

Abstract
Ocular developmental anomalies (ODA) such as anophthalmia/microphthalmia (AM) or anterior segment dysgenesis (ASD) have an estimated combined prevalence of 3.7 in 10,000 births. Mutations in SOX2 are the most frequent contributors to severe ODA, yet account for a minority of the genetic drivers. To identify novel ODA loci, we conducted targeted high-throughput sequencing of 407 candidate genes in an initial cohort of 22 sporadic ODA patients. Patched 1 (PTCH1), an inhibitor of sonic hedgehog (SHH) signaling, harbored an enrichment of rare heterozygous variants in comparison to either controls, or to the other candidate genes (four missense and one frameshift); targeted resequencing of PTCH1 in a second cohort of 48 ODA patients identified two additional rare nonsynonymous changes. Using multiple transient models and a CRISPR/Cas9-generated mutant, we show physiologically relevant phenotypes altering SHH signaling and eye development upon abrogation of ptch1 in zebrafish for which in vivo complementation assays using these models showed that all six patient missense mutations affect SHH signaling. Finally, through transcriptomic and ChIP analyses, we show that SOX2 binds to an intronic domain of the PTCH1 locus to regulate PTCH1 expression, findings that were validated both in vitro and in vivo. Together, these results demonstrate that PTCH1 mutations contribute to as much as 10% of ODA, identify the SHH signaling pathway as a novel effector of SOX2 activity during human ocular development, and indicate that ODA is likely the result of overactive SHH signaling in humans harboring mutations in either PTCH1 or SOX2.
Severe congenital ocular malformations, including the reduction of eye size and anterior chamber anomalies at birth, are rare, with an estimated combined prevalence of 3.7 in 10,000 births (Bermejo and Martinez-Frias 1998). Ocular developmental anomalies (ODA) such as anophthalmia/microphthalmia (AM) or anterior segment dysgenesis (ASD) are underscored by extensive genetic heterogeneity, phenotypic variability, and nonpenetrance.
Anophthalmia refers to the complete absence of ocular tissue in the orbit (true anophthalmia), or the absence of ocular tissue upon clinical examination (clinical anophthalmia). Microphthalmia corresponds to a globe with a total axial length that is at least two standard deviations below the mean for age (<19 mm in a one-year-old child, <21 mm in an adult) (Weiss et al. 1989; Verma and Fitzpatrick 2007). To date, more than 20 genes have been implicated in AM in humans. Mutations in SOX2 are the most frequent contributors to severe ODA (Fantes et al. 2003), yet account for a minority (10%–15%) of the genetic drivers (Verma and Fitzpatrick 2007), while other genes are more rarely involved. In AM cases, the genetic basis, either monogenic or chromosomal, is identified in ∼20%–40% of individuals who undergo genetic testing (Slavotinek 2011; Chassaing et al. 2014). ASD disorders encompass a wide variety of developmental conditions affecting the cornea, iris, and lens including corneal opacity, posterior embryotoxon, iris hypoplasia, corectopia or polycoria, and adhesions between the iris and cornea or lens and cornea (Reis and Semina 2011). Two clinically distinct ASD disorders have been recognized as separate entities based on unique combinations of diagnostic criteria: Axenfeld-Rieger anomaly (iris hypoplasia, posterior embryotoxon, iris hypoplasia, corectopia/polycoria, and/or irido-corneal adhesions) and Peters anomaly (corneal opacity, defects in the posterior layers of the cornea, and lenticulo-corneal and/or irido-corneal adhesions). Causative mutations are identified in ∼40% of individuals with Axenfeld-Rieger anomaly, mainly in two genes, PITX2 and FOXC1 (Pasutto et al. 2015). Despite the fact that thirteen genes have been associated with Peters anomaly, the genetic basis of this ocular anomaly remains unknown in the majority of cases (Weh et al. 2014).
Classical genetics approaches were successful to discover the first ODA genes. Linkage analysis studies allowed the identification of only few genes (Ferda Percin et al. 2000; Pasutto et al. 2007). The reason is most of the pedigrees available are small; therefore, the chromosomal regions that segregate with the disease are usually numerous and not reaching genome-wide significance. Positional candidate gene approaches pursuant to the identification of chromosomal abnormalities in ODA cases, or candidate gene approaches related to phenotype have yielded some gene discovery results such as SOX2 and OTX2 (Fantes et al. 2003; Ragge et al. 2005a). However, they have similarly reached an impasse toward identifying novel causal or contributory loci. In past years, the development of next-generation sequencing technologies has allowed the identification of several new ODA genes, including ALDH1A3 and RARB (Fares-Taie et al. 2013; Srour et al. 2013). To identify novel ODA loci, we harnessed the power of next-generation sequencing technologies and conducted targeted high-throughput sequencing of a subset of candidate genes with a potential role in eye development, in a cohort of 22 sporadic ODA patients.
Results
Targeted resequencing identifies a significantly enriched mutational burden in PTCH1
We selected 407 candidate genes (Supplemental Table 1) involved in ocular development for targeted exon liquid capture followed by massively parallel sequencing. We screened 22 unrelated individuals with ODA and mutation negative for SOX2, OTX2, RAX, and VSX2 and two positive control individuals (with known mutations in either STRA6 or in VSX2). These affected individuals had isolated ASD (n = 6), or AM that was isolated (n = 4), associated with ASD (n = 6), or coincident with coloboma (n = 6) (Supplemental Table 2). We identified ∼2500 variants in each patient; after stringent bioinformatic filtering that focused exclusively on alleles that were (1) absent from dbSNP132 (http://www.ncbi.nlm.nih.gov/projects/SNP/; Sherry et al. 2001), HapMap (http://hapmap.ncbi.nlm.nih.gov/; The International HapMap 3 Consortium et al. 2010), 1000 Genomes (http://www.1000genomes.org/; The 1000 Genomes Project Consortium 2015), and our in-house exomes; and (2) predicted in silico to be deleterious (Supplemental Table 3), we observed 0–5 variants per individual in a total of 46 loci. These genes harbored variation in the following locus-wide distribution: >1 variant/gene in 10 genes (Fig. 1A; Supplemental Tables 4, 5); 1 variant/gene in 36 genes; the remaining 361 genes were bereft of rare functional variants predicted to be deleterious (Supplemental Table 4, discussed in the Supplemental Note).
Figure 1.
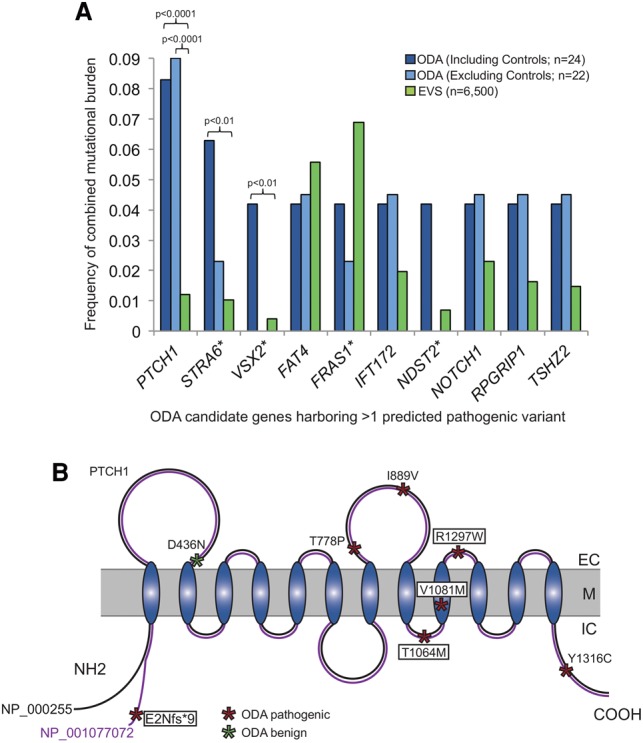
PTCH1 has a significantly enriched mutational burden in ocular developmental anomalies (ODA). (A) Mutational burden for all genes harboring >1 rare predicted pathogenic variants in the initial cohort of ODA compared to healthy control individuals from the Exome Variant Server (EVS). Frequency of combined mutational burden is shown for the 10 genes harboring multiple rare (<1% alternate allele frequency) functional variants (frameshift, nonsense, and splicing variants were considered as damaging; missense variants were classified as damaging or not based on PolyPhen-2) in either ODA cases including two positive controls (n = 22 unknown + 2 positive controls; dark blue bars), or ODA test cases alone (n = 22 unknown; light blue bars) vs. EVS controls (n = 6500; green bars). P-values are indicated for the only three genes with a significant enrichment in cases versus controls (χ2 test). (*) VSX2 and STRA6 are previously identified causal genes in the positive control ODA samples, C1 and C2, respectively; C1 also harbored variants in FRAS1 and NDST2. (B) Schematic representation of the PTCH1 receptor with its extracellular (EC), transmembrane (M), and intra-cellular domains (IC). The positions of the PTCH1 mutations identified in ODA are represented with asterisks, with the color indicating variant pathogenicity. The four variants enclosed with boxes were identified in the initial discovery cohort. p.E2Nfs*9 is specific to isoform NP_001077072 (purple), which is identical to NP_000255 (black) except for an alternate 66 amino acid region at the N terminus.
