

Abstract
The binding affinities at rat A1, A2a, and A3 adenosine receptors of a wide range of heterocyclic derivatives have been determined. Mono-, bi-, tricyclic and macrocyclic compounds were screened in binding assays, using either [3H]PIA or [3H]CGS 21680 in rat brain membranes or [125I]AB-MECA in CHO cells stably transfected with rat A3 receptors. Several new classes of adenosine antagonists (e.g. 5-oxoimidazopyrimidines and a pyrazoloquinazoline) were identified. Various sulfonylpiperazines, 11-hydroxytetrahydrocarbazolenine, 4H-pyrido[1,2-a]pyrimidinone, folic acid, and cytochalasin H and J bound to A3 receptors selectively. Moreover, cytochalasin A, which bound to A1 adenosine receptors with Ki value of 1.9 μM, inhibited adenylyl cyclase in rat adipocytes, but not via reversible A1 receptor binding.
Introduction
The A1, the A2a, the A2b and the A3 adenosine receptors are members of the G-protein-coupled superfamily and have now been defined both on the basis of pharmacological differences1 and on the basis of distinct amino acid sequences.2 Adenosine receptors mediate a wide variety of physiological functions3–5. These include: Inhibition of neurotransmitter release from nerve endings, vasoconstriction in the kidney, cardiac depression and inhibition of lipolysis via A1 receptors; vasodilatation, inhibition of platelet aggregation, and inhibition of lymphocyte function via A2 receptors; potentiation of histamine release from mast cells6 resulting in hypotension7 via A3 receptors. The A1 and A3 receptors cause inhibition of adenylyl cyclase and activation of phospholipase C. A1 receptors also couple to activation of potassium channels and inhibition of calcium channels. The A2a and A2b receptors activate adenylyl cyclase.
Numerous structure-activity relationship studies at A1 and A2 adenosine receptors, aimed at increasing potency and selectivity of agonists and antagonists, have been published (see ref. 1). Although xanthines are the classical adenosine antagonists, numerous classes of nonxanthine antagonists,1,8 mainly fused nitrogen-containing heterocyclic structures, have been reported. Xanthines have, however, proven to be either inactive at cloned rat A3 receptors, or relatively inactive at the sheep and human cloned A3 receptors. The lack of any selective antagonist for the rat A3 adenosine receptor9,27 prompted us to undertake a detailed examination of a variety of nonxanthine derivatives as ligands for A3 and other adenosine receptors. Ligands selective for A3 receptors10 have promise as agents for treating ischemia of the brain11 and heart,3 inflammation,5 and asthma.5 The present study is a survey of some known and some novel classes of nonxanthine adenosine ligands of widely varying structure.
Results and Discussion
Xanthines, the best known class of adenosine antagonists, contain fused 5:6 heterocyclic rings. Caffeine and theophylline, two well known xanthine antagonists have Ki values of approximately 40 and 15 μM, respectively, at both the A1 and A2a receptor.1 No significant antagonist activity has been observed for xanthines or nonxanthine derivatives at the rat A3 adenosine receptor. Thus, an A3 antagonist is actively being sought. In addition to screening this diverse group of compounds at the cloned A3 receptor, they were also tested at rat A1 and rat A2a receptors.
Table 1 shows the results of radioligand binding competition experiments at rat brain adenosine receptors for 110 cyclic compounds. Structures of selected compounds of interest, having Ki values at A1 receptors in the 10−6 to 10−4 M range, are shown in Figure 1. Potential antagonism of adenylyl cyclase inhibition mediated by A1 receptors in rat adipocytes (Table 2) and by cloned rat A3 receptors expressed in CHO cells9 was also examined. Several compounds that inhibited A1 receptor binding were shown to antagonize the inhibitory effects of N6-phenylisopropyladenosine (R-PIA) on adenylyl cyclase in adipocyte membranes (Table 2), with KB values of ≥1 μM, as calculated using Schild analysis (see below).9 Functional assays at A2a receptors were not carried out. At rat A3 receptors, although numerous compounds showed Ki values in the range of 10−5 to 10−4 M, two of these compounds (6 and 47), examined in the functional assay at cloned rat A3 receptors coupled to adenylyl cyclase in CHO cells, appeared to have no antagonist activity. The dose-response curve for IB-MECA39 was not shifted in the presence of 40 μM of either 6 or 47. In preliminary experiments, the amino-substituted pteroic acid derivative 68 alone inhibited forskolin-stimulated adenylyl cyclase in A3 transfected CHO cells, indicating possible agonist properties.
Table 1.
Affinities of heterocyclic derivatives in radioligand binding assays at rat brain A1, A2a, and A3 receptorsa–d.
