

Abstract
We describe here a forward genetic screen to investigate the biogenesis, mode of action, and biological function of miRNA-mediated RNA silencing in the model algal species, Chlamydomonas reinhardtii. Among the mutants from this screen, there were three at Dicer-like 3 that failed to produce both miRNAs and siRNAs and others affecting diverse post-biogenesis stages of miRNA-mediated silencing. The DCL3-dependent siRNAs fell into several classes including transposon- and repeat-derived siRNAs as in higher plants. The DCL3-dependent miRNAs differ from those of higher plants, however, in that many of them are derived from mRNAs or from the introns of pre-mRNAs. Transcriptome analysis of the wild-type and dcl3 mutant strains revealed a further difference from higher plants in that the sRNAs are rarely negative switches of mRNA accumulation. The few transcripts that were more abundant in dcl3 mutant strains than in wild-type cells were not due to sRNA-targeted RNA degradation but to direct DCL3 cleavage of miRNA and siRNA precursor structures embedded in the untranslated (and translated) regions of the mRNAs. Our analysis reveals that the miRNA-mediated RNA silencing in C. reinhardtii differs from that of higher plants and informs about the evolution and function of this pathway in eukaryotes.
RNA silencing in eukaryotes controls gene expression and protects against viruses and transposons (Baulcombe 2004). Small (s)RNAs of 20–31 nucleotides (nt) form RNA-induced silencing complexes (RISC) with proteins of the Piwi/Argonaute family (AGO) and they guide these effector proteins to their targets by complementary base-pairing (Meister 2013). AGO proteins achieve posttranscriptional gene silencing (PTGS) by target transcript degradation or translational repression and they promote transcriptional gene silencing (TGS) via chromatin/DNA modifications (Brodersen and Voinnet 2009; Castel and Martienssen 2013).
Corresponding to these various RNA silencing pathways, there are multiple types of sRNA that differ in their biogenesis mechanism or in their associated AGO isoform. These sRNAs include small interfering (si)RNAs, micro(mi)RNAs, and piwi-interacting (pi)RNAs (Ghildiyal and Zamore 2009). The siRNAs and miRNAs are produced by the action of RNase III Dicer (Dcr) or Dicer-like (DCL) proteins on fully- or near-complementary double-stranded (ds)RNA molecules (Carthew and Sontheimer 2009), whereas piRNAs are Dcr-independent and have single-stranded RNA precursors (Iwasaki et al. 2015).
The miRNAs of plants and animals are similar: They are 20–24 nt and derived from precursor RNAs with stem–loop structures (Brodersen and Voinnet 2009). However, there are also clear differences. The biogenesis of animal miRNAs, for example, involves processing of a primary miRNA transcript by various nucleases, including the microprocessor Drosha/DGCR8 to form a miRNA precursor that is then cleaved by Dcr in the cytoplasm (Bartel 2009). In contrast, plant miRNAs are processed in a nuclear DCL-mediated mechanism (Brodersen and Voinnet 2009). There are other differences based on the composition of the AGO complex, requirement for sequence complementarity between the miRNA and its target, and the ways that translation is suppressed. These differences prompted the speculation that miRNAs have evolved independently in plant and animal lineages (Axtell et al. 2011).
Most information about miRNAs is from multicellular organisms, although they are also present in unicellular organisms, including the green alga Chlamydomonas reinhardtii (Molnár et al. 2007; Zhao et al. 2007), protozoans Giardia lamblia (Saraiya and Wang 2008), Trichomonas vaginalis (Chen et al. 2009), Pentatrichomonas hominis (Huang et al. 2012), Symbiodinium microadriaticum (Baumgarten et al. 2013), Entamoeba histolytica (Mar-Aguilar et al. 2013), Trypanosoma brucei (Mallick et al. 2008), and Toxoplasma gondii (Braun et al. 2010). These organisms are descended from ancient ancestors of multicellular organisms, and they provide an opportunity to test hypotheses about the origin of miRNA pathways.