Among these 46 loci, PTCH1 carried the greatest mutational burden and was the most significantly enriched for rare putative pathogenic variants in the 22 individuals from the ODA cohort and in the two positive control individuals C1 and C2 in comparison to 13,006 control chromosomes in the Exome Variant Server (EVS) (P < 0.0001; Table 1; Fig. 1A,B) (http://evs.gs.washington.edu/EVS/; Fu et al. 2013) and remained nominally significant after correction for the 407 target gene set (P = 0.04). Notably, the only other genes harboring an enrichment of rare variants predicted to be pathogenic in the 24 patients were VSX2 and STRA6, both of which have been identified previously as rare ODA contributors (Ferda Percin et al. 2000; Chassaing et al. 2009) and were mutated in the positive control individuals C1 and C2, respectively (P < 0.01 vs. 13,006 EVS chromosomes) (Fig. 1A; Supplemental Table 5). Upon exclusion of each of C1 and C2 from the mutational burden analysis, PTCH1 was the only gene that carried a significant enrichment of pathogenic variants in the 22 ODA cases with previously unknown genetic etiology (P < 0.0001 vs. EVS) (Fig. 1A).
Table 1.
PTCH1 variants identified in ODA patients and/or studied in vivo using zebrafish experiments

Four individuals harbored rare PTCH1 heterozygous changes predicted to be deleterious (Table 1; Fig. 1B). One patient with microphthalmia, cataract, and sclerocornea (P5) had a frameshifting deletion (c.4delG, p.Glu2Asnfs*9) in exon 1 of PTCH1 isoform NP_001077072. Patient P20, affected with a bilateral Peters anomaly, harbored a heterozygous p.Tyr1316Cys change. Two additional unrelated patients with colobomatous microphthalmia, corpus callosum abnormality, and atrial septal defects (P8 and P15) harbored c.3191C>T (p.Thr1064Met) and c.3241G>A (p.Val1081Met) changes. With the exception of P5, for whom we were unable to perform segregation analysis, we determined that each of these three PTCH1 mutations was inherited from an asymptomatic parent (Table 1; Supplemental Table 4), consistent with incomplete penetrance.
Because of the significant enrichment of PTCH1 variants in our first-pass filtering strategy, and cognizant of the imperfect sensitivity and specificity of prediction algorithms, we returned to the set of rare variants remaining prior to in silico predictions. We found two additional heterozygous rare PTCH1 missense variants filtered out initially because they were considered benign by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/; Adzhubei et al. 2010). Individual P17, presenting with a bilateral Axenfeld-Rieger malformation, had a c.3889C>T (p.Arg1297Trp) mutation. Additionally, we identified a heterozygous p.Asp436Asn change in the control sample C2 (Table 1; Fig. 1B).
In vivo assay of variant pathogenicity shows that PTCH1 variants are deleterious
The significant enrichment of mutational burden in the 24 cases compared to 6500 EVS controls (P < 0.001), amino acid conservation, and in silico prediction evidence were suggestive but not conclusive with regard to the deleterious effect of the PTCH1 missense variants identified in the ODA cohort (Table 1; Supplemental Fig. 1). Moreover, the observed incomplete penetrance posed interpretive challenges. Therefore, we evaluated the effect of all discovered alleles in vivo. PTCH1 is a transmembrane-dependent receptor which functions with SHH as part of a dosage-sensitive pathway resulting in activation of downstream target genes, including the Smoothened (SMO) coreceptor, PTCH1 itself, and Gli transcription factors GLI1, GLI2, and GLI3 (Villavicencio et al. 2000). SHH signaling is a key regulator of somite patterning (Bumcrot and McMahon 1995), and numerous zebrafish models of the SHH pathway including shh (Schauerte et al. 1998), smo (Varga et al. 2001), suppressor of fused (sufu) (Wolff et al. 2003; Koudijs et al. 2005), patched 2 (ptch2, referred to previously as ptc1 [Koudijs et al. 2008]), and kif7 (Tay et al. 2005) display a visibly more obtuse angle of the somitic chevron compared to wild-type (WT) controls. We have shown previously that in vivo complementation studies of human mutations in SHH effector molecules using somite defects as a phenotypic readout are a robust assay to determine allele pathogenicity (Putoux et al. 2011). We therefore employed this strategy to test the ability of human mRNAs harboring the ODA PTCH1 missense mutations to rescue ptch1 MO-induced somite angle defects in comparison to that of WT.
Using reciprocal BLAST, we confirmed that the single ortholog of human PTCH1 is located on Danio rerio Chromosome 8 (73% identity, 80% similarity; human versus zebrafish protein); this gene, initially named ptc2, is mutated in the zebrafish leprechaun (lep) mutant (Koudijs et al. 2005) but will hereafter be referred to as ptch1. This locus is distinctly different from ptc1, the zebrafish ortholog of human PTCH2 that is mutated in the zebrafish blowout (blw) mutant (Koudijs et al. 2008), and is now referred to as ptch2. We obtained a previously published splice-blocking (sb) morpholino antisense oligonucleotide (MO) targeting the exon 3 splice donor site (Koudijs et al. 2008), and we detected a partial suppression of ptch1 expression by RT-PCR analysis of sb1 MO-injected whole-embryo lysates (Supplemental Fig. 2A). Even so, we observed somite phenotypes characteristic of Shh defects in WT embryos injected with 12 ng of ptch1 sb1 MO (81.5° degrees vs. 97.5° degrees for control vs. ptch1 sb1 MO; P < 0.0001) (Fig. 2; Supplemental Table 7). Importantly, co-injection of 100 pg of capped human PTCH1 WT mRNA resulted in a significant amelioration of the somite defect (85.9° vs. 97.5° for WT rescue vs. MO; P < 0.0001).
Figure 2.

