

Abstract
Bifidobacteria are members of the human gut microbiota, being numerically dominant in the colon of infants, while also being prevalent in the large intestine of adults. In this study, we determined and analyzed the pan-genome of Bifidobacterium adolescentis, which is one of many bacteria found in the human adult gut microbiota. In silico analysis of the genome sequences of eighteen B. adolescentis strains isolated from various environments, such as human milk, human feces and bovine rumen, revealed a high level of genetic variability, resulting in an open pan-genome. Compared to other bifidobacterial taxa such as Bifidobacterium bifidum and Bifidobacterium breve, the more extensive B. adolescentis pan-genome supports the hypothesis that the genetic arsenal of this taxon expanded so as to become more adaptable to the variable and changing ecological niche of the gut. These increased genetic capabilities are particularly evident for genes required for dietary glycan-breakdown.
Bifidobacteria are a common component of the microbiota of the human gastrointestinal tract (GIT) and, in particular, they are amongst the first bacterial colonizers of the intestine of neonates1,2,3. Bifidobacteria represent Gram positive, non-motile and non-spore-forming bacteria that belong to the phylum Actinobacteria4. Currently, genome sequences of one or more representatives of all 48 bifidobacterial species are available, which revealed a common saccharolytic genotype that is centred around a shared fermentative metabolic pathway particular to the Bifidobacterium genus and for this reason designated the bifid shunt. Furthermore, genome-based analyses revealed that bifidobacteria follow varying genetic strategies to adapt to their particular ecological niche(s), many of which relate to the mammalian GIT5,6. Among the bifidobacteria that colonize the human gut, strains of Bifidobacterium adolescentis appear to specifically colonize the gut of adult individuals and for this reason they represent a key bifidobacterial taxa of adult-associated bifidobacteria7. Recently, preliminary genetic and phenotypic characterization of the B. adolescentis species has been carried out, revealing their extensive capabilities to metabolize diet-derived glycans, in particular starch and starch-related/derived poly- and oligo-saccharides, such as amylopectin, pullulan, maltotriose and maltodextrin8.
Pan-genomes of two other human gut bifidobacterial species, Bifidobacterium bifidum9 and Bifidobacterium breve10, have previously been characterized. Notably, these analyses showed a closed pan-genome structure for both of these two species, revealing the presence of specific genetic strategies to establish and persist in the human gut, such as through the production of various types of pili11,12,13,14 or metabolic capabilities toward particular host-glycans9,15,16.
In contrast, the current genomic information available for B. adolescentis is rather limited, being represented by six genome sequences, of which three that are still fragmented in many contigs6,8,17. Thus, the genetic knowledge and understanding of the metabolic capabilities of this taxon is still in its infancy. In this study, we report on the genome sequences of twelve B. adolescentis strains that had been isolated from the adult gut or from rumen. Comparative genomic analyses of these sequences together with six other publicly available genome sequences of B. adolescentis species was performed. In addition, carbohydrate profiling of these strains was achieved involving various glycans including dietary- as well as host-derived glycans. Dietary changes are expected to impact on the ecological properties of the mammalian gut and thus on microbiota composition. The B. adolescentis taxon was shown to exhibit, through in silico and in vitro experiments, more extensive genetic flexibility and potential adaptive competitiveness to this highly variable ecological niche compared to other human bifidobacterial species.
Results
General genome features of B. adolescentis species
In order to evaluate the genetic content of the B. adolescentis species, we isolated eight strains in addition to four strains (obtained from international collections) belonging to this taxon from different ecological niches, including human feces, human milk, and bovine rumen (Table 1). The genome sequences of these strains were decoded through a Next Generation Sequencing (NGS) approach and subjected to comparative genomic analyses together with five other publicly available B. adolescentis genomes, corresponding to strains ATCC15703, 22L, BBMN23, 150 and L2-32. In addition, we included the chromosome sequence of B. stercoris JCM159185, since the latter microorganism has recently been re-classified as B. adolescentis JCM1591818. These twelve genome sequences of newly isolated and previously acquired B. adolescentis strains were sequenced to a coverage depth that ranged from 43.6-fold to 289.9-fold, which upon assembly resulted in thirty three to seven contigs, respectively (Table 1). Using the genome of the B. adolescentis type-strain ATCC15703 as a reference sequence, we were able to determine the presumed contig orientation and order for each genome draft. As outlined in Table 1, the number of predicted ORFs in each genome ranged from 1614 for B. adolescentis LMG11579 to 2215 for B. adolescentis 487B. In contrast to what has been observed for other bifidobacterial taxa for which three or more genomes are available, such as B. bifidum9, B. breve10, B. longum subsp. longum19 and Bifidobacterium animalis subsp. lactis20, the differences among the ORFomes from different B. adolescentis members were shown to be higher, suggesting that this bifidobacterial taxon exhibits a more extensive level of genetic diversity compared to that observed for other currently available bifidobacterial pan-genomes.