Here we focus on C. reinhardtii, which is from a lineage that diverged from the ancestor of land plants more than one billion years ago (Yoon et al. 2004). It has a complex RNA silencing machinery with three DCLs (DCL1-3) and three AGOs (AGO1-3) (Merchant et al. 2007; Casas-Mollano et al. 2008). These proteins are not encoded by orthologs of genes in higher plants, although it is well established that C. reinhardtii sRNAs, including miRNAs, are like those of higher plants in that they direct cleavage of their mRNA targets (Molnár et al. 2007; Zhao et al. 2007). To investigate the biogenesis, mode of action, and biological function of miRNAs in C. reinhardtii we have carried out a forward genetic screen in this genetically tractable organism.
Results
Isolation of RNA silencing mutants in C. reinhardtii
To characterize mechanisms and biological function of RNA silencing in C. reinhardtii we used a reporter system in which a nitrate-inducible artificial (a)miRNA was targeted to the 5′ region of the phytoene synthase (PSY) mRNA (ni-amiRNA-PSY) (Fig. 1A; Supplemental Fig. S1). The amiRNA was readily detectable by Northern blotting in cells using nitrate rather than ammonium as a source of nitrogen and, correspondingly, from qRT-PCR the PSY mRNA was less abundant in nitrate-grown cells (Supplemental Fig. S1A,C). From these data we conclude that the amiRNA down-regulated the PSY mRNA. We confirmed this conclusion by 5′-RACE detection of PSY mRNA cleavage products at the amiRNA target site (Supplemental Fig. S1D).
Figure 1.
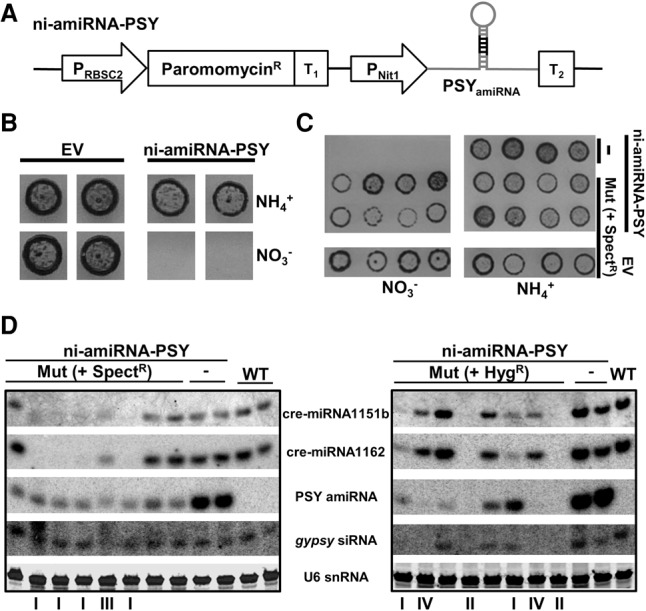
Screening and isolation of mutants affected in miRNA-mediated RNA silencing. (A) Schematic representation of the artificial miRNA construct used to transform the wild-type strain. Transgenic lines carrying this cassette were further screened by random insertional mutagenesis: (PRBSC2) RuBisCO small subunit (RBSC)2 promoter; (ParomomycinR) Streptomyces rimosus AphVIII coding gene; (T1) RBSC2 transcription terminator; (PNit1) nitrate reductase promoter; (PSYamiRNA) modified version of cre-miR1157 that carries a miRNA against the phytoene synthase; (T2) RLP12 transcription terminator. (B) Selective cell death of transgenic lines expressing the PSY amiRNA in the presence of nitrate, but not ammonium, as the sole nitrogen source. Transgenic lines carrying the empty amiRNA vector (EV) were used as control. (C) Growth in high light conditions of mutagenized (SpectR) and nonmutagenized (-) reporter lines (PSYamiRNA) in solid media containing either nitrate or ammonium as sole nitrogen source. Transgenic lines carrying the empty amiRNA vector (EV) and further transformed with the spectinomycin resistance cassette were used as control. (D) Detection by Northern blot of diverse small RNAs in total RNA samples from the indicated mutants and controls. These mutants were obtained by random insertional mutagenesis of either spectinomycin or hygromycin resistance cassettes. The mutants were grouped (I–IV) (Supplemental Table S1) based on the molecular phenotype. The two displayed mutants belonging to the group II correspond to the characterized mutant 47 (dcl3-2) and mutant 51 (dcl3-3).