PTCH1 variants identified in ODA patients are pathogenic. (A,B) Representative lateral images of uninjected control and ptch1 sb1 MO-injected live embryos taken at 36 h post-fertilization (hpf); dashed boxes are enlarged in the insets (right). Magnified panels show chevron-shaped somites (controls) and abnormal-shaped somites (morphants), caused by aberrant Hedgehog signaling in the zebrafish myotome. Dashed blue lines indicate measurement position (at the midpoint between the proximal hindgut and the anus) used for phenotypic scoring of embryo batches. (C) All six nonsynonymous PTCH1 variants identified in ODA cases were pathogenic as indicated by the inability of mutant mRNA to rescue the ptch1 MO-induced somite angle defects. PTCH1 p.Thr1052Met, a rare variant (minor allele frequency in controls 0.001; n = 13,006 chromosomes [EVS]) reported previously in HPE, is also pathogenic; p.Leu360Arg is a previously reported functional null. Rescue with a common PTCH1 p.Pro1315Leu encoding variant (rs357564; present in homozygosity in 8% of controls; n = 12,568 chromosomes in EVS) was not significantly (NS) different from wild type (WT), nor was a previously reported benign variant p.Pro1125Leu change, providing support for the specificity of the assay. p.Gly509Val is a positive control for dominant negative effects. The missense variant p.Asp436Asn identified in the ODA control C2 was benign in this assay. We measured 38–58 embryos per injection batch with blind scoring. Asterisks indicate statistical differences between mutant and WT rescue (P < 0.0001; Student's t-test). Error bars, SEM. See Supplemental Table 7 for somite measurement data.
We next evaluated the five missense mutations (p.Asp436Asn, p.The1064Met, p.Val1081Met, p.Arg1297Trp, p.Tyr1316Cys) identified among the 22 patients and two control ODA individuals. As a measure of the sensitivity and relevance of this assay, we also evaluated known deleterious variants that have been proposed to be pathogenic based on either genetic evidence or functional evaluation of SHH pathway readout. As such, we tested the effect of human PTCH1 mRNA carrying the known pathogenic p.Thr1052Met change, associated previously with a holoprosencephaly (HPE)-like phenotype (including bilateral microphthalmia) and normal MRI (Ribeiro et al. 2006) or with alobar HPE (Ming et al. 2002). We also evaluated p.Leu360Arg, reported previously as a loss-of-function variant that is unable to complement PTCH1 function in murine Ptch1−/− fibroblast cells (Bailey et al. 2003). Considering the possibility that the PTCH1 variants in our ODA cohort could function as dominant negative alleles, we tested p.Gly509Val, a highly conserved change in the sterol-sensing domain that confers a dominant negative effect in Drosophila (Hime et al. 2004). As negative controls for the assay, we evaluated a nonsynonymous change found commonly in population controls, p.Pro1315Leu, and a second C-terminal change, p. Pro1125Leu, shown previously to restore SHH signaling readout in vitro (Bailey et al. 2003). In contrast to the significant rescue of the morphant phenotype resulting from co-injection of either WT mRNA or the negative control mRNAs (p.Pro1125Leu and p.Pro1315Leu), the morphant somite angle defect was significantly worse than WT rescue (P < 0.0001 for each co-injection vs. WT rescue) and either partially ameliorated or not significantly different from ptch1 sb1 MO alone. We observed similar results for the p.Thr1052Met change (associated with HPE) and p.Leu360Arg (functional null) (Fig. 2; Supplemental Table 7). Notably, co-injection of ptch1 sb1 MO with the mRNA bearing the p.Asp436Asn change identified in control C2 showed phenotypic rescue similar to that of the WT and negative control mRNAs, demonstrating that this change is benign. Unlike the mRNA harboring the known dominant negative p.Gly509Val, injection of WT or mutant mRNA in the absence of MO resulted in no significant defects, arguing in favor of a loss-of-function rather than a dominant-negative effect for the ODA-associated alleles. Together, these results provided in vivo evidence that all rare missense variants identified in our ODA discovery cohort, as well as the mutation associated with HPE, have a detrimental effect on PTCH1 protein activity (Supplemental Table 7), while the negative control variants p.Pro1125Leu, p.Pro1315Leu, and the p.Asp436Asn change identified in a control patient had no detectable effect on protein function.
Pathogenic PTCH1 alleles are present in an independent ODA cohort
Encouraged by these observations, we conducted bidirectional Sanger sequencing of the coding regions of PTCH1 in an independent cohort of 48 samples with ODA. We identified two additional rare heterozygous PTCH1 missense variants: p.Ile899Val in a patient with bilateral Peters anomaly, and p.Thr778Pro in an autosomal dominant AM-ASD family, each of which was absent from EVS (Table 1; Fig. 1B; Supplemental Table 4). An in vivo functional assay of each of the two additional variants demonstrated that, similar to the alleles found in our original cohort, both changes resulted in partial loss of PTCH1 function (mean somite angle 90.0° and 94.7° for p.Thr778Pro and p.Ile899Val, respectively; P < 0.0001 for each MO plus mutant mRNA co-injection versus WT rescue) (Fig. 2; Supplemental Table 6). Combined, we identified a total of seven rare heterozygous PTCH1 variants (six missense and one frameshifting) in a total of 70 individuals with ODA (10%) (Fig. 1B).
In vivo suppression of ptch1 results in microphthalmia
Concomitant with aberrant somite formation, defects in the development of the visual system represent well-documented phenotypes in zebrafish SHH pathway component mutants. Importantly, zebrafish ENU mutant models of both patched homologs display ocular abnormalities: ptch1 (lep/ptc2) mutants have vitreo-retinal abnormalities (Bibliowicz and Gross 2009); and mutation of ptch2 (blw/ptc1) results in gross defects in eye morphology with variably penetrant coloboma (Karlstrom et al. 1996; Lee et al. 2008). To determine whether ptch1 suppression produced a phenotype relevant to our ODA cohort, we evaluated ptch1 sb1 MO-injected embryos for eye size defects by measuring eye area from lateral views at 3 d post-fertilization (dpf) and 5 dpf; consistent with previous observations, we observed no significant eye size differences between sb1 morphants versus controls (data not shown; Koudijs et al. 2005). This is likely due to incomplete knockdown of endogenous transcript (Supplemental Fig. 2A). Therefore, we designed two additional MOs including a translation blocker (tb) and a splice blocker (sb2) targeting the exon 5 splice donor site of ptch1. RT-PCR monitoring of endogenous ptch1 transcript in MO-injected embryos confirmed the higher efficiency of sb2 in suppressing WT ptch1 expression in comparison to sb1 (Supplemental Fig. 2B). Next, we injected WT embryos with either ptch1 sb2 or tb MOs; embryo batches were grown to 36 h post-fertilization (hpf) and scored first for abnormal somitic shape. Similar to ptch1 sb1 MO-injected embryos, either mRNA splicing (sb2) or translational suppression (tb) resulted in obtuse somite angles significantly broader than controls (85.2° vs. 95.4° [tb] or 97.4° [sb2] for control vs. ptch1 tb or sb2 MOs, respectively; P < 0.0001) (Supplemental Fig. 3).
We then aged embryos to 3 dpf and the eye area was assessed as a percentage of WT eye area (Fig. 3A). We observed a significant reduction in eye size in both ptch1 morphant batches (1.5 ng tb MO; 2 ng sb2 MO; n = 42–52 embryos/injection; repeated three times; P < 0.001), supporting the direct role of ptch1 in ocular development. Importantly, this phenotype was validated using a CRISPR/Cas9 F0 mutant model (Fig. 3B–D). Injection of a guide RNA targeting exon 6 of the ptch1 locus induces gene-disruptive insertion-deletions (70% of embryos targeted; embryos with >65% mosaicism were selected for phenotyping) and leads to a significant reduction in eye size and retina size compared to controls (P < 0.001) (Fig. 3B,C), as described for the ptch1lep mutant (Bibliowicz and Gross 2009).
Figure 3.