Table 1. Bifidobacterium adolescentis strain list.
| Strains | Source of isolation | Genome Size | No. of ORFs | Coverage | Contigs | Accession Number | Reference* |
|---|---|---|---|---|---|---|---|
| B. adolescentis 22L | Human milk | 2203222 | 1725 | / | / | NZ_CP007443.1 | Duranti et al.8 |
| B. adolescentis ATCC15703 | Intestine of adult | 2089645 | 1649 | / | / | AP009256.1 | NCBI database |
| B. adolescentis BBMN23 | Human feces | 2173720 | 1742 | / | / | NZ_CP010437.1 | NCBI database |
| B. adolescentis L2-32 | Intestine of adult | 2355465 | 1960 | / | / | NZ_AAXD00000000.2 | NCBI database |
| B. adolescentis 150 | Human feces | 2310538 | 1934 | / | / | NZ_LBHQ00000000.1 | Dyachkova et al.17 |
| B. adolescentis LMG18897 | Human feces | 2146787 | 1751 | 289x | 33 | LNKM00000000 | This study |
| B. adolescentis LMG11579 | Bovine rumen | 2061686 | 1614 | 290x | 7 | LNKL00000000 | This study |
| B. adolescentis LMG10733 | Intestine of adult | 2084232 | 1654 | 139x | 8 | LNKJ00000000 | This study |
| B. adolescentis LMG10734 | Intestine of adult | 2148629 | 1653 | 143x | 20 | LNKK00000000 | This study |
| B. adolescentis 42B | Human feces | 2212742 | 1855 | 120x | 15 | LNKB00000000 | This study |
| B. adolescentis 70B | Human feces | 2212129 | 1774 | 114x | 14 | LNKC00000000 | This study |
| B. adolescentis 487B | Human feces | 2597776 | 2215 | 76x | 11 | LNKD00000000 | This study |
| B. adolescentis 703B | Human feces | 2372808 | 2045 | 59x | 33 | LNKE00000000 | This study |
| B. adolescentis AD2-8 | Human feces | 2379716 | 2029 | 44x | 26 | LNKF00000000 | This study |
| B. adolescentis AL12-4 | Human feces | 2094907 | 1666 | 125x | 8 | LNKG00000000 | This study |
| B. adolescentis AL46-2 | Human feces | 2208560 | 1770 | 115x | 17 | LNKH00000000 | This study |
| B. adolescentis AL46-7 | Human feces | 2203026 | 1817 | 119x | 12 | LNKI00000000 | This study |
| B. stercoris/adolescentis JCM15918 | Adult feces | 2304613 | 1890 | / | / | JGZQ00000000 | Lugli et al.6 |
*The references were based on the decoding genomes project according to NCBI database.
Notably, functional classification of the B. adolescentis ORFome based on the eggNOG database21 was possible for 89.3% of the predicted ORFs. No function could be assigned to the remaining 10.7% and these identified ORFs were therefore annotated as hypothetical proteins. Furthermore, the eggNOG classification of the B. adolescentis genomes revealed that the majority of genes for which a function could be assigned are predicted to be involved in housekeeping functions, amino acid and carbohydrate metabolism, and associated transport activities (Fig. S1), similar to what had been observed for other members of the Bifidobacterium genus5,6,22. Our findings furthermore highlight that, being consistent with observations for other (sub)species of the genus Bifidobacterium, chromosomal sequences of the B. adolescentis species support the view that also this bifidobacterial taxon has adopted a saccharolytic life style.
Phylogenomic analyses of B. adolescentis species
The availability of the genome sequences of the type strains from all (currently described) 48 (sub)species of the Bifidobacterium genus allows a robust reconstruction of the phylogeny among members of this genus6. Thus, a comparative analysis was undertaken to identify orthologous genes among the bifidobacterial genome sequences of the strains belonging to the B. adolescentis species, as well as among the strains belonging to the B. adolescentis group2, which include Bifidobacterium dentium, Bifidobacterium pseudocatenulatum, Bifidobacterium catenulatum, Bifidobacterium moukalabense, Bifidobacterium kashiwanohense, Bifidobacterium ruminantium and B. stercoris/adolescentis. These analyses revealed the existence of 872 orthologous genes, which are shared among sequenced members of the B. adolescentis group. A concatenated protein sequence deduced from these orthologous genes of the B. adolescentis group was constructed in order to build a B. adolescentis group supertree (Fig. 1), which demonstrated that all 18 B. adolescentis strains are positioned on sub-branches of the same cluster. Interestingly, the B. adolescentis cluster includes a separate branch encompassing the genome of B. adolescentis LMG11579, which was isolated from a bovine rumen. This branch appears to be phylogenetically distinct from the other B. adolescentis strains that were isolated from various human samples, thus suggesting that B. adolescentis strain differences reflect the ecological origin of such investigated strains.
Figure 1. Genetic relationship within the B. adolescentis group.

The phylogenetic supertree was generated based on sequence similarities among 872 orthologous genes that are present in all analyzed strains.
