. Novel point mutations and mutational complexes in the enhancer II, core promoter and precore regions of hepatitis B virus genotype D1 associated with hepatocellular carcinoma in Saudi Arabia. Int J Cancer. 2013;133:2864–71. doi: 10.1002/ijc.28307. [DOI] [PubMed] [Google Scholar]
- 7.Abdo AA, Al-Jarallah BM, Sanai FM, Hersi AS, Al-Swat K, Azzam NA, et al. Hepatitis B genotypes: Relation to clinical outcome in patients with chronic hepatitis B in Saudi Arabia. World J Gastroenterol. 2006;12:7019–24. doi: 10.3748/wjg.v12.i43.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Miao X, Qi Z, Zeng W, Liang J, Liang Z. Hepatitis B virus X protein upregulates HSP90alpha expression via activation of c-Myc in human hepatocarcinoma cell line, HepG2. Virol J. 2010;7:45. doi: 10.1186/1743-422X-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007;13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purdy MA, Talekar G, Swenson P, Araujo A, Fields H. A new algorithm for deduction of hepatitis B surface antigen subtype determinants from the amino acid sequence. Intervirology. 2007;50:45–51. doi: 10.1159/000096312. [DOI] [PubMed] [Google Scholar]
- 11.Sayiner AA, Ozcan A, Sengonul A. Naturally occurring MHR variants in Turkish patients infected with hepatitis B virus. J Med Virol. 2008;80:405–10. doi: 10.1002/jmv.21104. [DOI] [PubMed] [Google Scholar]
- 12.Kim H, Lee SA, Kim DW, Lee SH, Kim BJ. Naturally occurring mutations in large surface genes related to occult infection of hepatitis B virus genotype C. PLoS One. 2013;8:e54486. doi: 10.1371/journal.pone.0054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khedive A, Sanei-Moghaddam I, Alavian SM, Saberfar E, Norouzi M, Judaki M, et al. Hepatitis B virus surface antigen (HBsAg) mutations are rare but clustered in immune epitopes in chronic carriers from Sistan-Balouchestan Province, Iran. Arch Iran Med. 2013;16:385–9. [PubMed] [Google Scholar]
- 14.Bian T, Yan H, Shen L, Wang F, Zhang S, Cao Y, et al. Change in hepatitis B virus large surface antigen variant prevalence 13 years after implementation of a universal vaccination program in China. J Virol. 2013;87:12196–206. doi: 10.1128/JVI.02127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu DM, Li XH, Mom V, Lu ZH, Liao XW, Han Y, et al. N-glycosylation mutations within hepatitis B virus surface major hydrophilic region contribute mostly to immune escape. J Hepatol. 2014;60:515–22. doi: 10.1016/j.jhep.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Baclig MO, Alvarez MR, Gopez-Cervantes J, Natividad FF. Unique surface gene variants of hepatitis B virus isolated from patients in the Philippines. J Med Virol. 2014;86:209–16. doi: 10.1002/jmv.23717. [DOI] [PubMed] [Google Scholar]
- 17.Sayan M, Buğdacı MS. HBV vaccine escape mutations in a chronic hepatitis B patient treated with nucleos (t) ide analogues. Mikrobiyol Bul. 2013;47:544–9. doi: 10.5578/mb.5442. [DOI] [PubMed] [Google Scholar]
- 18.Avellón A, Echevarria JM. Frequency of hepatitis B virus 'a' determinant variants in unselected Spanish chronic carriers. J Med Virol. 2006;78:24–36. doi: 10.1002/jmv.20516. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Wu G, Richards E, Jia S, Zeng C. Universal primers for HBV genome DNA amplification across subtypes: A case study for designing more effective viral primers. Virol J. 2007;4:92. doi: 10.1186/1743-422X-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Pan CQ, Zhang JX. Natural history and clinical consequences of hepatitis B virus infection. Int J Med Sci. 2005;2:36–40. doi: 10.7150/ijms.2.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allain JP, Candotti D. ISBT HBV Safety Collaborative Group. Hepatitis B virus in transfusion medicine: Still a problem? Biologicals. 2012;40:180–6. doi: 10.1016/j.biologicals.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 22.Carman WF. The clinical significance of surface antigen variants of hepatitis B virus. J Viral Hepat. 1997;(Suppl 1):11–20. doi: 10.1111/j.1365-2893.1997.tb00155.x. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz-Tachiquín ME, Valdez-Salazar HA, Juárez-Barreto V, Dehesa-Violante M, Torres J, Muñoz-Hernández O, et al. Molecular analysis of hepatitis B virus “a” determinant in asymptomatic and symptomatic Mexican carriers. Virol J. 2007;4:6. doi: 10.1186/1743-422X-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127:164–76. doi: 10.1016/j.virusres.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 25.Kramvis A, Kew MC. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J Viral Hepat. 2005;12:456–64. doi: 10.1111/j.1365-2893.2005.00624.x. [DOI] [PubMed] [Google Scholar]
- 26.Sánchez-Tapias JM, Costa J, Mas A, Bruguera M, Rodés J. Influence of hepatitis B virus genotype on the long-term outcome of chronic hepatitis B in western patients. Gastroenterology. 2002;123:1848–56. doi: 10.1053/gast.2002.37041. [DOI] [PubMed] [Google Scholar]
- 27.Mayerat C, Mantegani A, Frei PC. Does hepatitis B virus (HBV) genotype influence the clinical outcome of HBV infection? J Viral Hepat. 1999;6:299–304. doi: 10.1046/j.1365-2893.1999.00174.x. [DOI] [PubMed] [Google Scholar]