Suppression of ptch1 in vivo results in microphthalmia. (A) Representative lateral images of uninjected control and ptch1 tb, ptch1 sb2, or sox2 MO-injected embryos at 3 d post-fertilization (dpf) (top). Dashed yellow line indicates area of measurement used for quantification of eye size; morphant measurements were normalized relative to control (bottom). (B) Representative lateral images of uninjected control and ptch1 guide RNA (gRNA)/Cas9 injected embryos at 3 dpf (top). ptch1 CRISPR F0 embryos were scored for eye size and retina size (bottom). (C) At 1 dpf, a representative sampling of 10 founders and two uninjected controls were selected and subjected to T7 endonuclease 1 (T7E1) assay. The appearance of T7E1 fragments < 100 bp (marked by asterisks) indicate positive gRNA targeting of exon 6 in the ptch1 locus. No T7E1 fragments were detected in uninjected control embryos. Of the 10 founders subjected to T7E1 assay, seven showed the presence of T7E1 fragments, indicating that ∼70% of founders have insertion/deletions (indels) in the exon 6 region of ptch1. (D) Multiple sequence alignment of ptch1 reference sequence to ptch1-CRISPR variants generated from PCR amplification, subsequent TA cloning, and sequencing of ptch1-gRNA/Cas9 injected embryo #6. Black bold font marks the guide target, and the PAM recognition motif is underlined. Seven PCR-cloned sequences are shown, representing three wild-type variants and all four changes detected. TA cloning and sequencing of each founder embryo indicated 10%–65% mosaicism in individual fish (n = 5 assessed).
PTCH1 is regulated directly by the most frequently mutated ODA protein, SOX2
Pursuant to the elevated prevalence of ODA patients with pathogenic PTCH1 variants, we wondered if this locus might be linked mechanistically to SOX2, as observed previously for other genes implicated in disorders of ocular development (Kamachi et al. 2001; Danno et al. 2008). We asked whether PTCH1 might be regulated transcriptionally by SOX2. First, using RNA in situ hybridization, we found that robust embryonic Ptch1 expression in the neural retina and lens persists to later stages in the adult mouse (Fig. 4), as observed in humans (Bakrania et al. 2008), and overlaps the known expression pattern of SOX2 (Hever et al. 2006). Using a physiologically relevant model of genetically modified murine stem cells overexpressing Rax (Tabata et al. 2004), we suppressed Sox2 and tested the abundance of Ptch1 message; in biological triplicate experiments, we found that Ptch1 is up-regulated significantly upon Sox2 suppression (P < 0.001) (Fig. 5A), suggesting that the Ptch1 locus might be under the transcriptional regulation of SOX2. We therefore performed chromatin immunoprecipitation (ChIP)-seq on CCE-Rx cells using an antibody against SOX2; we identified a peak in intron 15 of Ptch1, which was confirmed using targeted ChIP-qPCR on five independent samples. Importantly, amplification of the Ptch1 intron 22 negative region was equivalent when precipitated with either nonspecific IgG or SOX2 antibody, while amplification of the intron 15 region showed greater than fivefold enrichment in chromatin immunoprecipitated by the SOX2-specific antibody (P < 0.01) (Fig. 5B,C).
Figure 4.
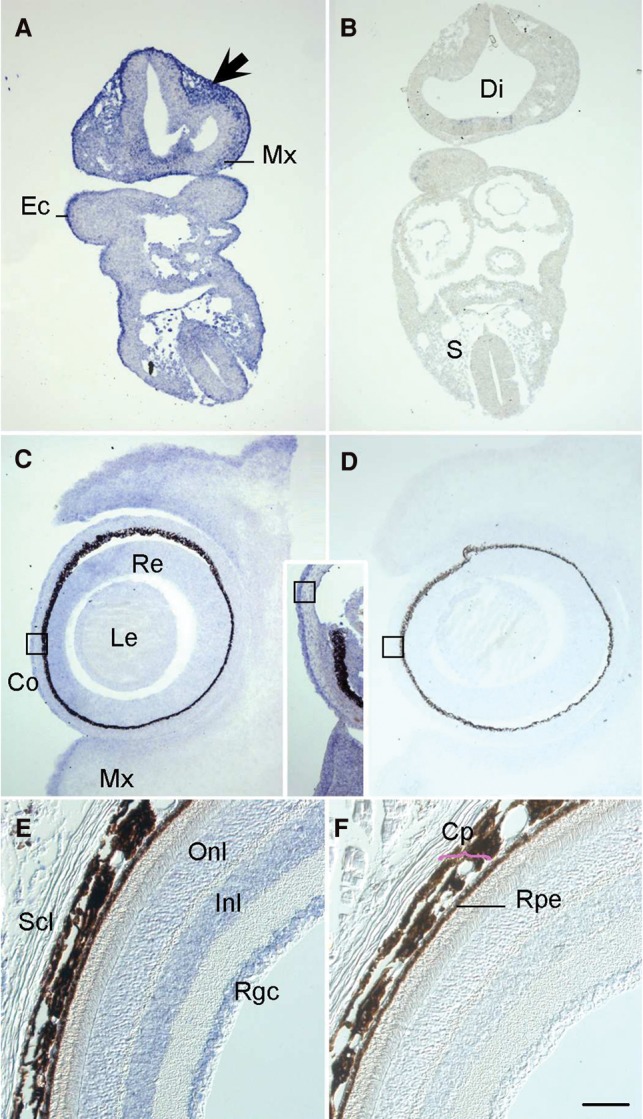
Ptch1 transcripts are present in periocular mesenchyme and the neural retina throughout eye morphogenesis and into post-natal life. (A) On embryonic day (E)9.5 in transverse mouse embryo sections, antisense riboprobes labeled with digoxygenin demonstrate Ptch1 expression in cephalic mesectoderm of neural crest origin in the head (arrow) and maxillary arch (Mx); in the basal diencephalon (Di) and basal neural tube at trunk levels, and in the somitic sclerotome (S) and basal spinal cord. Ectodermal expression is constant at all embryonic stages examined (Ec). By E11.5, the distal diencephalic infundibulum transcribes Ptch1 (data not shown) as does the subectodermal mesenchyme of the future eyelids and palate. (C) Mesenchymal Ptch1 expression continues at E13.5, particularly in the superior and inferior palpebrae (eyelids; sPa, iPa); the lateral neural retina (Re) and differentiated outer cells of the lens (Le) begin to also transcribe Ptch1, which continues throughout these structures at E15.5 (inset). By this stage, initially generalized expression in the developing cornea has become restricted to the epithelium (Co, boxes in C/inset/D). (E) In adult mouse eyes on post-natal day 50, transcripts are found within the outer and inner nuclear layers (Onl, Inl), corresponding to photoreceptor and Müller cell bodies, and within the retinal ganglion cell layer (Rgc), testifying to a post-natal role in retinal maintenance. Transcripts not observed within the stroma of the anterior chamber or the sclera (Scl). (F) Rpe, retinal pigmented epithelium; Cp; choroid plexus. Scale bar A–D, 400 µm; E, F, 200 µm. Hybridization with a sense-oriented Ptch1 probe as negative control in B, D, F.
Figure 5.

SOX2 regulates Ptch1 expression directly. (A) Targeted quantitative PCR after transfection of CCE-RX cells with a scrambled siRNA (siSc, n = 9) or a siRNA targeting Sox2 (siSox2, n = 9). This experiment showed that decreased expression of Sox2 leads to increased expression of Ptch1. (B) Results of ChIP-seq performed on CCE-RX cells using an antibody against SOX2. The Ptch1 gene structure is represented underneath the DNA fragments sequenced after ChIP, with higher peaks corresponding to more enrichment. A peak was identified in intron 15 of Ptch1 (underlined in black), while an example of an unenriched region is shown in intron 22 (underlined in gray). (C) Results obtained by ChIP-seq were confirmed using targeted ChIP-qPCR on five independent samples. Amplification of the intron 22 negative region (in gray) was equivalent whether using nonspecific IgG or a SOX2 antibody, while amplification of the intron 15 region (in black) showed greater than fivefold enrichment in chromatin immunoprecipitated by the SOX2-specific antibody. (A,C) Asterisks indicate statistical differences between the different conditions. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, Mann-Whitney U test. Error bars, SEM. (D) Quantitative (q)PCR of Shh targets ptch1 and gli1 after sox2 or ptch1 tb morpholino injection in zebrafish embryos from three biological replicates. p1 and p2 indicate primer sets targeting two different regions of ptch1. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, unpaired Student's t-test.