Pan-genome and core-genome analysis of B. adolescentis species
The genome sequences of the 18 B. adolescentis strains were used to calculate the total gene repertoire encountered in this species. Thus, we evaluated the pan-genome and core-genome of the B. adolescentis taxon based on the clusters of orthologous genes (COGs) following a previously described method23. We identified a B. adolescentis pan-genome that consists of 4448 COGs (Fig. 2a). The pan-genome size, when plotted on a log-log scale as a function of the number of analyzed genomes, shows that the power trend line has not reached a plateau. Indeed, the number of genes discovered by sequential addition of genome sequences was reduced from 334-230 COGs in the first three genome additions, to 97-90 COGs in the final three additions. These findings indicate the occurrence of an open pan-genome within the B. adolescentis species, and suggest that full knowledge on the genetic diversity of this species has not yet been obtained23. This is in contrast with the determined pan-genomes of other human isolated bifidobacterial species, such as B. bifidum9, B. breve10 or B. longum subsp. longum19, which all displayed a closed structure (Fig. 2b–d). Moreover, the genome sequences of other bifidobacterial species showed that the ORF numbers of B. bifidum and B. breve genomes range from 1689 to 1835 and from 1748 to 1915, respectively10,24. In contrast, among representatives of the B. adolescentis species the size differences between individual ORFomes are larger, ranging from 1614 to 2215 (Table 1). The functional classification of the genes that do not belong to the B. adolescentis core-genome revealed that a large part of these genes have an unknown function, followed by genes involved in transport and metabolism of carbohydrates (Fig. S2). This indicates that genomes of the B. adolescentis species possess a higher level of genetic diversity as compared to those of other human-derived bifidobacteria. Furthermore, analysis of the set of predicted COGs allowed the identification of 1238 genes shared by all analysed B. adolescentis genomes, representing the core-genome of the B. adolescentis species (Fig. 2e). Examination of functional classification distribution among this core-genome, based on the eggNOG database21, suggests that a large proportion of these identified core genes are associated with housekeeping functions, and amino acid and carbohydrate metabolism, and corresponding transport (Fig. 2f).
Figure 2. Pan-genome and core-genome of the B. adolescentis species.

The pan-genome of B. adolescentis species (panel a) as well as B. longum, B. breve and B. bifidum species (panel b–d) and core-genome of B. adolescentis species (panel e) are represented as variation of their gene pool sizes upon sequential addition of genomes analysed. Panel (f) shows functional assignment of the B. adolescentis core genome based on the eggNOG database. Each letter stands for the following function: J, Translation, ribosomal structure and biogenesis, A, RNA processing and modification, K, Transcription, L, Replication, recombination and repair, D, Cell cycle control, cell division, chromosome partitioning, V, Defense mechanism, T, Signal transduction mechanisms, M, Cell wall/membrane/envelope biogenesis, O, Posttranslational modification, protein turnover, chaperones, C, Energy production and conversion, G, Carbohydrate transport and metabolism, E, Amino acid transport and metabolism, F, Nucleotide transport and metabolism, H, Coenzyme transport and metabolism, I, Lipid transport and metabolism, P, Inorganic ion transport and metabolism, Q, Secondary metabolites biosynthesis, transport and catabolism, R, General function prediction only, S, Function unknown.
Core-genome sequences of B. adolescentis species
Comparative genome analyses based on the 18 B. adolescentis genomes revealed the occurrence of shared orthologous genes as well as unique genes. As described above and employing the criteria outlined in the Materials and Methods, in silico analyses identified that 1238 genes are common among these strains, of which 369 were also common among other (sub)species of the genus Bifidobacterium. Furthermore, a varying number of unique genes, ranging from nine for B. adolescentis ATCC15703 to 251 for B. adolescentis 487B, were detected (Fig. 3a), representing the presumed Truly Unique Genes (TUG) of each B. adolescentis strain. Moreover, in silico analysis of the functional classification of TUGs failed to identify any correspondence between the predicted function of TUGs and the particular ecological origin of the B. adolescentis strain in question. Using an in silico approach to predict average nucleotide identity (ANI) values between B. adolescentis genomes25, we showed a highly syntenic genome structure among members of this species, with associated ANI values ranging from 97.77% to 99.93%, validating that B. adolescentis genomes belong to the same species6 (Table S1). Furthermore, this highly syntenic structure of B.adolescentis genome sequences was confirmed by comparative analyses using Dot-plot alignments (Fig. 3b). These analyses showed an apparent genome inversion in the chromosome of B. adolescentis 22L that was validated by PCR (data not shown). Furthermore, the analysis of this region revealed the presence of TUGs that appeared to have been acquired by horizontal gene transfer (HGT; see also below) as well as genes that are included in the core genome. These genes encode hypothetical proteins, putative phage proteins, several carbohydrate transport systems, enzymes that are involved in carbohydrate metabolism and the F1F0-ATPase cluster.
Figure 3. Comparative analysis of B. adolescentis genomes.

Panel (a) displays the Venn diagram representing the unique and orthologues genes between the 18 B. adolescentis genomes. Panel (b) shows the synteny plot alignment between the 18 B. adolescentis genomes.
Finally, the analysis of the core genome sequences of the B. adolescentis taxon allowed the identification of three core genes that are uniquely present in this species (i.e. absent in any of the other analysed bifidobacterial genomes), thus representing unique B. adolescentis core genes and encoding a Major Facilitator Superfamily (MFS) transporter, an acyltransferase and a hypothetical protein.