The amiRNA-producing cells died in light in the presence of nitrate (Fig. 1B) most likely due to silencing of PSY mRNA by the amiRNA and to the consequent lack of the photoprotective function of PSY (McCarthy et al. 2004). Consistent with this interpretation the cell death was dependent on the light intensity (Supplemental Fig. S1B), and it did not occur in cells using ammonium rather than nitrate as nitrogen source where the amiRNA promoter is repressed (Fig. 1B). We therefore used the light-induced cell death to screen for mutants in amiRNA silencing pathways.
Two independent amiRNA lines (named A4-1 and E9-3) were mutagenized by random genomic insertion of either spectinomycin or hygromycin resistance cassettes. The mutagenized cells grew well on solid medium with ammonium as nitrogen source but, unlike cultures of wild-type cells expressing the amiRNA, there were some cells that grew in nitrate (Fig. 1C). We hypothesized that amiRNA silencing of PSY had failed in these nitrate-tolerant cells due to a mutation either in the amiRNA gene, in the amiRNA biogenesis pathways, or in the effector machinery of amiRNA silencing.
To further characterize 48 of these nitrate-tolerant lines, we used Northern blotting with probes for the PSY amiRNA, cre-miR1151b, cre-miR1162, and for a siRNA from a gypsy transposon locus (Fig. 1D). Of these lines, 22 were depleted in the PSY amiRNA but without any effect on the endogenous sRNAs. These mutants are likely to affect the amiRNA gene and were not analyzed further. In the other lines, the amiRNA and endogenous sRNAs were reduced to different extents: Group I mutants had reduced levels of miRNAs but not the siRNA; group II were depleted for all tested sRNAs; group III sRNAs were slightly less abundant than in wild-type cells and they were heterodispersed in size; and group IV sRNAs were depleted for the gypsy siRNA and amiRNA and had reduced levels of endogenous miRNAs (Fig. 1D; Supplemental Table S1). From these data, we conclude that there may be separate but overlapping pathways for miRNA- and siRNA-mediated silencing. The mutant cells grew well, and we further conclude that these RNA silencing pathways are not required for normal growth of the algal cells in solid or liquid media in normal laboratory conditions.
Mapping of DCL3 mutants
Because mutant strains in group II displayed the most severe molecular phenotype, we decided to characterize them in detail. Restriction enzyme site-directed amplification (RESDA-)PCR revealed that three group II mutations were in DCL3. The mutagenic inserts were in exon 29 (mutant 51) (Figs. 1D, 2A); the 3′ UTR with a deletion that extended to the 5′ end of its neighbour gene Cre07.g345900 (mutant 47) (Fig. 1D); and exon 6 (mutant 37) (Supplemental Fig. S2A). The PSY mRNA was at wild-type levels in these lines (Supplemental Fig. S2B) and, corresponding to the absence of the amiRNA, we could not detect the miRNA cleavage products of the PSY mRNA (Supplemental Fig. S2C).
Figure 2.
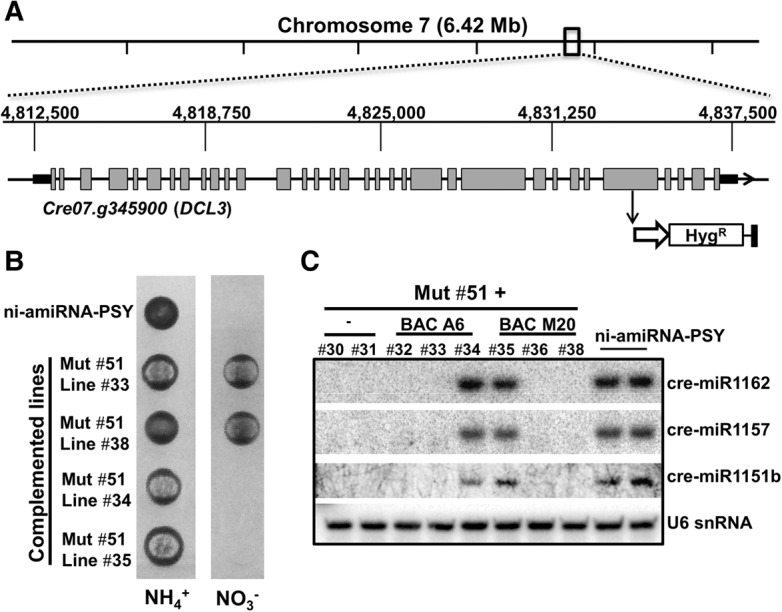
Mapping and complementation of group II mutant 51. (A) Location of the mutagenic hygromycin resistance cassette in mutant 51. (B) Phenotype of the indicated parental line and both complemented and noncomplemented lines (biological triplicates) in the presence of either nitrate or ammonium under high light conditions. (C) Detection by Northern blot of the indicated miRNAs in total RNA samples from the C. reinhardtii strains analyzed in B.