Next, we validated the putative regulation of ptch1 by SOX2 in an in vivo context. First, we obtained a sox2 tb-MO (Kamachi et al. 2008), and we evaluated eye size of zebrafish morphants. Consistent with both the ocular defects observed in humans with SOX2 mutations (Fantes et al. 2003; Chassaing et al. 2014), as well as the ptch1 sb2 and tb-injected embryos, we observed a significant reduction in eye area upon translational suppression of sox2 (P < 0.001) (Fig. 3A). Further, we noted a significant defect in optic closure in sox2 morphants (62% in morphants compared to 3.5% in controls, P < 0.001), differing from ptch1 morphant batches, which showed a modest proportion of embryos with coloboma (23.8% for ptch1 sb2, P = 0.04; 28% in ptch1 tb P = 0.012). Next, we monitored downstream SHH pathway activity in sox2 morphants with qPCR analysis of ptch1 and gli1. We observed significantly augmented ptch1 expression in whole-embryo lysates from sox2 morphants in comparison to controls at 2 dpf (P < 0.01) (Fig. 5D). Notably, ptch1 and gli1 expression was also increased in ptch1 tb-MO-injected embryos, suggesting that diminished PTCH1 protein results in overactivity of SHH signal transduction. These data are reminiscent of the transcriptional signature observed in vertebrate limb patterning in which SHH activation ensues after PTCH1 inactivation (Butterfield et al. 2009). Since PTCH1 is a regulator of its own expression through SHH pathway activity (Jeong and McMahon 2005), and SOX2 likely targets multiple SHH effectors (Zhao et al. 2012), deciphering the direct versus indirect roles of SOX2 protein on PTCH1 expression remains challenging. However, taken together, these data suggest that PTCH1 expression can be regulated directly by SOX2 (as indicated by ChIP-seq) and that overactivity of SHH signaling conferred through the loss of function of either gene results in the same phenotypic consequences of ODA.
Discussion
The SHH signaling pathway is associated strongly with ocular development in models ranging from insects to mammals that reflect ∼600 million years of selection (Macdonald et al. 1995; Chiang et al. 1996; Nasrallah and Golden 2001; Takabatake et al. 2002; Bakrania et al. 2008; Christiansen et al. 2012). The ptch1 zebrafish mutant displays an incompletely penetrant lens malformation phenotype (Koudijs et al. 2005); these mutants display abnormalities at the vitreo-retinal interface resulting in misshapen retinas and a reduced pupil size (Bibliowicz and Gross 2009). In addition, an ENU mutagenesis screen in zebrafish for visual system mutants identified a splice-acceptor site mutation in ptch2 that results in ocular colobomas (Lee et al. 2013), and ptch1;ptch2 double mutants have a severe ocular phenotype with absent lens development at 24 hpf and completely absent eyes at 48 hpf (Koudijs et al. 2008), phenocopying AM. Eye defects in ptch1tj222 mutants could be suppressed by pharmacologically inhibiting the Hedgehog pathway with cyclopamine, providing evidence in support of a direct involvement of SHH signaling in the manifestation of the phenotype. Last, optic morphogenesis and gene expression patterns have been compared in blind cavefish and sighted surface fish embryos, both morphological variants of the same Astyanax mexicanus species (Yamamoto et al. 2004). In contrast to surface fish embryos, cavefish embryos develop small eye primordia, which later arrest in development, degenerate, and sink into the orbits, recapitulating human secondary anophthalmia. An expansion of the SHH signaling domain in the presumptive ocular neuroepithelium resulted in hyperactivation of downstream genes, lens apoptosis, and arrested eye growth and development in cavefish embryos (Yamamoto et al. 2004). These features could be mimicked in surface fish by shh overexpression, and eye development was restored partially in cavefish embryos by using cyclopamine (Yamamoto et al. 2004). Recently, it has been demonstrated that mutations in SOX11 and SMAD7 may lead to various ODA through the activation of the SHH pathway (Zhang et al. 2013; Pillai-Kastoori et al. 2014). In humans, PTCH1 mutations have been associated previously with basal cell nevus syndrome (BCNS) (MIM#109400) and with holoprosencephaly (HPE7) (MIM#610828). Of note, ODA such as AM or ASD are part of both the BCNS (Bree and Shah 2011) and HPE (Pineda-Alvarez et al. 2011) phenotypes.
Our results show that the mutations identified in our ODA cohort lead to a loss of function of PTCH1 and increased SHH signaling. We also show that inhibition of SOX2 can drive overactivity of the SHH signaling pathway to produce the same ocular phenotypic outcome. Observations from our transient ptch1 zebrafish models exemplify the dosage sensitivity of PTCH1 in SHH signal transduction. The subeffective sb1 results in normal eye size but a somite defect that can be rescued by co-injection with human PTCH1 mRNA; however, the efficient sb2 and tb MOs produced both a somite defect as well as a reduction in eye area in 3-dpf zebrafish embryos. Overexpression of human PTCH1 mRNA in zebrafish embryos did not lead to an ocular phenotype (data not shown), suggesting that overexpression of this SHH target gene alone may not be sufficient to cause ODA. Moreover, although we demonstrated that Sox2 regulates ptch1 expression, it is probably not the primary mechanism governing eye development anomalies due to SOX2 mutations, since SOX2 is also involved in regulation of numerous genes including ocular developmental effectors such as PAX6, OTX2, and RAX (Hever et al. 2006; Danno et al. 2008).
Various ocular phenotypes (microphthalmia, colobomatous microphthalmia, and anterior segment dysgenesis) with incomplete penetrance are evident among our ODA cohort harboring PTCH1 variants. Such a myriad of ocular phenotypes has already been associated with mutations in most of the genes involved in ODA (Slavotinek 2011), especially genes involved in the SHH pathway such as SHH, GLI2, and PTCH1 (Roessler et al. 2003; Ragge et al. 2005b; Bakrania et al. 2010). The wide phenotypic spectrum within our ODA cohort, coupled to the incomplete penetrance observed among families, suggests that additional factors contribute to the phenotype. Such factors may include the other 45 genes harboring rare variants in the initial cohort of 22 ODA samples. However, their rarity in cases and undetectable enrichment of variation in comparison to controls suggests that their contribution will be modest; increased sample numbers and robust experimental models to test epistasis are required to demonstrate their involvement, if any, in ODA. Finally, phenotypic variability in ODA may also be explained by environmental factors, as suggested by the observation that mice heterozygous for a Ptch1 null mutation had a fourfold higher risk to develop congenital malformations (including microphthalmia and anophthalmia) compared to WT mice when exposed to ionizing radiation during organogenesis (Hahn et al. 1998). More recently, mice heterozygous for a Ptch1 null mutation were shown to develop spontaneous cataracts and increased susceptibility to cataract induction by early post-natal exposure to ionizing radiation compared to their WT counterparts (De Stefano et al. 2015).
In summary, high-throughput sequencing of candidate genes in ODA identified PTCH1 as a significant contributor to congenital ocular malformations (10% in our cohort), placing it similar to its transcriptional regulator, SOX2, in terms of genetic burden to this phenotypic category (Fantes et al. 2003). Importantly, this study highlights the importance of a multifaceted approach toward identifying genetic contributors to traits such as ODA that are hallmarked by incomplete penetrance and genetic heterogeneity, especially when the cohort size is modest due to low disease frequency in the population. This study exemplifies how, together, a combined candidate gene sequencing approach, in vivo functional assessment of allele pathogenicity, and placement to a known disease gene network provides robust interpretive data that would not have been possible to achieve through genetic studies alone.
Methods
Candidate gene selection
We selected 407 candidate genes based on evidence for putative involvement in ODA (Supplemental Table 1). These genes were (1) linked to normal or abnormal ocular development in vertebrates and/or invertebrates in the literature, or (2) likely to be regulated by the SOX2 transcription factor according to transcriptomic and ChIP-seq analysis.