Mobilome of the chromosomes of B. adolescentis species
The mobilome represents the total number of genes that may have been acquired by HGT and its identification was performed using the software suite COLOMBO v3.8 implemented by SIGI-HMM26 and DarkHorse software27. The obtained results were merged, revealing that 13% of all ORFs were predicted to represent HGT-acquired genes. Furthermore, the observed percentage of ORFs putatively acquired by HGT events and predicted to be TUGs ranged from 3.09% in the genome of B. adolescentis ATCC15703 to over 40% in the chromosomes of B. adolescentis 70B and B. adolescentis AD2-8, respectively (Table S1). The predicted mobilome of B. adolescentis highlights a rich arsenal of insertion sequences (IS) and an abundance of prophage-like elements. Additional putative mobile elements identified in the B. adolescentis genomes are represented by CRISPR loci and a Restriction Modification (R/M) system. Other variable regions were shown to include an EPS gene cluster and finally a type IVa pilus biosynthesis gene cluster (Fig. S2). The type IVa pilus biosynthesis gene cluster was previously described in B. adolescentis 22L8 and its presence is also identified in B. adolescentis LMG10734 (locus tags LMG10734_1251-LMG10734_1263), B. adolescentis LMG18897 (locus tags LMG18897_1585-LMG18897_1597), B. adolescentis 15O (locus tags 15O_1475-15O_1485), B. adolescentis 42B (locus tags 42B_1313-42B_1323), B. adolescentis 70B (locus tags 70B_1301-70B_1311), B. adolescentis 487B (locus tags 487_1645-487_1655), B. adolescentis AD2-8 (locus tags AD28_1414-AD28_1424) and B. adolescentis AL46-2 (locus tags AL46-2_1300-AL46-2_1310). The function of this cluster is unknown, yet in other microbes it has been linked to motility, conjugation, adherence and DNA uptake28. Overall, these findings suggest that the genetic diversity found in B. adolescentis species is driven by various processes including conjugation, transformation, as well as phage-mediated transduction, which is consistent with previous publications29,30,31,32,33.
The glycobiome of B. adolescentis species
Thanks to their saccharolytic enzyme arsenal bifidobacteria are in a position to metabolize a wide range of carbohydrates (as a carbon and energy source), ranging from dietary- as well as host-derived glycans8,22,34,35. In order to extend our knowledge on the carbohydrate fermentation capabilities of B. adolescentis species, we performed an in silico prediction, in accordance to Carbohydrate-Active enZYmes Database (CAZy) database36, involving all 18 sequenced B. adolescentis genomes. This analysis revealed that the pan-genome of B. adolescentis contains genes encoding predicted carbohydrate-active enzymes, including 36 glycosyl hydrolase (GH) families, 12 glycosyl transferase (GT) families and four carbohydrate esterase (CE) families (Fig. S3). Interestingly, all 18 analyzed B. adolescentis genomes encompass genes predicted to encode enzymes involved in the uptake and utilization of plant-derived carbohydrates such as starch and starch-like oligo/polysaccharides, like maltodextrin, maltotriose, amylose, glycogen and pullulan37. These enzymes were previously identified to represent a distinctive characteristic of B. adolescentis 22L genome, supporting a superior growth performance of this strain compared to most other bifidobacteria when cultivated on starch or starch-like oligo/polysaccharides8. Consistent with this work, our analysis revealed a high abundance of genes belonging to the GH13 family (ranging from 14 in B. adolescentis 70B, B. adolescentis 703B, B. adolescentis ATCC15703 and B. adolescentis LMG10733, to 18 in B. adolescentis LMG10734 and B. adolescentis AL46-2), which encompasses enzymes with predicted α-glucosidase, amylase, pullulanase, and cyclomaltodextrinase activities. In silico analyses also highlighted the presence of a number of GHs (e.g., GH10, GH29, and GH59) that appear to be present in just a single strain of B. adolescentis species (Fig. S3a), and their annotation suggests that they may encode hydrolytic activities and thus carbohydrate-related metabolic abilities that are unique to such strains. We have further predicted the evolution of the GH arsenal of B. adolescentis pan-genome using BlastGraph38. This analysis allows the generation of a tree based on information regarding the presence or absence of COG families in each of the bifidobacterial strains assayed through the use of the dollo-parsimony algorithm39. Notably, this analysis predicted the early acquisition of GH43 and GH13 family members during the evolution of the B. adolescentis species (Fig. 4), which are crucial for the degradation, respectively, of plant polysaccharides36, and glycans with α-glucosidic linkages such as plant-derived starch or glycogen. Interestingly, B. adolescentis LMG11579, which is the only B. adolescentis strain so far isolated from bovine rumen, was predicted to neither have gained nor lost members of this GH13 COG. Furthermore, exploration of the functional evolution of this species showed widespread acquisition of COGs classified as GH23 and GH25 at multiple nodes. These GH families encode lysozymes that are often carried by phage genomes and expressed during the lytic phase, thus their acquisition during the evolution of B. adolescentis species may be linked to frequent interactions with phages populations (http://www.cazy.org/Glycoside-Hydrolases.html). In order to validate the above mentioned genomic-based analyses, we carried out growth experiments of B. adolescentis strains on 33 carbohydrates including both plant- and host-derived glycans, as unique carbon sources (Fig. 5). As displayed in Fig. 5 all tested strains are able to ferment a common set of sugars, such as galactose, glucose, galactooligosaccharides (GOS), lactose, lactulose, maltodextrin, maltose, maltotriose, melibiose, and raffinose. In contrast, fermentation capabilities for other sugars including fructooligosaccharides (FOS), fructose, glycogen, mannitol, pullulan, ribose, sorbitol, trehalose, turanose and xylose were shown to be variable among the strains tested. Furthermore, in contrast to other B. adolescentis strains, B. adolescentis 703B and B. adolescentis JCM15918 strains did not exhibit any appreciable growth on starch (Fig. 5).
Figure 4. Reconstruction of gene gain and loss events among the analyzed B. adolescentis strains.