| Ki (μM) or % inhibitiond | |||||
|---|---|---|---|---|---|
| Compound | Sourcef | Name | (A1)a | (A2a)b | (A3)c |
| Non-Fused Rings | |||||
| 1 | F | N,N′-(2-Chloro-5-cyano-1,3-phenylene)dioxamic acid (Lodoxamide) | n.d. | n.d. | 17% |
| 2 | T | N,N-Diethylaminoethyl-2,2-diphenylvalerate (SKF-525A, Proadifen) | 11±6%d | e | 49.3 ±10.3 |
| 3 | A | 2,6-bis[(4S)-Isopropyl-2-oxazolin-2-yl]pyridine | 90.9±5.8 | 19±5%d | 46.2 ± 0.3 |
| 4 | A | 6-(4-Chlorophenyl)-4,5-dihydro-2-(2-hydroxybutyl)-3(2H)-pyridazinone | 75.1±20.8 | e | 22.4 ± 10.0 |
| Fused Bicyclics (6:5) | |||||
| 5 | A | (−)-5-Bromo-4-chloro-3-indolyl-β-D-glucoside | 25±7%d | e | 37.0 ±9.4 |
| 6 | A | 5-Bromo-4-chloro-3-indolyl-β-D-galactoside | 18±14%d | e | 18.1 ±3.8 |
| 7 | R | 5-Chloro-2(3H)-benzoxazolone (Chlorzoxazone) | 21 ±3% | 15±4% | 34±5% |
| 8 | R | Calcimycin | 29± 4% | e | 24±8% |
| 9 | A | 1-Methyl-2-phenylbenzimidazole | [23]j | [99]k | 19±8% (10−5) |
| 10 | R | 4-[3-[(1,1-Dimethylethyl)amino]-2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one hydrochloride ((±)-CGP-12177A) | 23±3% | 40±3% | 109±5 |
| 11 | Bo | 2-[2-Methoxy-4-(methylsulfinyl)phenyl]-1H-imidazo[4,5-b]pyridine (Sulmazole, ARL115) | [52]j | 22.7±8.6 | 15±0% (10−5) |
| 12 | D | 1,4,6-Trimethyl-1H-pyrazolo-pyridine | [>100]j | e | 22±0% (10−5) |
| 13 | D | Ethyl 4-phenylamino-1-methyl-pyrazolopyridine-5-carboxylate | [1.1]i | [2.4]i | 43±2% |
| 14 | D | Ethyl 4-benzylamino-1-methyl-pyrazolopyridine-5-carboxylate | [1.0]i | [2.5]i | 29±3% (10−5) |
| 15 | D | Ethyl 4-phenylethylamino-1-methyl-pyrazolopyridine-5-carboxylate | [4.3]i | [12]i | 26±6% |
| 16 | D | Ethyl 4-ethoxy-1-methyl-pyrazolopyridine-5-carboxylate | [22]i | [19]i | 15 ± 2% |
| 17 | M | Ethyl 1,3-dimethyl-4-(4-methoxyphenoxy)-1H-pyrazolo-[3,4-b]pyridine | 9.39±0.95 | 11.7 ± 2.0 | e |
| 18 | M | Ethyl 1,3-dimethyl-4-(4-fluorophenoxy)-1H-pyrazolo-[3,4-b]pyridine-5-carboxylate | 6.95±1.10 | 9.24 ± 1.04 | e |
| 19 | M | Ethyl 4-[3,5-dichlorophenoxy]1,3-dimethyl-1H-pyrazolo [3,4-b]pyridine-5-carboxylate | 22.8 ± 6.1 | e | e |
| 20 | C | 5-(Hydroxymethyl)-4,5,6,7-(tetrahydro)imidazo-[4,5-c]pyridine-6-carboxylic acid | e | 16 ± 2%d | 86.1 ± 3.4 |
| 21 | C | 5-Oxo-(5,6-dihydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 73.6±24.6 | 24 ± 2%d | n.d. |
| 22 | C | 1-Chloro-5-oxo-(5,6-dihydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 60.6 ± 20.7 | 85.1 ± 1.4 | 36.8 ± 1.0 |
| 23 | C | 1-Chloro-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 26 ± 3%d | 14 ± 7%d | n.d. |
| 24 | C | 1,3-Dichloro-5-oxo-(5,6 dihydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 10.3 ± 2.7 | 16.5 ± 3.0 | 53.0 ± 13.0 |
| 25 | C | 5-Oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine | e | 22 ± 1 %d | 53.5 ± 15.8 |
| 26 | C | 1,3-Diiodo-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 8.48 ± 0.84 | 35.9 ± 5.9 | n.d. |
| 27 | C | 1-Iodo-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | e | 27 ± 4%d | n.d. |
| 28 | C | 1-Bromo-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | 11 ±4%d | e | n.d. |
| 29 | C | 6-t-Butyloxycarbonyl-8-chloro-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | e | 24±7% | n.d. |
| 30 | C | 5-Oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid benzyl ester | e | 35±8%d | n.d. |
| 31 | C | 5-Oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidine-7-carboxylic acid methyl ester | e | e | n.d. |
| 32 | C | 7-(Methoxycarbonyl)-2-phenylmethyl-5-oxo-(5,6,7,8-tetrahydro)imidazo[1,5-c]pyrimidinium bromide | 440 | e | n.d. |
| 33 | E | 4-Amino-5,6,-dimethyl-2-phenyl-7H-pyrrolo-[2,3-d]pyrimidine | [1.49]g | [21.3]l | 15% (10−5) |
| 34 | E | 4-Amino-5,6,-dimethyl-2-phenyl-7H-7-(phenyl)pyrrolo-[2,3-d]pyrimidine | [0.036]g | [14.3]l | 25 ± 2% (10−5) |
| 35 | M | Ethyl 6-(4-chlorophenyl)-4-methyl pyrazolo[1,5-a] pyrimidine 3-carboxylate | 12 ± 1% | e | e |
| 36 | M | 5,7-bis(Trifluoromethyl)-3-cyano-2-(methylthio)pyrazolo-[1,5-a]pyrimidine | e | e | e |
| 37 | G | Anhydro-2-phenyl-6,8-diethyl-5-hydroxy-7-oxo-1,3,4-thiadiazolo[3,2-a]pyrimidinium hydroxide | [80]h | 23±6% | 55.6 ± 17.1 |