Patients
Seventy ODA patients (22 in the discovery cohort, 48 in the replication cohort) and two positive controls with known mutations were enrolled in this study. A signed informed consent was obtained from each participant, which adhered to the tenets of the Declaration of Helsinki and was approved by the local Ethics Committee (CPP Sud-Ouest and Outre-Mer II). Ocular phenotypes of the 22 patients included in the first cohort were isolated ASD (n = 6), isolated AM (n = 4), AM with ASD (n = 6), or AM with coloboma (n = 6) (reviewed in Supplemental Table 2). Ocular phenotypes of the 48 patients included in the second cohort were isolated ASD (n = 14), isolated AM (n = 17), AM with ASD (n = 9), or AM with coloboma (n = 8).
Targeted enrichment and high-throughput DNA sequencing
A custom-made SureSelect oligonucleotide probe library was designed to capture the exons of 407 candidate genes (Supplemental Table 1). The probe library also aimed to capture 880 kb of potential regulatory sequences (i.e., noncoding region located within 20 kb of the 407 genes and conserved among species). A total of 56,059 probes, covering 2.46 Mb, were designed and synthesized. Sequence capture, enrichment, and elution were performed according to the manufacturer's instructions (SureSelect, Agilent). Each eluted-enriched DNA sample was then sequenced on an Illumina GAIIx as paired-end 75-bp reads (Integragen). Sequence reads were aligned to the reference human genome (UCSC hg19) using commercially available software (CASAVA1.7, Illumina) and the ELANDv2 alignment algorithm. The mean coverage was 325× with 96.9% of the targeted sequences over 10× and 93.4% over 25×.
Filtering strategy
All variants reported were filtered to ensure an optimal prioritization of candidate mutations (Supplemental Table 3). We first filtered out variants that did not meet the quality criteria (array confidence < 0.3, sequence read depth < 10, and sequence base quality < 10). We then filtered out all variants present in the local, in-house exome sequencing database (200 exomes) as well as in dbSNP132, 1000 Genomes Project, and the HapMap Project databases. Only exonic and splice-site variants were retained; we removed synonymous variants and variants predicted to be benign by the PolyPhen-2 software. The presence of the final selected variants was confirmed by Sanger sequencing.
Mutational burden analysis
We determined the mutational burden for the 10 genes identified in the targeted resequencing of ODA samples harboring multiple rare (<1% alternate allele frequency) functional variants (frameshift, nonsense, and splicing variants were considered as damaging; missense variants were classified as damaging or not based on PolyPhen-2) in ODA cases (n = 22 unknown + 2 positive controls) vs. Exome Variant Server controls (n = 6500). EVS was accessed in November 2013, and a χ2 test was used for comparisons.
PTCH1 molecular screening
To support further the involvement of PTCH1 lesions in ODA, we screened this locus in a new cohort of 48 patients by direct bidirectional Sanger sequencing. Primers used to amplify the 23 coding exons and intron-exon splice junctions are listed in Supplemental Table 7.
Zebrafish embryo microinjection and manipulation
We obtained a previously published (Koudijs et al. 2005) morpholino antisense oligonucleotide (MO; GeneTools) targeting the splice donor site of ptch1 exon 3 (sb1) and designed two additional MOs to suppress endogenous ptch1 expression; a translation blocker and a splice blocker targeting exon 5 (sb2). We also obtained a previously described sox2 tb (Kamachi et al. 2008; Supplemental Table 6). One nanoliter of the indicated cocktail was injected into wild-type (WT) zebrafish embryos at the one- to four-cell stage (n = 38–58 embryos/injection, repeated at least twice for MO plus mRNA injections and seven times for ptch1 sb1 alone; with masked scoring).
For somite evaluation, embryos were reared at 28.5°C and imaged live at 36 h post-fertilization. To generate human PTCH1 WT and mutant mRNA, we first obtained a full-length open reading frame (ORF) construct (clone ID: 100016192; OpenBiosystems). We generated a stop codon and subsequently introduced additional nonsynonymous changes using site-directed mutagenesis (QuikChange; Agilent). We then transferred sequenced-confirmed PTCH1 ORFs into the pCS2+ plasmid (LR clonase II; Life Technologies), linearized with NotI, and performed in vitro transcription with the SP6 mMessage mMachine kit (Ambion). For rescue experiments, 12 ng of MO and 100 pg of PTCH1 mRNA were injected, respectively. Live embryo imaging of lateral views was conducted on a Nikon AZ100 microscope at 6× magnification facilitated by NIS Elements software. Somite angle measurements were taken at the midpoint between the proximal hindgut and the anus and recorded from the resulting images using ImageJ software. For eye size measurements, embryos were fixed in 4% PFA overnight at 3 dpf, and lateral images were acquired on an SMZ745T microscope facilitated by NIS Elements software. The area of the retina and pupil was quantified using ImageJ software. Embryo batches were compared for statistical significance using Student's t-test.
Generation of CRISPR/Cas9 ptch1 mutants
ptch1 gRNA targeting the antisense strand of ptch1 exon 6 was produced by synthesizing and annealing two oligonucleotides, gRNA3 F: TAGGGAAGCCCATCGGATCGAAGT and gRNA3 R: AAACACTTCGATCCGATGGGCTTC. The annealed oligos were then ligated to a BsmBI-digested T7cas9sgRNA2 vector overnight at room temperature (NEB). Two microliters of the reaction were used for transformation. Prior to transcription, the gRNA vector was linearized with BamHI. gRNA was transcribed using the MEGAshortscript T7 kit (AM1354, Life Technologies) and purified using alcohol precipitation. A total of 150 pg of ptch1 gRNA and 300pg of Cas9 protein (PNA Bio) were co-injected into single cells of one-cell stage embryos. For the T7 endonuclease I assay, genomic DNA was prepared from 1-dpf embryos by digestion in 5 µg/mL proteinase K for 90 min at 65°C, followed by 15 min at 95°C. A short stretch of the genomic region (∼150 bp) flanking the ptch1 gRNA target site was PCR-amplified from the genomic DNA (Fwd: TAGTGGCAAACCCCCATTAC, Rev: CCTTGACCCACATCTGCTTT). The PCR amplicon was then denatured slowly and reannealed to facilitate heteroduplex formation. The reannealed amplicon was then digested with 5 units of T7 endonuclease I (New England Biolabs) at 37°C for 90 min. The samples were resolved by electrophoresis and ethidium bromide staining in a 3.0% agarose gel. PCR products were cloned (TOPO-TA cloning, Life Technologies), and individual colonies were sequenced to determine insertion-deletion sites and to estimate the extent of mosaicism in F0 founders.
Chromatin immunoprecipitation (ChIP)
We performed quantitative ChIP-seq in murine stem cells genetically modified to overexpress Rax (retina and anterior neural fold homeobox) (CCE-Rx cells, a kind gift from S. Watanabe) (Tabata et al. 2004). These cells have the ability to differentiate into retinal ganglion cells and were cultured using the standard procedures (Tabata et al. 2004). Two × 106 resuspended CCE-Rx cells were cultured in LIF-free medium on 10-cm bacterial plates. Forty-eight hours later, CCE-Rx embryoid bodies were treated with formaldehyde for 10 min, chromatin was prepared, and ChIP was performed according to the Upstate (Millipore) protocol, using 10 μg of anti-SOX2 antibody (sc-17320, Santa Cruz Biotechnology) or mouse IgG (PP54, Millipore) as a control. ChIP-seq libraries were prepared and sequenced using the standard Illumina protocol. Peaks were called with SeqMonk using the contig generator function. Inside intron 15 of Ptch1, a peak was identified in the SOX2-immunoprecipitated sample. The online JASPAR database (http://jaspar.genereg.net/; Mathelier et al. 2014) confirmed the presence of a putative SOX2 binding site within the peak sequence. Amplicons corresponding to the Ptch1 intron 15 SOX2 ChIP-seq peak and to a region not predicted to bind SOX2 in intron 22 (see Supplemental Table 8 for primers) were selected for validation. These amplicons were tested in five independent samples immunoprecipitated either with SOX2 or mouse IgG antibodies.