A tree was constructed using information related to the presence or absence of COGs for the whole B. adolescentis pan-genome. Each node is marked by a pie diagram showing the acquired COGs (in red) and the COGs derived from the previous node (in green). Furthermore, the number of members of GH families that had been acquired is indicated next to each diagram.
Figure 5. Evaluation of carbohydrate utilization by B. adolescentis strains.
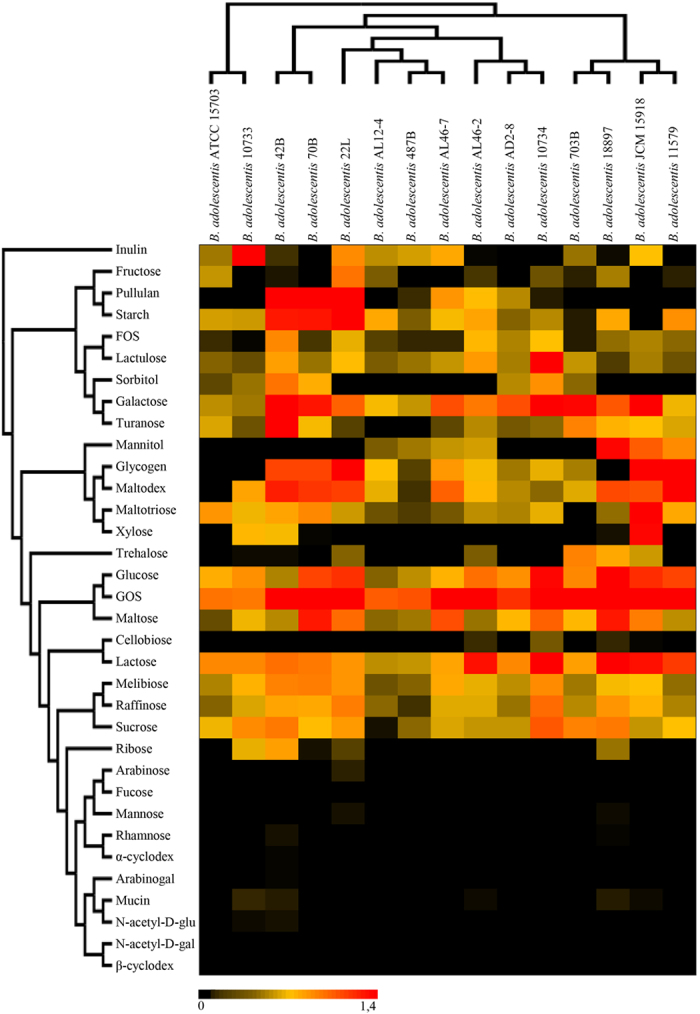
The heat map shows the growth performance of B. adolescentis strains on different carbon sources. Cultures were grown in biologically independent triplicates. The different shading represents the optical density reached by the various culture.
Notably, none of the B. adolescentis strains assayed here was shown to be capable of utilizing mucin, N-acetyl-D-galactosamine, N-acetyl-D-glucosamine or fucose, which indicates that the tested B. adolescentis strains possess limited metabolic capabilities to with regards to (monosaccharide components of) host-derived glycans (Fig. 5). Such metabolic features are commonly found among bifidobacterial taxa associated with the early stages of life in mammals such as B. bifidum, B. longum subsp. infantis and B. breve9,15,16,40,41. It is not unexpected that these metabolic characteristics are absent and/or have been lost by a bifidobacterial species that is typically found in the adult-colon such as B. adolescentis. This is also further supported by the absence in the B. adolescentis pan-genome of genes encoding sialidases, sialic acid metabolism, fucosidases, the relative paucity of beta-galactosidase-encoding genes and the absence of homologues of the LNB/GNB utilization cluster. This suggests that this species has specialized itself towards the utilization of plant-derived glycans, in particular (resistant) starch and starch-like polysaccharides, that are normally present in high abundance in the diet of adults.
Discussion
The increasing number of publicly available bifidobacterial genomes offers the possibility to understand the evolution and the genomic diversity within this genus5,6. In this study, we analyzed the genomic diversity of 12 different B. adolescentis strains combined with six additional, publicly available B. adolescentis genomes. These results allowed the identification of an open pan-genome of the B. adolescentis species, which is in contrast to what has been observed for other human gut bifidobacterial species such as B. bifidum, B. breve, B. longum subsp. longum and B. animalis subsp. lactis9,10,19,20. This indicates that the B. adolescentis taxon exhibits greater genetic diversity compared to other human gut bifidobacterial (sub)species. Furthermore, the pan-genome of the B. adolescentis species is enriched in genes that are predicted to be involved in the metabolism of dietary, plant-derived glycans, in particular starch and starch-like oligo- and poly-saccharides, except in the case of two strains, B. adolescentis 703B and B. adolescentis JCM15918. In contrast, the B. adolescentis genomes do not appear to encode genes involved in the metabolism of host-derived glycans such as mucin and human milk oligosaccharides. These results supports the notion that the (human-associated) B. adolescentis species has evolved towards an ecological niche where plant-polysaccharides are present in high abundance, such as the large intestine of adult human beings. However, caution should be taken when drawing inferences based in silico predictions and in vitro experiments. Thus, future in vivo analyses performed in murine models or clinical trials will be needed in order to validate the results described in this study.