| 38 | D | 2-Phenyl-4,6-dimethyl thiazolopyrimidine-5,7-dione | [79]j | [130]k | 31 ±7% (10−5) |
| 39 | C | 8-(Heptatluoropropyl)adenine | e | 27±3%d | 99.5 ±11.5 |
| 40 | C | 8-(Heptafluoropropyl)guanine | e | 24±1%d | 77.7 ±27.1 |
| 41 | R | 6-[(1-Methyl-4-nitro-1H-imidazol-5-yl)thio]-1H-purine (Azathioprine) | 18±1% | 28±5% | 116±3 |
| 42 | P | 8-Methyl-6-(1-piperidinlyl)-1,2,4-triazolo[4,3-b]pyridazine (MDL 257) | [>100] | [0.9]k | 32±6% (10−5) |
| 43 | P | 6- (Morpholinyl)-1,2,4-triazolo [4,3-b]pyridazine (MDL 850) | [22] | 11.2±2.5 | 21±9% (10−5) |
| 44 | Mu | 7,9-Dibenzyl-1-methylxanthinium chloride | 20% | 23±6% | 25±4% (10−5) |
| 45 | G | Anhydro-1,6-dimethyl-5-hydroxy-6-propyl-1,3,4-triazolo[3,2a]pyrimidinium hydroxide | [≫250]h | e | 20±10% (10−5) |
| 46 | R | 1-Methylisoguanosine | n.d. | 1.97±0.56 | 25.7 ± 0.1 |
| Fused Bicyclics (6:6) | |||||
| 47 | A | Esculin | 27±5%d | 32±5%d | 101 ± 29 |
| 48 | F | Disodium chromoglycate | n.d. | n.d. | 19% |
| 49 | A | 8-(β-D-Glucopyranosyloxy)-7-hydroxyl-6-methoxycoumarin | 15±7%d | e | 104±6 |
| 50 | A | Hesperidine | 22±3%d | e | 263 ± 34 |
| 51 | R | 5,7-Dichloro-4-hydroxy-quinoline-2-carboxylic acid (5,7-Dichlorokynurenic acid) | 22±5% | e | 38±3% |
| 52 | T | 1-(5-Chloronaphthalenesulfonyl) piperazine (ML-9) | e | e | 96.2 ± 21.0 |
| 53 | T | 1-(5-Iodonaphthalenesulfonyl) piperazine (ML-7) | e | e | 119 ± 67 |
| 54 | T | 1-(5-Isoquinolinesulfonyl) piperazine (HA-100) | e | e | 24.5±10.0 |
| 55 | T | 1-(5-Isoquinolinesulfonyl)-2-methylpiperazine (H-7) | e | e | 50.9 ± 15.2 |
| 56 | T | 1-(5-Isoquinolinesulfonyl)-3-methylpiperazine | e | e | 59.0 ± 21.8 |
| 57 | A | 1-(5-Isoquinolinesulfonyl) homopiperazine (HA-1077) | 23±9%d | e | 43.3 ±13.9 |
| 58 | T | 1-[5-(8-Chloro-isoquinoline)sulfonyl]piperazine (HA-156) | e | e | 40.4 ± 12.9 |
| 59 | R | 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) | 25±2% | e | 36±9% |
| 60 | R | 6-Chloro-2H-1,2,4-benzothiadiazine-7-sulfonamide 1,1-dioxide (Chlorothiazide) | e | 24±5% | 34±4% |
| 61 | R | 7-Benzyl-1-ethyl-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid (Amfonelic acid) | 25.2±7.5 | e | 157±3 |
| 62 | W | 4H-Pyrido[1,2-a]-pyrimidin-4-one | e | e | 48.3 ± 4.8 |
| 63 | D | 1,3-Dipropyllumazine | [20]j | 24±7% | 46.1±3.8 |
| 64 | D | 1,3-Dimethyl-7-phenyllumazine | 32.9±7.1 | 107±8 | 20±1% (10−5) |
| 65 | R | Neopterin | e | e | n.d. |
| 66 | A | 2, 6-Diamino-6-(hydroxymethyl)-pteridine | 14±7%d | e | 135 ± 21 |
| 67 | A | 2, 4-Diamino-6,7-diisopropyl-pteridine | 2.51 ± 0.47 | 11.9±3.2 | 71 ± 14% |
| 68 | A | 4-[N-(2,4-Diamino-6-pteridinylmethyl)amino]benzoic acid | e | e | 48.2±7.5 |
| 69 | A | 4-[N-(2,4-Diamino-6-pteridinylmethyl)-N-methylamino]benzoic acid | 27±1 %d | e | 48.5 ± 10.0 |
| 70 | A | Folic acid | e | 20.3% | 28.4 ± 9.9 |
| 71 | A | Dihydrofolic acid | 33.8±2.3 | e | 45.9 ± 18.0 |
| 72 | A | Dipyridamole | 22±17%d | 54±2% | 19.0 ± 0.7 |
| Fused Bicyclic (>6) | |||||
| 73 | W | 1-Aza-8,9-benzcyclononadi-2,7-one | 22.5% | e | 70.5 ± 0.7 |
| 74 | Br | 7-Methyl-4-propyl-4,5,6,7-tetrahydro-6H-imidazo[4,5e][1,4]diazepine-5,8-dione | 19±2% | 16±7% | 27±3% (10−5) |
| 75 | Br | 7-Benzyl-1,4-dipropyl-4,5,6,7-tetrahydro-6H-imidazo[4,5e][1,4]diazepine-5,8-dione | 13.4±2.1 | 2.18±0.12 | e(10−5) |
| Cytochalasins (Macrocyclics) | |||||
| 76 | A | Cytochalasin A | 1.91±0.43 | 29±9% | see text |
| 77 | A | Cytochalasin B | 27.0±3.8 | e | 47.5±21.8 |
| 78 | A | Cytochalasin C | e | e | 18±2% (10−5) |
| 79 | A | Cytochalasin D | e | e | 53.7 ± 11.8 |
| 80 | A | Cytochalasin E | e | 10±1% | 155 ± 56 |
| 81 | A | Cytochalasin H | e | 16±7% | 23.0 ± 7.1 |
| 82 | A | Cytochalasin J | e | 19±8% | 27.9 ± 11.9 |
| Fused Tricyclics (5:6:5) | |||||
| 83 | P | 1,7-Dihydro-3,5-dimethylbenzo[1,2-c:5,4-c′]dipyrazole (MDL 26,020) | [8.0]m | [25.6]k | 12±2% (10−5) |
| 84 | P | 1,7-Diethyl-1,7-dihydro-3,5-dimethylbenzo[1,2-c:5,4-c′]dipyrazole (MDL 26,629) | [27]m | [56]k | e(10−5) |
| 85 | P | 1-Hydro-3,6-dimethylbenzo-[1, 2-c: 5, 4-c′]dipyrazole (MDL 26,687A) | [74] | [17]k | 55.7 ± 22.4 |
| Fused Tricyclics (6:5:6) | |||||