Quantitative RT-PCR
Quantitative PCR analysis was performed in CCE-Rx cells and zebrafish embryos to confirm Ptch1 regulation by SOX2. CCE-Rx cells were cultured using the standard procedures (Tabata et al. 2004) and transfected as previously described (Ko et al. 2009) either with an siRNA targeting Sox2 mRNA or with a scrambled siRNA (Stealth siRNA MSS277200 and Negative Control Medium GC, respectively, Invitrogen). Quantitative real-time PCR analyses on complementary DNA transcribed from total RNA showed that the remaining Sox2 expression was 50% at 24 h post-transfection and 75% at 48 h post-transfection. Forty-eight hours after transfection of either the siSOX2 (n = 9) or the siScramble (n = 9), total RNA was isolated using the GenElute Mammalian Total RNA Miniprep kit (Sigma-Aldrich). Samples were used to analyze Ptch1 expression levels in the siSox2 samples compared to controls (Supplemental Table 8 for primers). Zebrafish embryos injected with 1.5 ng ptch1 tb MO or 2.5 ng sox2 tb MO were grown for 48 h before isolation of total RNA using TRIzol (Invitrogen) following the manufacturer's protocol. cDNA was synthesized from 1 μg of total RNA using SuperScript III (Invitrogen). Real-time PCR was performed with Power SYBR Green PCR Master Mix on a 7900HT (Applied Biosystems) and analyzed with a Sequence Detection System software package version 2.3 (Applied Biosystems) (Supplemental Table 8 for primers).
In situ hybridization
In situ hybridization was carried out according to standard protocols (Goodrich et al. 1996) on paraffin sections using a murine digoxygenin-labeled Ptch1 riboprobe (Chotteau-Lelievre et al. 2006) synthesized in antisense and sense (negative control) orientations.
Data access
The data from transcriptomic and ChIP-seq from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE74600. All variants identified during this study have been submitted to the ClinVar database (www.ncbi.nlm.nih.gov/clinvar; Landrum et al. 2016), and ClinVar accession numbers are listed in Table 1 (PTCH1 variants identified in ODA patients) and in Supplemental Table S4 (final prioritized variants in patients with ODA).
Supplementary Material
Acknowledgments
The authors thank the families, Christine Peres, Beatrice Atlan, and Jason Willer for their technical assistance. The authors also thank the following physicians: C. Baumann, M. Mathieu-Dramard, B. Duban-Bedu, C. Francannet, P. Jalbert, S. Julia, B. Leheup, S. Lyonnet, S. Mercier, M. Privat, P. Ribaï, and A. Toutain. This work was supported by grants from the Clinical Research Hospital Program from the French Ministry of Health (PHRC 09 109 01) and from Retina France.
Author contributions: N.C., E.E.D, H.C.E., S.F., and P.C. designed and directed the study. N.C., E.E.D, N.K., H.C.E., S.F., and P.C. wrote the manuscript. C.V-D., L.P., C.C., D.L., M.R., J-L.D., H.D., and J.K. collected samples and provided the subjects’ clinical information. N.C., A.C., V.D., A.D., and S.L. performed ChIP and transcriptomic analyses and confirmation of NGS results. H.C.E. performed HIS analyses. V.D. performed PTCH1 molecular screening. E.E.D, K.L.M, A.R.N, and N.K. performed zebrafish studies.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196048.115.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey EC, Zhou L, Johnson RL. 2003. Several human PATCHED1 mutations block protein maturation. Cancer Res 63: 1636–1638. [PubMed] [Google Scholar]
- Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Martin A, Williams S, Lindley V, et al. 2008. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet 82: 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Ugur Iseri SA, Wyatt AW, Bunyan DJ, Lam WW, Salt A, Ramsay J, Robinson DO, Ragge NK. 2010. Sonic hedgehog mutations are an uncommon cause of developmental eye anomalies. Am J Med Genet A 152A: 1310–1313. [DOI] [PubMed] [Google Scholar]
- Bermejo E, Martinez-Frias ML. 1998. Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet 75: 497–504. [PubMed] [Google Scholar]
- Bibliowicz J, Gross JM. 2009. Expanded progenitor populations, vitreo-retinal abnormalities, and Müller glial reactivity in the zebrafish leprechaun/patched2 retina. BMC Dev Biol 9: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bree AF, Shah MR. 2011. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A 155A: 2091–2097. [DOI] [PubMed] [Google Scholar]
- Bumcrot DA, McMahon AP. 1995. Somite differentiation. Sonic signals somites. Curr Biol 5: 612–614. [DOI] [PubMed] [Google Scholar]
- Butterfield NC, Metzis V, McGlinn E, Bruce SJ, Wainwright BJ, Wicking C. 2009. Patched 1 is a crucial determinant of asymmetry and digit number in the vertebrate limb. Development 136: 3515–3524. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Vigouroux A, Calvas P. 2009. Mutations in the newly identified RAX regulatory sequence are not a frequent cause of micro/anophthalmia. Genet Test Mol Biomarkers 13: 289–290. [DOI] [PubMed] [Google Scholar]
- Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, Boute-Benejean O, Dollfus H, Duban-Bedu B, Gilbert-Dussardier B, et al. 2014. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin Genet 86: 326–334. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. 1996. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383: 407–413. [DOI] [PubMed] [Google Scholar]
- Chotteau-Lelievre A, Dolle P, Gofflot F. 2006. Expression analysis of murine genes using in situ hybridization with radioactive and nonradioactively labeled RNA probes. Methods Mol Biol 326: 61–87. [DOI] [PubMed] [Google Scholar]
- Christiansen AE, Ding T, Bergmann A. 2012. Ligand-independent activation of the Hedgehog pathway displays non-cell autonomous proliferation during eye development in Drosophila. Mech Dev 129: 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danno H, Michiue T, Hitachi K, Yukita A, Ishiura S, Asashima M. 2008. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate Rax expression. Proc Natl Acad Sci 105: 5408–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano I, Tanno B, Giardullo P, Leonardi S, Pasquali E, Antonelli F, Tanori M, Casciati A, Pazzaglia S, Saran A, et al. 2015. The Patched 1 tumor-suppressor gene protects the mouse lens from spontaneous and radiation-induced cataract. Am J Pathol 185: 85–95. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, et al. 2003. Mutations in SOX2 cause anophthalmia. Nat Genet 33: 461–463. [DOI] [PubMed] [Google Scholar]
- Fares-Taie L, Gerber S, Chassaing N, Clayton-Smith J, Hanein S, Silva E, Serey M, Serre V, Gerard X, Baumann C, et al. 2013. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am J Hum Genet 92: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferda Percin E, Ploder LA, Yu JJ, Arici K, Horsford DJ, Rutherford A, Bapat B, Cox DW, Duncan AM, Kalnins VI, et al. 2000. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet 25: 397–401. [DOI] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, et al. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493: 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. 1996. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 10: 301–312. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. 1998. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med 4: 619–622. [DOI] [PubMed] [Google Scholar]
- Hever AM, Williamson KA, van Heyningen V. 2006. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet 69: 459–470. [DOI] [PubMed] [Google Scholar]
- Hime GR, Lada H, Fietz MJ, Gillies S, Passmore A, Wicking C, Wainwright BJ. 2004. Functional analysis in Drosophila indicates that the NBCCS/PTCH1 mutation G509V results in activation of smoothened through a dominant-negative mechanism. Dev Dyn 229: 780–790. [DOI] [PubMed] [Google Scholar]