Methods
Bacterial strains, growth conditions and chromosomal DNA extraction
The strains used in this study are listed in Table 1. B. adolescentis cultures were incubated in an anaerobic atmosphere [2.99% (vol/vol) H2, 17.01% (vol/vol) CO2, and 80% (vol/vol) N2] in a chamber (Concept 400, Ruskin) in de Man-Rogosa-Sharpe (MRS) (Scharlau Chemie) supplemented with 0.05% (wt/vol) L-cysteine hydrochloride and incubated at 37 °C for 16 h. Bacterial DNA was extracted as described previously42 and subjected to further phenol/chloroform purification using a previously described protocol43.
Genome sequencing and data assembly and bioinformatics analyses
All genomes used for this study were determined by GenProbio srl (Parma, Italy) using the MiSeq Illumina (Illumina, USA). Genomic libraries were constructed employing the TruSeq DNA PCR-Free LT Kit (Illumina) and using 2.5 μg of genomic DNA, which was fragmented with a Bioruptor NGS ultrasonicator (Diagenode, USA) followed by size evaluation using Tape Station 2200 (Agilent Technologies). Library samples were loaded into a Flow Cell V3 600 cycles (Illumina) according to the technical support guide, and generated reads were depleted of adapter sequences, quality filtered and assembled through the MEGAnnotator pipeline44. The overall generated sequencing output per genome ranged from 78 Mb to 515 Mb, approximately corresponding to a 43.6- to 289.9-fold genomic coverage, respectively.
Protein-encoding open reading frames (ORFs) were predicted using a combination of the methods Prodigal45 and BLASTX46. Results of the two gene finder programs were then automatically annotated on the basis of BLASTP47 analysis using B. adolescentis ATCC15703 as the reference genome (NCBI Accession Number: AP009256). Functional assignment was performed and manually edited based on similarity searches against the non-redundant protein database provided by the National Centre for Biotechnology Information. Artemis48 was employed to inspect the results of the ORF finding efforts and associated BLASTP results, and used for manual editing in order to check, or if necessary, redefine the start of every predicted coding region, or to remove or add coding regions. Furthermore, the revised gene-protein set was searched against the Swiss-Prot (http://www.expasy.ch/sprot//TrEMBL), PRIAM (http://priam.prabi.fr/), protein family (Pfam; http://pfam.sanger.ac.uk/), TIGRFAMs (http://www.jcvi.org/cms/research/projects/tigrfams/overview/), Interpro (INTERPROSCAN; http://www.ebi.ac.uk/Tools/InterProScan/), Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/), and COG (http://www.ncbi.nlm.nih.gov/COG/) databases, in addition to BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Functional assignments were defined by manual processing of the combined results. Manual corrections to automated functional assignments were completed on an individual gene-by-gene basis as needed.
Ribosomal RNA genes were detected on the basis of BLASTN searches and annotated manually. Transfer RNA genes were identified using tRNAscan-SE49. Restriction/modification (R/M) systems were searched on the basis of the REBASE database50, Carbohydrate-active enzymes were identified based on similarity to the carbohydrate-active enzyme (CAZy) database entries36, transporter classification was performed according to the TC-DB scheme51 and Enzyme Commission (EC)/Gene Onthology (GO) annotation was assigned using annot8r52. Variances in GC content were profiled by the DNA segmentation algorithm hosted at http://tubic.tju.edu.cn/GC-Profile/53, atypical codon usage regions were mapped using the factorial correspondence analysis through the assistance of the GCUA software54.
Comparative genomics
Each predicted proteome of a given B. adolescentis strain was searched for orthologues against the total proteome of B. adolescentis species, where orthology between two proteins was defined as the best bidirectional FASTA hits55. Identification of orthologues, paralogues, and unique genes was performed following a preliminary step involving the comparison of each protein against all other proteins using BLAST analysis47 (cutoff: E value of <1 × 10−4 and 30% identity over at least 80% of both protein sequences), and the resulting output was then clustered into protein families using MCL (graph theory-based Markov clustering algorithm)56. Following this approach, unique protein families encoded by the analyzed B. adolescentis genomes were classified. Protein families shared between all genomes, named core gene families, were defined by selecting the families that contained at least one single protein member for each genome.
Each set of orthologous proteins was aligned using CLUSTAL_W57, and phylogenetic trees were constructed using the maximum-likelihood in PhyML58. The supertree was built using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Pan-genome calculation
For all genomes used in this study, a pan-genome calculation was performed using PGAP59. The ORF content of each genome was organized in functional gene clusters using the gene family (GF) method. A pan-genome profile and a core genome profile were built using all possible BLAST combinations for each genome being sequentially added. Finally, using the pan-genome profile of shared orthologues between the B. adolescentis genomes, a pan-genome tree was constructed. This tree was visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Prediction of gene acquisition and loss
Prediction and tree visualization of glycosyl hydrolase-encoding genes that had either been acquired or lost was performed with BlastGraph38. Data from BLASTP46 comparisons of all pan-genome-deduced proteins to each other were used as the input, where the clustering cut-off value was set at 50% identity over at least 50% of both protein sequences.
Carbohydrate growth assays
Fifteen B. adolescentis strains were grown on semi-synthetic MRS medium supplemented with 0.5% (wt/vol) of a particular sugar and optical densities (OD at 600 nm) were recorded using a plate reader (BioTek, Winooski, VT, USA). The plate reader was run in discontinuous mode, with absorbance readings performed in 30 minutes intervals for 48 hours, where each reading was preceded by 30 s shaking at medium speed. Cultures were grown in biologically independent triplicates and the resulting growth data were expressed as the mean of these replicates. Carbohydrates were purchased from Sigma and Carbosynth (Berkshire, UK).