| 86 | R | Methyl 6,7-dimethoxy-4-ethyl-β-carboline-3 carboxylate (DMCM) | 1.57±0.32 | 3.33±0.67 | 49.2 ± 16 |
| 87 | E | 4-Hydroxy-5,6,7,8-tetrahydro-9-phenyl-9H-pyrimido[4,5-b]indole | [2.07]g | 1.49±0.51 | 15±1% (10−5) |
| 88 | W | 11-Hydroxytetrahydrocarbazolenine | e | 20% | 21.9±6.5 |
| 89 | G | Anhydro-1-cyclopropylmethyl-3-ethyl-2-hydroxy-4-oxo-pyrimido[2,1a] benzothiazolium hydroxide | [37]h | 6.15±0.69 | 53.4 ± 16.1 |
| 90 | E | 4-Hydroxy-9-phenyl-9H-pyrimido[4,5-b]indole | [0.88]g | [1.44]l | 18% (10−5) |
| Fused Tricyclics (6:6:5) | |||||
| 91 | - | lin-Benzohypoxanthine | 21.2±4.9 | 0.992 | 26±8% |
| 92 | Pf | 1,3-Dimethyl-7-phenyl-6H-imidazo-(4,5-g)lumazine | e | e | n.d. |
| 93 | P | 2,11-Dihydro-11-(4-morpholinyl)-6H-pyrimido [2,1-b]quinazolin-6-one (MDL 43400A) | [>100] | [160]k | 12±1% (10−5) |
| 94 | Pf | 1,3,6-Trimethyl-7-(3,4-dichlorophenyl)-imidazo-(4,5-g)lumazine | n.d | n.d. | 15% (10−5) |
| 95 | M | Ethyl 5-chloropyrazolo[1,5-a]quinazoline-3-carboxylate | 5.36±0.36 | 4.06±0.50 | e |
| Fused Tricyclics (6:6:6) | |||||
| 96 | A | Alloxazinen | n.d. | n.d. | 32±3% |
| 97 | T | Roseoflavin | 28.9±4.0 | e | 84±8 |
| 98 | B | Riboflavin | 12.7±2.9 | e | see text |
| 99 | S | Flavin adenine dinucleolide (FAD) | 33.8±12.0 | 18.3±2.5 | e |
| 100 | S | Fluorescein | 76.1±6.8 | 34±5% | 37±2% |
| 101 | A | Carminic acid | 21±9%d | e | 117 ± 22 |
| Fused Tricyclics (>6) | |||||
| 102 | T | Doxepin | 66.4±16.8 | e | 72±14% |
| 103 | W | R,S-6-Hydroxy-7,8,9,10-tetrahydro-6H-cyclohept-[b]indole | e | n.d. | 68.2 ± 23.8 |
| 104 | W | 5,6,8,9,10,11-Hexahydro-7H-cyclohepta[c]-quinolin-6-one | 45±7% | e | n.d. |
| Fused Rings (Misc.) | |||||
| 105 | M | Cedrol | 28± 2% | 34±10% | 50 ± 0% |
| 106 | M | Cedrene | 18± 3% | e | 26± 1% |
| 107 | A | Reserpine | e | 25.2% | n.d. |
| 108 | T | Podophyllotoxin | 31±8%d | e | 79±11% |
| 109 | T | 4′-Dimethylepipodophyllotoxin | 18±5%d | e | 70±15% |
| 110 | D | Mitragynineo | >100 | 75.6±28.0 | 55±2% |
n.d.: not determined.
Displacement of specific [3H]PIA binding, unless noted, in rat brain membranes expressed as Ki ± S.E.M. in μM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding, unless noted, in rat striatal membranes, expressed as Ki ± S.E.M. in μM (n = 3–6).
Displacement of specific [125I]AB-MECA binding, unless noted, in membranes of CHO cells stably transfected with the rat A3-cDNA, expressed as Ki ± S.E.M. in μM (n = 3–5).
A percent value indicates the percent displacement of radioligand at the concentration (M) given in parentheses or at 10−4M, if none specified.
≤10% displacement of radioligand.
A = Aldrich (Milwaukee, WI); B = BioRad; Bo, Boehringer-Ingelheim (Germany); Br = Prof. Peter Bridson (Univ. Memphis); C = Dr. Louis Cohen (NIH), D = Dr. John W. Daly (NIH); E = Prof. Kurt Eger (Univ. Tübingen, Germany); F = Dr. John Fozard (Sandoz, Geneva); G = Prof. Richard Glennon (Medical College of Virginia, Richmond); M = Maybridge (Trevillett, UK); Mu = Dr. Christa Müller (Univ. Tübingen); P = Dr. Norton Peet (Marion Merrell Dow, Cincinnati OH); Pf = Prof. Wolfgang Pfleiderer (Univ. Konstanz); R = RBI (Natick MA); S = Sigma (St. Louis MO), T = Toronto Research Chemicals (Toronto); W = Dr. B. Witkop (NIH).
ref. 33.
ref. 34.
ref. 12.
ref. 8.
KB versus NECA-stimulation of adenylate cyclase in human platlets.
Ki versus [3H]NECA in rat striatal membranes.
ref. 35.
ref. 40.
ref. 38.
Fig. 1.


Structures of selected adenosine receptor ligands.
Table 2.
Antagonism of A1 receptor-mediated inhibition of adenylyl cyclase in rat adipocycte membranes.a
| Compoundb | KB ± SEM (μM) |
|---|---|
| 4 | 600 |
| 18 | 17.1 ± 3.6 |
| 24 | 10.8 ± 3.0 |
| 26 | 600 |
| 61 | 600 |
| 64 | 46.7 ± 11.9 |
| 67 | 3.94 ± 0.79 |
| 77 | 81 |
| 86 | 1.36 ± 0.48 |
| 95 | 12.7 ± 2.95 |
Calculated from shift in dose-response curve to R-PIA (see Figure 3), using the Schild equation8 (n=3–5, or for single determination).
Compound 71 (50 μM) caused a slight shift of the R-PIA dose-respose curve to the left. Compound 91 (50 μM) antagonized the effects of R-PIA only weakly, thus a KB could not be determined.