- The International HapMap 3 Consortium, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, et al. 2010. Integrating common and rare genetic variation in diverse human populations. Nature 467: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, McMahon AP. 2005. Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development 132: 143–154. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Tanouchi A, Sekido R, Kondoh H. 2001. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev 15: 1272–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamachi Y, Okuda Y, Kondoh H. 2008. Quantitative assessment of the knockdown efficiency of morpholino antisense oligonucleotides in zebrafish embryos using a luciferase assay. Genesis 46: 1–7. [DOI] [PubMed] [Google Scholar]
- Karlstrom RO, Trowe T, Klostermann S, Baier H, Brand M, Crawford AD, Grunewald B, Haffter P, Hoffmann H, Meyer SU, et al. 1996. Zebrafish mutations affecting retinotectal axon pathfinding. Development 123: 427–438. [DOI] [PubMed] [Google Scholar]
- Ko BS, Chang TC, Shyue SK, Chen YC, Liou JY. 2009. An efficient transfection method for mouse embryonic stem cells. Gene Ther 16: 154–158. [DOI] [PubMed] [Google Scholar]
- Koudijs MJ, den Broeder MJ, Keijser A, Wienholds E, Houwing S, van Rooijen EM, Geisler R, van Eeden FJ. 2005. The zebrafish mutants dre, uki, and lep encode negative regulators of the hedgehog signaling pathway. PLoS Genetics 1: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudijs MJ, den Broeder MJ, Groot E, van Eeden FJ. 2008. Genetic analysis of the two zebrafish patched homologues identifies novel roles for the hedgehog signaling pathway. BMC Dev Biol 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. 2016. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Willer JR, Willer GB, Smith K, Gregg RG, Gross JM. 2008. Zebrafish blowout provides genetic evidence for Patched1-mediated negative regulation of Hedgehog signaling within the proximal optic vesicle of the vertebrate eye. Dev Biol 319: 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Cox BD, Daly CM, Lee C, Nuckels RJ, Tittle RK, Uribe RA, Gross JM. 2013. An ENU mutagenesis screen in zebrafish for visual system mutants identifies a novel splice-acceptor site mutation in patched2 that results in Colobomas. Invest Ophthalmol Vis Sci 53: 8214–8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Wu CI, Luo CC. 1984. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol 21: 58–71. [DOI] [PubMed] [Google Scholar]
- Macdonald R, Barth KA, Xu Q, Holder N, Mikkola I, Wilson SW. 1995. Midline signalling is required for Pax gene regulation and patterning of the eyes. Development 121: 3267–3278. [DOI] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. 2014. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42: D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. 2002. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet 110: 297–301. [DOI] [PubMed] [Google Scholar]
- Nasrallah I, Golden JA. 2001. Brain, eye, and face defects as a result of ectopic localization of Sonic hedgehog protein in the developing rostral neural tube. Teratology 64: 107–113. [DOI] [PubMed] [Google Scholar]
- Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nurnberg G, Brasch F, Schirmer-Zimmermann H, Tolmie JL, Chitayat D, et al. 2007. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet 80: 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasutto F, Mauri L, Popp B, Sticht H, Ekici A, Piozzi E, Bonfante A, Penco S, Schlotzer-Schrehardt U, Reis A. 2015. Whole exome sequencing reveals a novel de novo FOXC1 mutation in a patient with unrecognized Axenfeld–Rieger syndrome and glaucoma. Gene 568: 76–80. [DOI] [PubMed] [Google Scholar]
- Pillai-Kastoori L, Wen W, Wilson SG, Strachan E, Lo-Castro A, Fichera M, Musumeci SA, Lehmann OJ, Morris AC. 2014. Sox11 is required to maintain proper levels of Hedgehog signaling during vertebrate ocular morphogenesis. PLoS Genet 10: e1004491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Alvarez DE, Solomon BD, Roessler E, Balog JZ, Hadley DW, Zein WM, Hadsall CK, Brooks BP, Muenke M. 2011. A broad range of ophthalmologic anomalies is part of the holoprosencephaly spectrum. Am J Med Genet A 155A: 2713–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, Uz E, Buzas D, Gomes C, Patrier S, et al. 2011. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 43: 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, et al. 2005a. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 76: 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragge NK, Salt A, Collin JR, Michalski A, Farndon PA. 2005b. Gorlin syndrome: The PTCH gene links ocular developmental defects and tumour formation. Br J Ophthalmol 89: 988–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, Semina EV. 2011. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol 22: 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro LA, Murray JC, Richieri-Costa A. 2006. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A 140: 2584–2586. [DOI] [PubMed] [Google Scholar]
- Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. 2003. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci 100: 13424–13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauerte HE, van Eeden FJ, Fricke C, Odenthal J, Strahle U, Haffter P. 1998. Sonic hedgehog is not required for the induction of medial floor plate cells in the zebrafish. Development 125: 2983–2993. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM. 2011. Eye development genes and known syndromes. Mol Genet Metab 104: 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour M, Chitayat D, Caron V, Chassaing N, Bitoun P, Patry L, Cordier MP, Capo-Chichi JM, Francannet C, Calvas P, et al. 2013. Recessive and dominant mutations in retinoic acid receptor β in cases with microphthalmia and diaphragmatic hernia. Am J Hum Genet 93: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata Y, Ouchi Y, Kamiya H, Manabe T, Arai K, Watanabe S. 2004. Specification of the retinal fate of mouse embryonic stem cells by ectopic expression of Rx/rax, a homeobox gene. Mol Cell Biol 24: 4513–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabatake Y, Takabatake T, Sasagawa S, Takeshima K. 2002. Conserved expression control and shared activity between cognate T-box genes Tbx2 and Tbx3 in connection with Sonic hedgehog signaling during Xenopus eye development. Dev Growth Differ 44: 257–271. [DOI] [PubMed] [Google Scholar]
- Tay SY, Ingham PW, Roy S. 2005. A homologue of the Drosophila kinesin-like protein Costal2 regulates Hedgehog signal transduction in the vertebrate embryo. Development 132: 625–634. [DOI] [PubMed] [Google Scholar]
- Varga ZM, Amores A, Lewis KE, Yan YL, Postlethwait JH, Eisen JS, Westerfield M. 2001. Zebrafish smoothened functions in ventral neural tube specification and axon tract formation. Development 128: 3497–3509. [DOI] [PubMed] [Google Scholar]
- Verma AS, Fitzpatrick DR. 2007. Anophthalmia and microphthalmia. Orphanet J Rare Dis 2: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villavicencio EH, Walterhouse DO, Iannaccone PM. 2000. The Sonic hedgehog–Patched–Gli pathway in human development and disease. Am J Hum Genet 67: 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weh E, Reis LM, Happ HC, Levin AV, Wheeler PG, David KL, Carney E, Angle B, Hauser N, Semina EV. 2014. Whole exome sequence analysis of Peters anomaly. Hum Genet 133: 1497–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AH, Kousseff BG, Ross EA, Longbottom J. 1989. Simple microphthalmos. Arch Ophthalmol 107: 1625–1630. [DOI] [PubMed] [Google Scholar]
- Wolff C, Roy S, Ingham PW. 2003. Multiple muscle cell identities induced by distinct levels and timing of hedgehog activity in the zebrafish embryo. Curr Biol 13: 1169–1181. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Stock DW, Jeffery WR. 2004. Hedgehog signalling controls eye degeneration in blind cavefish. Nature 431: 844–847. [DOI] [PubMed] [Google Scholar]
- Zhang R, Huang H, Cao P, Wang Z, Chen Y, Pan Y. 2013. Sma- and Mad-related protein 7 (Smad7) is required for embryonic eye development in the mouse. J Biol Chem 288: 10275–10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zevallos SE, Rizzoti K, Jeong Y, Lovell-Badge R, Epstein DJ. 2012. Disruption of SoxB1-dependent Sonic hedgehog expression in the hypothalamus causes septo-optic dysplasia. Dev Cell 22: 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.