Nucleotide sequence accession numbers
The sequences reported in this study have been deposited in the GenBank database (Accession Number SAMN04231308, SAMN04231309, SAMN04231310, SAMN04231311, SAMN04231312, SAMN04231313, SAMN04231314, SAMN04231315, SAMN04231316, SAMN04231317, SAMN04231318 and SAMN04231319).
Additional Information
How to cite this article: Duranti, S. et al. Evaluation of genetic diversity among strains of the human gut commensal Bifidobacterium adolescentis. Sci. Rep. 6, 23971; doi: 10.1038/srep23971 (2016).
Supplementary Material
Acknowledgments
We thank GenProbio srl for financial support of the Laboratory of Probiogenomics. This work was financially supported by a PostDoc fellowship (Fondazione Caritro) to SD. DvS is member of The APC Microbiome Institute funded by Science Foundation Ireland (SFI), through the Irish Government’s National Development Plan (Grant number SFI/12/RC/2273).
References
- Turroni F. et al. Diversity of bifidobacteria within the infant gut microbiota. Plos One 7, e36957, 10.1371/journal.pone.0036957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F. et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl Environ Microbiol 75, 1534–1545, 10.1128/AEM.02216-08 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M. et al. From bacterial genome to functionality; case bifidobacteria. Int J Food Microbiol 120, 2–12, 10.1016/j.ijfoodmicro.2007.06.011 (2007). [DOI] [PubMed] [Google Scholar]
- Ventura M. et al. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71, 495–548, 10.1128/MMBR.00005-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C. et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl Environ Microbiol 80, 6290–6302, 10.1128/AEM.02308-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli G. A. et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl Environ Microbiol 80, 6383–6394, 10.1128/AEM.02004-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F., van Sinderen D. & Ventura M. Genomics and ecological overview of the genus Bifidobacterium. Int J Food Microbiol 149, 37–44, 10.1016/j.ijfoodmicro.2010.12.010 (2011). [DOI] [PubMed] [Google Scholar]
- Duranti S. et al. Genomic characterization and transcriptional studies of the starch-utilizing strain Bifidobacterium adolescentis 22L. Appl Environ Microbiol 80, 6080–6090, 10.1128/AEM.01993-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duranti S. et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ Microbiol 17, 2515–2531, 10.1111/1462-2920.12743 (2015). [DOI] [PubMed] [Google Scholar]
- Bottacini F. et al. Comparative genomics of the Bifidobacterium breve taxon. BMC genomics 15, 170, 10.1186/1471-2164-15-170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F. et al. Role of sortase-dependent pili of Bifidobacterium bifidum PRL2010 in modulating bacterium-host interactions. Proc Natl Acad Sci USA 110, 11151–11156, 10.1073/pnas.1303897110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F. et al. Expression of sortase-dependent pili of Bifidobacterium bifidum PRL2010 in response to environmental gut conditions. FEMS Microbiol Lett 357, 23–33, 10.1111/1574-6968.12509 (2014). [DOI] [PubMed] [Google Scholar]
- O’Connell Motherway M. et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc Natl Acad Sci USA 108, 11217–11222, 10.1073/pnas.1105380108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroni E. et al. Genetic analysis and morphological identification of pilus-like structures in members of the genus Bifidobacterium. Microb Cell Fact 10 Suppl 1, S16, 10.1186/1475-2859-10-S1-S16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F. et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci USA 107, 19514–19519, 10.1073/pnas.1011100107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni F., Milani C., van Sinderen D. & Ventura M. Genetic strategies for mucin metabolism in Bifidobacterium bifidum PRL2010: an example of possible human-microbe co-evolution. Gut microbes 2, 183–189 (2011). [DOI] [PubMed] [Google Scholar]
- Dyachkova M. S. et al. Draft Genome Sequences of Bifidobacterium angulatum GT102 and Bifidobacterium adolescentis 150: Focusing on the Genes Potentially Involved in the Gut-Brain Axis. Genome Announc 3, 10.1128/genomeA.00709-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killer J., Sedlacek I., Rada V., Havlik J. & Kopecny J. Reclassification of Bifidobacterium stercoris Kim et al. 2010 as a later heterotypic synonym of Bifidobacterium adolescentis. Int J Syst Evol Micr 63, 4350–4353, 10.1099/ijs.0.054957-0 (2013). [DOI] [PubMed] [Google Scholar]
- O’Callaghan A., Bottacini F., O’Connell Motherway M. & van Sinderen D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction-modification systems. BMC genomics 16, 832, 10.1186/s12864-015-1968-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C. et al. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl Environ Microbiol 79, 4304–4315, 10.1128/AEM.00984-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S. et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acid Res 42, D231–D239, 10.1093/nar/gkt1253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C. et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci Rep. 5, 15782, 10.1038/srep15782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial ‘pan-genome’ (vol 102, pg 13950, 2005). Proc Natl Acad Sci USA 102, 16530–16530, 10.1073/pnas.0508532102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duranti S. et al. Insights from genomes of representatives of the human gut commensal Bifidobacterium bifidum. Environ Microbiol 17, 2515–2531, 10.1111/1462-2920.12743 (2015). [DOI] [PubMed] [Google Scholar]