Numerous fused ring compounds and a few nonfused ring compounds were examined in binding assays in the present study. Among nonfused ring compounds examined, only compounds 3 and 4 were weak competitors at A1 receptors. A number of fused 5:6 heterocyclic ring compounds showed moderate receptor affinity. The effects of certain pyrazolopyridines12, compounds 17 – 19, in binding were studied. They had Ki values around 10 μM at both A1 and A2a receptors. No binding activity was detected at A3 receptors. Related pyrazolopyridines, such as tracazolate, have been reported to have anxiolytic activity, and the A1 and A2a receptor antagonist activity of a large series of pyrazolopyridines has been reported.12
Imidazopyrimidine-7-carboxylic acid derivatives, compounds 24 and 26, have Ki values of 10 and 8 μM, respectively at A1 receptors. This class of histidine derivatives has been prepared in connection with potential antimalarial activity.13 Compound 42 is a triazolopyridazine bronchodilator36 that did not bind appreciably to adenosine receptors.
We have determined the Ki value of a nucleoside analogue, 1-methylisoguanosine, 46, at A2a and A3 receptors to be 2.0 μM and 25 μM, respectively. This compound was reported previously to bind to adenosine receptors as an agonist.14
The sulfonylpiperazine derivatives 52–58 displayed no binding activity at A1 and A2a receptors. However, they did displace radioligand binding weakly at A3 receptors. These compounds are members of a well-known class of inhibitors of protein kinase C activity, acting in the concentration range of 0.1 – 1 μM.15
A number of fused 6:6 bicyclic heterocycles displayed affinity at adenosine receptors. The Ki value of amfonelic acid, 61, at the A1 receptor was 25 μM. Amfonelic acid has been reported to induce seizures,16 and at that time it was not realized that the same compound binds to adenosine receptors with an affinity similar to that of caffeine, another proconvulsant agent. 4H-Pyrido[1,2-a]pyrimidin-one, 62, bound selectively to rat A3 receptors with a Ki value of 48 μM. A phenyllumazine derivative, 64, which could be considered an analogue of the A1/A2 antagonist 8-phenyltheophylline,1 bound to A1 receptors with a Ki value of 33 μM.
Various pteridine derivatives were shown to act as antagonists at adenosine receptors.8 We have found that the presence of chained alkyl groups (e.g. isopropyl) at the 6- and 7- positions enhance the potency of binding to A1 receptors in this series. Thus, compound 67 has a Ki value of 2.5 μM. Pteridine derivatives 68 and 69, related to the anticancer and antiinflammatory drug methotrexate, displaced radioligand from A3 receptors with Ki values of 48 μM. Another related heterocycle, dihydrofolic acid, 71, bound to A1 receptors with a Ki value of 34 μM, while curiously the more planar folic acid, 70, itself, was inactive at A1 or A2a receptors. The adenosine uptake blocker dipyridamole, 72, bound to A3 receptors with a Ki value of 19 μM.
Surprisingly, several of the cytochalasins showed considerable affinity at adenosine receptors. Cytochalasin A, 76, was the most potent of the series with a Ki value of 1.9 μM at A1 receptors. Cytochalasin A caused an increase in the amount of [125I]AB-MECA bound in membranes of A3-transfected CHO cells (150±16% of control at 300 μM, n = 4). In the presence of 100 μM NECA, the level of radioligand binding was 59±8% of control, thus the additional binding likely occurs at a non-A3 receptor site. Cytochalasin B, 77, displayed a Ki value of 27 μM at A1 receptors. The other cytochalasins, 78–82, were totally inactive in binding at A1 receptors. None of the cytochalasins bound appreciably at A2a receptors. Given the pharmacological difference between cytochalasins A and B and their subtle differences in molecular structure, the region of the molecule which is responsible for binding to or modulating binding to the adenosine receptors is not apparent. The cytochalasins are macrocyclic compounds that are fungal metabolites, used as tools in cytological research and in characterization of polymerization properties of actin.17
A number of benzodipyrazole derivatives, 83 and 84, studied previously at adenosine receptors35 lacked affinity at A3 receptors. A related compound, 85, had a Ki of 56 μM at A3 receptors.
Methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM), 86, a β-carboline derivative, displayed considerable affinity for adenosine receptors, with Ki values of 1.6 and 3.3 μM at A1 and A2a receptors, respectively. DMCM acts as a proconvulsant, by virtue of its activity as a benzodiazepine inverse agonist.18 Previously β-carboline itself and several derivatives8 were shown to have weak affinity at the A1 receptor and to act as antagonists at that receptor, and this was proposed to be related to their proconvulsant activity. Since the potency of DMCM is even greater than other β-carbolines examined,8 it becomes an even more relevant aspect of the biological effects. DMCM was used to induce seizures in a study of the anticonvulsant effect of adenosine agonists,19 apparently without knowledge of its potency in adenosine antagonism. Another carbazole derivative, 11-hydroxytetrahydrocarbazolenine, 88,22 was A3 selective in binding with a Ki value of 22 μM.
A series of lin-benzoxanthine derivatives was shown to have adenosine antagonist activity.20 lin-Benzohypoxanthine, 91,21 formalistically an elongated derivative of unsubstituted hypoxanthine, showed considerable affinity at adenosine receptors, although it was nonselective. An imidazoquinazolinone derivative, 93,37 lacked affinity at adenosine receptors. Another 6:6:5 fused tricyclic, compound 95, a pyrazoloquinazoline derivative, showed an affinity of 4–5 μM at A1 and A2a receptors, with no displacement of radioligand from A3 receptors.