Final confirmation of DCL3 mutation was by complementation of mutant 51 with bacterial artificial chromosomes (BACs) (BAC A6 and BAC M20) carrying the genomic sequence corresponding to DCL3 (Cre07.g345900). After transformation of mutant 51 only two independent colonies had the extra copy of DCL3 in their genome. Importantly, these complemented lines were light sensitive when PSY amiRNA was induced with nitrate (Fig. 2B), and they regained the capacity to produce endogenous miRNAs (Fig. 2C). Henceforth, we refer to the original lines isolated from the screen as carrying dcl3-1 (mutant 37), dcl3-2 (mutant 47), and dcl3-3 (mutant 51).
C. reinhardtii DCL3 has the typical DCL domain organization except that, like the other DCLs in this alga, it lacks a PAZ domain that could be detected by primary and secondary structure prediction algorithms (Supplemental Fig. S3A). This protein is also exceptional among other DCL proteins in that it has a proline rich region (39/52 residues) on the amino terminal side of the RNase III motifs although a similar domain is also found in a related protein, Drosha. Drosha also has RNAse III motifs and it is involved in the first steps of the animal miRNA biogenesis pathway (Supplemental Fig. S3A,B).
DCL3 and sRNA biogenesis
The Northern blot analysis indicated a requirement of DCL3 for biogenesis of both siRNAs and miRNAs (Fig. 1D). To extend this analysis on a genome-wide basis, we sequenced sRNAs from two wild-type parental lines and two dcl3 lines (dcl3-1 and dcl3-3). Consistent with previous reports (Molnár et al. 2007; Zhao et al. 2007), the sRNAs from lines expressing the amiRNA were mostly 20–22 nt long with a clear peak at 21 nt that was absent in the dcl3 mutants. As observed previously, the 21-nt sRNAs had a bias toward U or A as first nucleotide (Molnár et al. 2007; Zhao et al. 2007) and those with a 5′ U were clearly reduced in dcl3 mutants (Fig. 3A). The heterogeneity of both 20- and 21-nt-long small RNAs was also diminished in dcl3 mutants, as observed in the analysis of nonredundant reads (Fig. 3B).
Figure 3.

Effect of dcl3 mutation on C. reinhardtii small RNA population. (A) Size-distribution histograms of sRNAs from the parental line A4-1 and its derivative dcl3-1 mutant expressed as the number of counted reads of a given size per million (CPM) of reads matching the C. reinhardtii genome. The percentage of 21-nt sRNAs with their 5′ nucleotide identities is also shown. (B) Size-distribution histograms of nonredundant sRNAs from the parental line A4-1 and its derivative dcl3-1 mutant expressed as CPM of reads matching the C. reinhardtii genome. Two additional replicates per sample, as well as three replicates from the E9-3 parental and dcl3-3 lines, showed the same result.
To identify the DCL3-dependent sRNA loci we aligned libraries of sRNA sequence from wild-type and dcl3 lines to the reference genome of C. reinhardtii. There were 5152 sRNA loci identified in all samples, of which 4313 (83.7%) were differentially expressed between the wild-type parental cells and the dcl3 mutant lines. The majority of these, 3366 (65.3%), were expressed at a higher level in the parental lines than in the mutant.
To evaluate the effect of dcl3 loss of function on miRNA production, taking into account a controversy about the number of miRNA genes in C. reinhardtii (Nozawa et al. 2012; Taylor et al. 2014), we carried out a stringent de novo prediction of miRNAs from all the identified sRNA loci present in both wild-type and mutant-derived samples (see Methods). This prediction indicated the presence of 18 canonical miRNA loci in C. reinhardtii, named in this paper as “high confidence miRNAs” (Table 1). These high confidence miRNAs include seven of the nine miRNAs identified by Taylor et al. (2014), as well as other previously reported/predicted miRNAs. Northern blot confirmed the production, as well as DCL3-dependency, of three of four novel high-confidence miRNAs found by our prediction tool (Table 1; Supplemental Fig. S4). Twenty-four additional loci specified precursor RNAs with miRNA-like features, but lacking a miRNA*, with more than one major sRNA species per arm, or with a variable 5′ end. These candidate miRNA loci were assigned to “medium confidence miRNAs” (Table 1). Only 16 of the 50 miRNA precursors currently annotated in miRBase v.21 were identified by our stringent prediction and, in agreement with a previous analysis (Taylor et al. 2014), it is likely that the others are misannotated siRNA loci.