- Richter M. & Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106, 19126–19131, 10.1073/pnas.0906412106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waack S. et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC bioinformatics 7, Artn 142 10.1186/1471-2105-7-142 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podell S. & Gaasterland T. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol 8, Artn R16 10.1186/Gb-2007-8-2-R16 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam S., Chen Z., Roos D. S. & Pohlschroder M. Identification of surprisingly diverse type IV pili, across a broad range of gram-positive bacteria. Plos One 6, e28919, 10.1371/journal.pone.0028919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottacini F. et al. Discovery of a conjugative megaplasmid in Bifidobacterium breve. Appl Environ Microbiol 81, 166–176, 10.1128/AEM.02871-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez W. & O’Sullivan D. J. Developing an efficient and reproducible conjugation-based gene transfer system for bifidobacteria. Microbiol-Sgm 159, 328–338, 10.1099/mic.0.061408-0 (2013). [DOI] [PubMed] [Google Scholar]
- Ventura M. et al. Prophage-like elements in bifidobacteria: insights from genomics, transcription, integration, distribution, and phylogenetic analysis. Appl Environ Microbiol 71, 8692–8705, 10.1128/AEM.71.12.8692-8705.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M. et al. Comparative analyses of prophage-like elements present in bifidobacterial genomes. Appl Environ Microbiol 75, 6929–6936, 10.1128/AEM.01112-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli G. A. et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ Microbiol 10.1111/1462-2920.13154 (2015). [DOI] [PubMed] [Google Scholar]
- Turroni F. et al. Analysis of predicted carbohydrate transport systems encoded by Bifidobacterium bifidum PRL2010. Appl Environ Microbiol 78, 5002–5012, 10.1128/AEM.00629-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokusaeva K., Fitzgerald G. F. & van Sinderen D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr 6, 285–306, 10.1007/s12263-010-0206-6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M. & Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acid Res 42, D490–495, 10.1093/nar/gkt1178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maarel M. J., van der Veen B., Uitdehaag J. C., Leemhuis H. & Dijkhuizen L. Properties and applications of starch-converting enzymes of the alpha-amylase family. J Biotechnol 94, 137–155 (2002). [DOI] [PubMed] [Google Scholar]
- Ye Y. B., Wei B., Wen L. & Rayner S. BlastGraph: a comparative genomics tool based on BLAST and graph algorithms. Bioinformatics 29, 3222–3224, 10.1093/bioinformatics/btt553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris J. S. Phylogenetic Analysis Under Dollo’s Law. Syst Zool 26, 77–78 (1977). [Google Scholar]
- Sela D. A. et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci USA 105, 18964–18969, 10.1073/pnas.0809584105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Moyano S. et al. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of Bifidobacterium breve. Appl Environ Microbiol 79, 6040–6049, 10.1128/AEM.01843-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M., Reniero R. & Zink R. Specific identification and targeted characterization of Bifidobacterium lactis from different environmental isolates by a combined multiplex-PCR approach. Appl Environ Microbiol 67, 2760–2765, 10.1128/AEM.67.6.2760-2765.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. & David W. Russell. Molecular cloning a laboratory manual. (Cold Spring Harbor Laboratory Press, 2001). [Google Scholar]
- Lugli G. A., Milani C., Mancabelli L., van Sinderen D. & Ventura M. MEGAnnotator: a user-friendly pipeline for microbial genomes assembly and annotation. FEMS Microbial Lett. 10.1093/femsle/fnw049 (2016). [DOI] [PubMed] [Google Scholar]
- Hyatt D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC bioinformatics 11, 119, 10.1186/1471-2105-11-119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish W. & States, D. J. Identification of protein coding regions by database similarity search. Nat Genet 3, 266–272, 10.1038/ng0393-266 (1993). [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, 10.1016/S0022-2836(05)80360-2 (1990). [DOI] [PubMed] [Google Scholar]
- Rutherford K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000). [DOI] [PubMed] [Google Scholar]
- Lowe T. M. & Eddy S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acid Res 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. J., Vincze T., Posfai J. & Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acid Res 43, D298–299, 10.1093/nar/gku1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch W. & Saier M. H. The Transporter Classification (TC) system, 2002. Crit Rev Biochem Mol 37, 287–337, 10.1080/10409230290771528 (2002). [DOI] [PubMed] [Google Scholar]
- Schmid R. & Blaxter M. L. annot8r: GO, EC and KEGG annotation of EST datasets. BMC bioinformatics 9, Artn 180 10.1186/1471-2105-9-180 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F. & Zhang C. T. GC-Profile: a web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acid Res 34, W686–W691, 10.1093/nar/gkl040 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney J. O. G. C. U. A.: General codon usage analysis. Bioinformatics 14, 372–373, 10.1093/bioinformatics/14.4.372 (1998). [DOI] [PubMed] [Google Scholar]
- Pearson W. R. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol 132, 185–219 (2000). [DOI] [PubMed] [Google Scholar]
- van Dongen S. Graph clustering by flow simulation. PhD thesis, University of Utrecht, The Netherlands (2000).
- Thompson J. D., Gibson T. J. & Higgins D. G. Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics Chapter 2, Unit 2 3, 10.1002/0471250953.bi0203s00 (2002). [DOI] [PubMed] [Google Scholar]
- Guindon S. & Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- Zhao Y. et al. PGAP: pan-genomes analysis pipeline. Bioinformatics 28, 416–418, 10.1093/bioinformatics/btr655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.