Fused 6:6:6 tricyclic derivatives were also examined. Alloxazine, 96, has been found to be 9-fold selective for A2b (Kg 2.3 μM) vs A2a receptors.40 The naturally occurring flavins contain the same ring structure as alloxazine with an appended carbohydrate mioety. Riboflavin,23 98, a vitamin and enzyme cofactor having an attached open chain ribose moiety, and roseoflavin, 97, the homologous derivative with a shorter carbohydrate chain (C3), bound to A1 receptors with Ki values of 13 and 29 μM, respectively, with no detectable binding at A2a receptors. Unexpectedly, the binding of the A3 receptor radioligand was dramatically enhanced (Figure 3), with 355±35% of control binding in membranes of A3-transfected CHO cells at 100 μM riboflavin. Since the additional binding occurred also in the presence of 100 μM NECA (280±40% of control binding), it does not represent selective binding enhancement at A3 receptors, as has been shown for benzoylthiophene derivatives (allosteric enhancers) at A1 receptors.24 Instead, riboflavin apparently causes enhanced binding of [125I]AB-MECA to a nonreceptor site on the membranes. The nature of this site has not been explored, but perhaps it is related to an enzyme at which riboflavin acts as a cofactor. The adenosine conjugate FAD, 99, was somewhat weaker than riboflavin in binding to A1 receptors, yet bound with increased affinity at A2a receptors. Fluorescein dye, 100, also bound weakly to adenosine receptors with a Ki value of 76 μM at the A1 subtype.
Fig. 3.
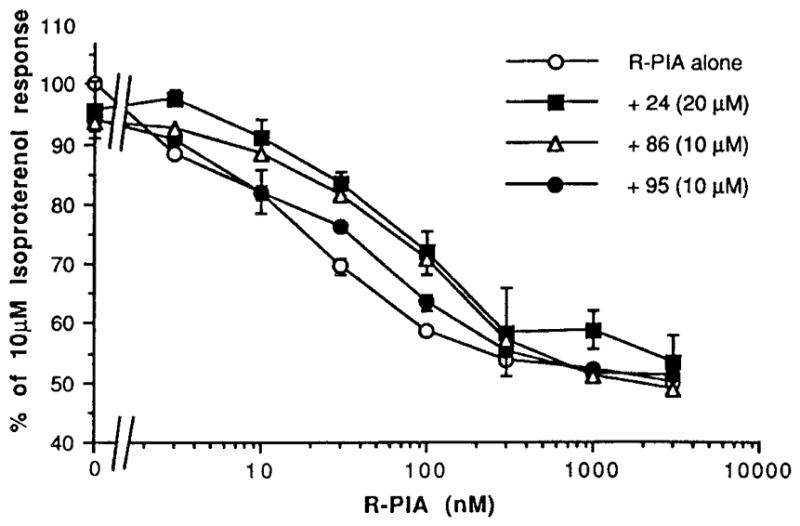
Effects on A1-agonist-induced inhibition of adenylyl cyclase in rat adipocyte membranes.
The antidepressant drug doxepin,25 102, is a dibenzoxepin derivative. This compound showed a Ki value of 66 μM at A1 receptors. Other psychotropic drugs, such as barbiturates, were previously shown to bind weakly to adenosine receptors.26
Functional assays at rat adipocyte A1 receptors were carried out (Table 2, and Figures 3 and 4). The compounds that bound with highest affinity were examined for the ability to antagonize the inhibition of adenylyl cyclase elicited by R-PIA. It was found that DMCM, 86, and the pteridine derivative 67, were the most potent antagonists, with micromolar KB values. The cyclized histidine derivative (a 5-oxoimidazopyrimidine) 24, the pyrazoloquinazoline 95, the pyrazolopyridine 18, and the phenyllumazine derivative 64 were also weak antagonists. lin-Benzohypoxanthine, 91, slightly antagonized the effects of R-PIA. Cytochalasin A, 76 (10 μM), shifted the R-PIA dose response curves to the left and alone inhibited adenylyl cyclase in the adipocyte membranes with an IC50 of 6 μM (Figure 4). However, the inhibition by cytochalasin A was not reversed in the presence of the selective A1-antagonist l,3-dipropyl-8-cyclopentylxanthine, 1 μM (data a not shown). Thus, cytochalasin A appears to have a direct inhibitory effect not mediated through A1 receptors. Doxepin, 102 (50 μM), clearly shifted the R-PIA dose response curve to the left, however alone did not inhibit adenylyl cyclase. Thus doxepin is not an agonist at A1 receptors, but it may act to enhance agonist-induced effects. If so, it is conceivable that this may occur at an allosteric site. Dihydrofolic acid, 71, produced a slight left shift of the R-PIA dose response curve, but 71 alone had no effect on adenylyl cyclase in adipocyte membranes.
Fig. 4.
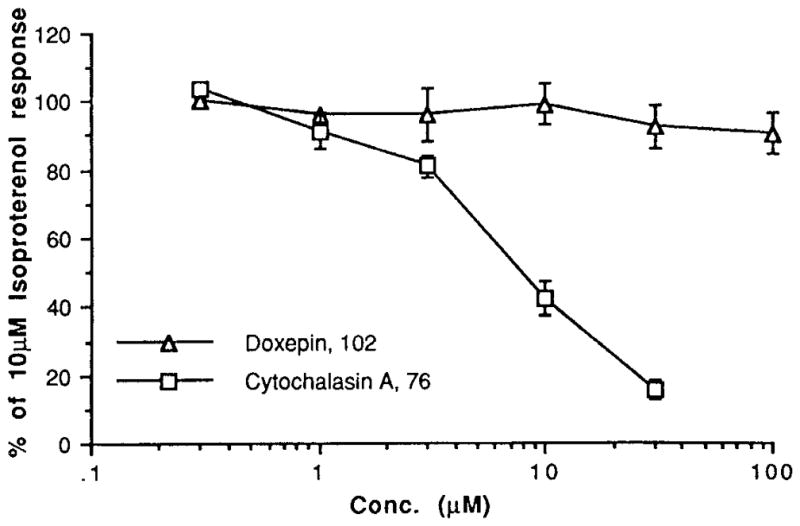
Inhibition of adenylyl cyclase in rat adipocyte membranes.
Conclusions
A selective antagonist at the rat A3 receptor is lacking. Several xanthine analogs that act as potent antagonists at rat, rabbit, and human A1 and A2 receptors only weakly displaced the binding of radioligand from cloned rat A3 receptors.27 The present study has not identified any effective A3 antagonist in the rat, however, it has provided leads for future structural modification.