Table 1.
Stringent de novo prediction of miRNA precursors in C. reinhardtii

The miRNAs or candidate miRNAs from all class loci were less abundant in dcl3-1 and dcl3-3 cells than in the corresponding parental lines (Table 1; Supplemental Table S2). Many (61.1%) of the high confidence miRNAs were derived from introns (nine miRNAs) or UTRs (two miRNAs) of mRNA precursors. The medium confidence miRNA candidates were also from mRNA precursors (75%) but they corresponded to UTRs (11 miRNAs) more than introns (seven miRNAs). The remaining miRNAs in both classes fell into a more canonical class derived from noncoding RNAs (Table 1).
We refer to the non-miRNAs as siRNAs and we classified the genomic siRNA loci into three major classes corresponding to protein-coding genes, transposable elements, and repeat elements. We further classified transposons and repeat associated siRNAs based on the output of RepeatMasker (Supplemental Table S2). All types of siRNA were predominantly dependent on DCL3, including gypsy siRNAs (Fig. 1). However, there were some protein-coding genes and non-LTR transposons (SINEX, RE, RTE) at which siRNA production was as great or greater in the dcl3 mutants than in the wild-type parents (DE dcl3>wt and NDE in Supplemental Table S2). These DCL3-independent siRNAs, as well as the marginal amount of miRNAs produced in dcl3-1 and dcl3-3 (Table 1), were presumably generated either by DCL1 or DCL2.
Processing of intron-derived miRNAs in Chlamydomonas
Intron-derived (id-)miRNAs are not unique to C. reinhardtii, they are also found in animals. The maturation of id-miRNAs in animals referred to as miRtrons (Ruby et al. 2007) is linked to intron splicing. To investigate this possibility in C. reinhardtii, we assembled a spectinomycin resistance gene with a miRNA-containing intron embedded in the coding sequence (spect/intron(mi)). The intron was from a C. reinhardtii gene (Cre12.g537671) and it contained the stem–loop RNA that is the precursor of the high confidence cre-miR1157 but with the miRNA sequence modified to target the mRNA of the tryptophan synthase beta-subunit (Maa7) (Fig. 4A). Silencing of Maa7 confers resistance to 5-fluoro indole (5-FI) (Rohr et al. 2004). Control constructs either lacked an intron (spect) or had an intron without the miRNA stem–loop (spect/intron) (Fig. 4A).
Figure 4.

The cre-miR1157 is an intron-derived miRNA. (A) Schematic representation of constructs carrying the cre-miR1157 intron inserted into the spectinomycin resistance gene coding sequence. The cre-miR1157 intron was modified to either lack the miRNA stem–loop or carry an artificial miRNA against Maa7 in spect/intron and spect/intron(mi) plasmids, respectively: (P) Hybrid RBSC2/HSP70A promoter; (SpectinomycinR) recoded Escherichia coli-derived aadA coding gene; (T) RBSC2 transcription terminator; (SpeI) unique cleavage site for SpeI restriction enzyme; (Maa7 amiRNA) modified version of cre-miR1157 that carries a miRNA against Maa7. (B, top) Growth of the indicated transgenic lines in solid media carrying spectinomycin with/without 5-Fluorindole (5-Fl). (Bottom) Detection by Northern blot of the artificial miRNA against Maa7 in total RNA samples from the indicated lines (three independent lines per construct). (C) Schematic representation of constructs used to test the requirement of splicing for the expression of id-miRNA. The GT × AT point mutations in the exon/intron junction are indicated. These plasmids also carry the ParomomycinR cassette (equivalent to the cassette showed in Fig. 1A) to allow the primary selection of transgenic lines in paromomycin. (D) Growth of lines transformed with the indicated plasmids in solid media containing either paromomycin (test for plasmid integration), spectinomycin (test for splicing events), or 5-Fl (test for amiRNA production).