Sulfonylpiperazines, 52–58, and cytochalasin H and J, 81 and 82, respectively, bind to A3 receptors without binding to the other subtypes. Moreover, compounds 2, 6, 25, 62, 68, 70, 72, and 88 also were somewhat selective in binding to A3 receptors. The pteridine derivative, 67, was identified as a slightly A1 selective (binding) antagonist. Compounds 26, 76, 77, 97, and 98 were also weakly A1 selective. Compounds 11, 43, 75, and 89 were identified as slightly A2a selective ligands.
The most significant new findings are the discovery of several new classes of adenosine antagonists (e.g. 5-oxoimidazopyrimidines and a pyrazoloquinazoline), and that nonpurine heterocycles (e.g. 68 at A3 receptors and 76 at A1 receptors) inhibit adenylyl cyclase, possibly through activation of adenosine receptors. It will be necessary to explore the mechanism of the inhibition of adenylyl cyclase, since action at the P site would also have this effect.39 Previously it was observed1 that only purine nucleosides were known to activate adenosine receptors.
Experimental
Compound 91 was synthesized as described by Leonard and coworkers.21
Cell culture and radioligand binding
CHO cells stably expressing the A3 receptor9,28 were grown in F-12 medium containing 10% FBS and penicillin/streptomycin (100 U/mL and 100 μg/mL respectively) at 37 °C in a 5% CO2 atmosphere, and membrane homogenates were prepared as reported.28
Binding of [125I]4-amino-3-iodobenzyladenosine-5′-N-methyluronamide ([125I]-AB-MECA) to the CHO cell membranes was performed as described.29 Assays were performed in 50/10/1 buffer in glass tubes and contained 100 μL of the membrane suspension, 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of inhibitor. Inhibitors were routinely dissolved in DMSO and were then diluted with buffer; final DMSO concentrations never exceeded 1%. Incubations were carried out in duplicate for 1 hour at 37 °C, and were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). Tubes were washed three times with 3 mL of buffer. Radioactivity was determined in a Beckman gamma 5500B counter. Nonspecific binding was determined in the presence of 40 μM R-PIA. Ki -values were calculated according to Cheng-Prusoff,30 assuming a Kd for [125I]AB-MECA of 1.55 nM.29
Binding of [3H]PIA (Amersham, Arlington Heights, IL) to A1 receptors from rat brain membranes and of [3H]CGS 21680 (DuPont NEN, Boston MA) to A2a receptors from rat striatal membranes was performed as described previously.31, 32 Adenosine deaminase (3 U/mL) was present during the preparation of brain membranes, in which an incubation at 30°C for 30 min was carried out, and during the incubation with radioligand. At least six different concentrations spanning three orders of magnitude, adjusted appropriately for the IC50 of each compound, were used. The IC50 values that were computer-generated using a nonlinear regression formula on the InPlot program (GraphPAD, San Diego CA), were converted to apparent Ki values using Kd values of 1.0 and 14 nM for [3H]PIA and [3H]CGS 21680 binding, respectively, and the Cheng-Prusoff equation.30
Adenylyl cyclase measurements in rat adipocyte membranes and in A3-transfected CHO cells were carried out as described.8,28
Figure 2.
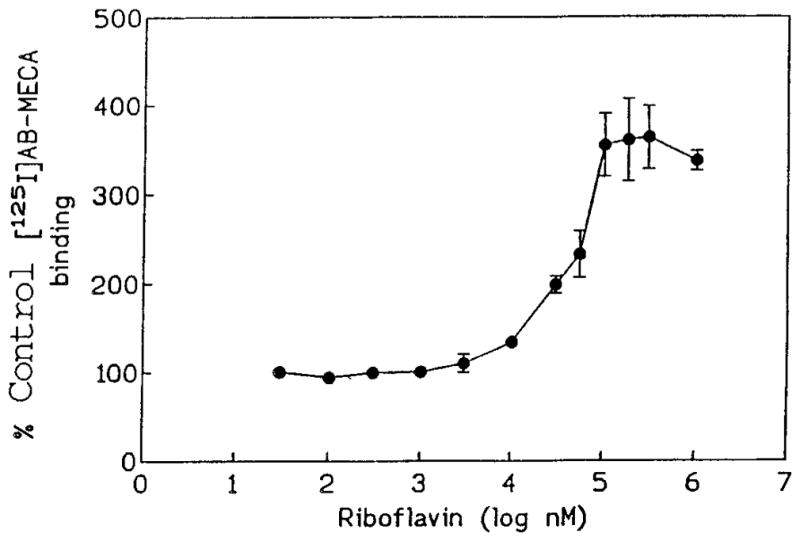
Enhancement of radioligand (0.4 nM) binding in A3-transfected CHO cell membranes by riboflavin, 98.
Acknowledgments
We thank the Cystic Fibrosis Foundation (N.M.), Gilead Sciences of Foster City, CA (M.G.) for financial support. We thank Mary Pound for technical assistance. G.L.S. is supported by NHLBI Grant RO1HL35134 from the National Institutes of Health. In addition, we thank Dr. Norton Peet (Marion Merrell Dow Research Inst., Cincinnati OH), Professor W. Pfleiderer (University of Konstanz, Germany), Professor Peter Bridson (University of Memphis, TN), Professor Richard Glennon (Medical College of Virginia, Richmond), Dr. Christa Müller and Prof. Kurt Eger (Universität Tübingen, Germany), and Dr. Bernhard Witkop (NIH) for the gift of compounds.
Abbreviations
- AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide
- CGS 21680
2-[4-[(2-carboxyethyl)phenyl]ethyl8amino]-5′-N-ethylcarboxamido-adenosine
- CHO
Chinese hamster ovary
- DMCM
methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- DMSO
dimethylsulfoxide
- PIA
R-N6-phenylisopropyladenosine
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
Dedicated to Prof. Yoshihisa Mizuno on the occasion of his 75th birthday.
References
- 1.van Galen PJM, Stiles GL, Michaels G, Jacobson KA. Med Res Rev. 1992;12:423–471. doi: 10.1002/med.2610120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson M. In: Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Bellardinelli L, Pelleg A, editors. Kluwer; Boston: 1995. pp. 5–13. [Google Scholar]
- 3.Lasley RD, Bünger R, Mentzer RM. In: Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Bellardinelli L, Pelleg A, editors. Kluwer; Boston: 1995. pp. 351–360. [Google Scholar]
- 4.Rudolphi KA, Schubert P. In: Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Bellardinelli L, Pelleg A, editors. Kluwer; Boston: 1995. pp. 391–397. [Google Scholar]
- 5.Beaven MA, Ramkumar V, Ali H. Trends Pharmacol Sci. 1994;15:13–14. doi: 10.1016/0165-6147(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 6.Ramkumar V, Stiles GL, Beaven MA, Ali H. J Biol Chem. 1993;268:16871–16890. [PubMed] [Google Scholar]
- 7.Carruthers AM, Fozard J. Eur J Pharmacol. 1993;250:185–188. doi: 10.1016/0014-2999(93)90641-t. [DOI] [PubMed] [Google Scholar]
- 8.Daly JW, Hong O, Padgett WL, Shamim MT, Jacobson KA, Ukena D. Biochem Pharmacol. 1988;37:655–664. doi: 10.1016/0006-2952(88)90139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Proc Natl Acad Sci USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Lubitz DKJE, Lin RCS, Popik P, Carter MF, Jacobson KA. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daly JW, Hutchinson KD, Secunda SI, Shi D, Padgett WL, Shamin MT. Med Chem Res. 1994;4:293–306. [Google Scholar]
- 13.Jain R, Cohen LA. manuscript in preparation. [Google Scholar]
- 14.Davies LP, Baird LJ, Hall JG. Neuropharmacology. 1987;26:493–7. doi: 10.1016/0028-3908(87)90033-5. [DOI] [PubMed] [Google Scholar]
- 15.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 16.Ewing AG, Bigelow JC, Wightman RM. Science. 1983;221:169. doi: 10.1126/science.6857277. [DOI] [PubMed] [Google Scholar]
- 17.Yahara I, Harada F, Sekita S, Yoshihira K, Natori S. J Cell Biol. 1982;92:69–78. doi: 10.1083/jcb.92.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croucher M, DeSarro G, Jensen L, Meldrow B. Eur J Pharmacol. 1984;104:55–60. doi: 10.1016/0014-2999(84)90368-6. [DOI] [PubMed] [Google Scholar]
- 19.Klitgaard H, Knutsen LJS, Thomsen C. Eur J Pharmacol. 1993;242:221–228. doi: 10.1016/0014-2999(93)90245-d. [DOI] [PubMed] [Google Scholar]
- 20.Schneller SW, Ibay AC, Christ WJ, Bruns RFJ. Med Chem. 1989;32:2247–2254. doi: 10.1021/jm00130a004. [DOI] [PubMed] [Google Scholar]
- 21.Leonard NJ, Morrice AG, Sprecker MA. J Org Chem. 1975;40:356–363. doi: 10.1021/jo00891a021. [DOI] [PubMed] [Google Scholar]
- 22.Witkop B, Patrick JB. J Am Chem Soc. 1950;73:2196–2200. [Google Scholar]
- 23.Christensen HN. Nutr Rev. 1993;51:149–150. doi: 10.1111/j.1753-4887.1993.tb03092.x. [DOI] [PubMed] [Google Scholar]
- 24.Bruns RF, Fergus JH. Mol Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- 25.Elonen E, Mattila MJ, Saarnivaara L. Eur J Pharmacol. 1974;1:178–88. doi: 10.1016/0014-2999(74)90130-7. [DOI] [PubMed] [Google Scholar]
- 26.Lohse MJ, Klotz KN, Jakobs KH, Schwabe U. J Neurochem. 1985;45:1761–1770. doi: 10.1111/j.1471-4159.1985.tb10532.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim HO, Ji XD, Melman N, Olah ME, Stiles GL, Jacobson KA. J Med Chem. 1994;37:4020–4030. doi: 10.1021/jm00049a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Galen PJM, van Bergen AH, Gallo-Rodriquez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- 29.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 31.Schwabe U, Trost T. Naunyn-Schmiedeberg’s Arch Pharmacol. 1980;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 32.Jarvis MF, Schulz R, Hutchison AJ, Do UH, Sills MA, Williams M. J Pharmacol Exp Ther. 1989;251:888–93. [PubMed] [Google Scholar]
- 33.Müller CE, Hide I, Daly JW, Rothenhausler K, Eger K. J Med Chem. 1990;33:2822–2828. doi: 10.1021/jm00172a023. [DOI] [PubMed] [Google Scholar]
- 34.Glennon RA, Tejani-Butt SM, Padgett W, Daly JW. J Med Chem. 1984;27:1364–1367. doi: 10.1021/jm00376a027. [DOI] [PubMed] [Google Scholar]
- 35.Peet NP, Dickerson GA, Abdallah AH, Daly JW, Ukena D. J Med Chem. 1988;31:2034–2039. doi: 10.1021/jm00118a032. [DOI] [PubMed] [Google Scholar]
- 36.Peet NP, Malecha J, LeTourneau ME, Sunder SJ. Heterocyclic Chem. 1989;26:257–264. [Google Scholar]
- 37.Abdallah A, Burnell JW, Cregge RJ. Drug Devel Res. 1986;7:185–193. [Google Scholar]
- 38.Macko E, Weisbach JA, Douglas B. Arch Int Pharmacodyn Ther. 1972;198:145–161. [PubMed] [Google Scholar]
- 39.Jacobson KA, Siddiqi SM, Olah ME, Ji Xd, Melman N, Bellamkonda K, Meshulam Y, Stiles GL, Kim HO. J Med Chem. 1995;38:1720–1735. doi: 10.1021/jm00010a017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brackett LE, Daly JW. Biochem Pharmacol. 1994;47:801–814. doi: 10.1016/0006-2952(94)90480-4. [DOI] [PubMed] [Google Scholar]