The id-miRNA was spliced efficiently from these RNAs because the spec/intron(mi) construct conferred spectinomycin resistance as efficiently as the spec and spec/intron controls (Supplemental Fig. S5A). RT-PCR further confirmed correct splicing of the id-miRNA (Supplemental Fig. S5B,C), and a sRNA Northern blot (Fig. 4B) showed, as predicted, production of the mature Maa7 amiRNA. The id-miRNA was fully functional as it silenced the Maa7 mRNA so that the spec/intron(mi) cells were resistant to 5-Fl. Cells with the control constructs without the id-miRNA did not produce the amiRNA, and they were fully susceptible to 5-FI (Fig. 4B).
Finally, to analyze the requirement for splicing in miRNA biogenesis, we generated an id-miRNA construct with a mutation in the splice donor site (Fig. 4C, spec/Δintron(mi)). This construct conferred resistance to 5-FI but, as expected, not to spectinomycin (Fig. 4D). From our results in Figure 4 and Supplemental Figure S5, it is clear that the presence of the id-miRNA does not prevent the intron processing and that, unlike animal miRtrons, the intron processing is not required for miRNA biogenesis.
Differential gene expression in dcl3 mutants
To identify mRNA targets of miRNAs, we used RNA-seq of the transcriptome in dcl3 mutant and parental lines. There were 118 annotated genes with statistically significant differences (equal to or greater than 0.9 likelihood) in abundance between the dcl3-1 and dcl3-3 mutants and the corresponding wild-type parental cells (Supplemental Table S3).
The 118 DCL3-sensitive RNAs were in several classes corresponding to the following:
Noncoding RNAs with miRNA precursors (five genes);
Noncoding RNAs with siRNA precursors (64 genes);
mRNAs with miRNA precursors in the exons corresponding to the coding sequence (one gene) and 3′ UTR (eight genes);
mRNAs with siRNA precursors in the exons corresponding to the 5′ UTR (five genes), coding sequence (three genes), and 3′ UTR (21 genes); and
mRNAs with fold back RNA structures producing no clear siRNAs (nine genes).
The predicted and confirmed miRNA-targeted mRNAs from C. reinhardtii (Molnár et al. 2007; Zhao et al. 2007) were conspicuously absent from the list of differentially expressed RNAs (Supplemental Table S3). These RNAs were equally abundant in the RNA-seq data sets of wild-type and dcl3 mutant lines (Fig. 5A) despite the presence of the miRNA guided mRNA cleavage products only in the RNA samples from the wild-type strains (Fig. 5B,C). Presumably the miRNA-directed cleavage products are present at only low abundance in these samples.
Figure 5.

The effect of miRNA on mRNA accumulation. (A) Steady-state accumulation levels of previously reported miRNA targets (Molnár et al. 2007; Zhao et al. 2007) assessed as the number of normalized reads (y-axis) in RNA-seq data. Error bars for three independent samples are shown. The target genes with their corresponding miRNA are indicated. These miRNAs were predicted as either high confidence miRNAs (cre-miR1162, cre-miR1151a/b) or medium confidence miRNA (miR-C82) (see Table 1), with the exception of cre-miR909 that is a hairpin-derived siRNA also depleted in the dcl3 mutant background. (B) 5′RACE to test the specific cleavage of CPLD52 (Cre13.g608000) mediated by cre-miR1162. The asterisk indicates an unspecific PCR product. The PCR products were sequenced, and the right panel shows the 5′ terminus of these cleavage products aligned to the 5′ to 3′ mRNA sequence and the 3′ to 5′ miRNA. G:U base pairs are indicated by a circle. (C) 5′RACE to test the specific cleavage of OMT2 (Cre17.g713200) mediated by the miR-C82 (Zhao et al. 2007) with the 5′ terminus of these cleavage products aligned to the 5′ to 3′ mRNA sequence and the 3′ to 5′ miRNA as in B.
The primary effect of DCL3 on mRNA accumulation must be by direct cleavage of the mature mRNA as shown for two examples in Figure 6. These are mRNAs for which the exonic reads are more abundant in the dcl3 mutant rather than wild-type samples (Fig. 6; Supplemental Table S3). The miRNA reads corresponding to the respective 3′ UTRs are conversely more abundant in wild-type samples (Fig. 6; Table 1). The other 45 mRNAs accumulating at higher level in the dcl3 mutants correspondingly were from mRNAs containing miRNA/siRNA-like stem–loop structures in their coding or noncoding exons (Supplemental Table S3).
Figure 6.

The effect of DCL3 on mRNAs with miRNA hairpin-like structures in the 3′ UTR. Cre16.g694950 (serine/threonine kinase) (A) and Cre24.g755697 (aminoglycoside 3′-phosphotransferase) (B) have the respective cre-miR1169 and cre-miR1172 precursors in their 3′ UTR. A schematic representation of both genes is shown with their exons (gray boxes) and introns (black solid lines) at the bottom of each panel. Light gray (A4-1 parental line) and black (dcl3-1) hills represent sRNA and mRNA read counts. Both panels show the results for one replicate of A4-1 parental line and its dcl3-1 derivative knock out mutant. Overaccumulation (X-fold) of the indicated mRNA in dcl3-1 regarding the A4-1 parental line is indicated. Two additional samples from this parental and mutant line combination and a biological triplicate from E9-3 parental and dcl3-3 derivative lines showed the same trend in both miRNA and mRNA accumulation (Table 1; Supplemental Table S3).
Most mRNAs with id-miRNAs, with the exception of the mRNA linked to cre-miR1154, were not affected by dcl3 mutation (Supplemental Fig. S6). Based on these examples and results with the cre-miR1157 precursor (Fig. 4; Supplemental Fig. S5), we conclude that the DCL3 cleavage must be separate from mRNA splicing.
Discussion
From our genetic analysis we have identified the DCL3 protein of C. reinhardtii as being responsible for sRNA biogenesis and mRNA accumulation. Our findings reinforce the idea that the miRNA silencing system in this alga is distinct from that of land plants and it may have features in common with the functional equivalent in animals. It is clear, however, from the phenotype of dcl3 mutants that, unlike animals and land plants, miRNA silencing in C. reinhardtii is not required for normal growth and development. Our findings have implications for understanding the evolution and biological function of miRNA silencing in eukaryotes.
miRNA silencing in C. reinhardtii is not typical of higher plants
DCL3 in C. reinhardtii has two features that are characteristic of similar proteins in nonplant organisms. The first of these is the absence of a PAZ domain (Supplemental Fig. S3) as with Dicer from the human parasite Toxoplasma gondii. This protozoan protein, together with the three DCLs from C. reinhardtii, forms a clade that is independent of both higher plant and animal DCLs (Fig. 1B in Braun et al. 2010).
The PAZ domain mediates the cleavage site selection in the miRNA precursor and size specification of the miRNA (MacRae et al. 2007) and, in its absence, it is likely that other proteins carry out these functions. Perhaps the large domain replacing PAZ in C. reinhardtii DCL3 is the anchoring site for such accessory functions in miRNA biogenesis. The proteins encoded by uncharacterized group II mutant loci are candidates for these accessory factors (Supplemental Table S1).
The second nonplant feature of DCL3 is a proline-rich domain on the amino terminal side of the two RNase III domains. There is a similar domain in an equivalent position in Drosha, the animal miRNA processor, which has an RNase III and lacks a PAZ domain (Ha and Kim 2014). These similarities prompt the hypothesis that C. reinhardtii DCL3 is both a Dcr and a Drosha with roles at several stages in miRNA biogenesis. The higher plant DCL1 is similarly involved in miRNA processing at early stages in addition to the final pre-miRNA cleavage (Brodersen and Voinnet 2009) but, unlike C. reinhardtii DCL3, it does not have any specific Drosha feature.
The miRNA genes of C. reinhardtii, like the DCL3 protein, also have nonplant characteristics. The most striking of these features is their overlap with protein-coding genes (Table 1). This is a frequent feature of animal miRNAs whereas higher plant miRNAs are, with only few exceptions, from noncoding RNA precursors. It is estimated in animals that ∼40% of the entire miRNA population are from introns (Kim et al. 2009) whereas, in plants, there are only three experimentally validated id-miRNAs (one and two in Arabidopsis thaliana and Oryza sativa, respectively) (Rajagopalan et al. 2006; Zhu et al. 2008; Joshi et al. 2012). In one of these examples the Arabidopsis DCL1 strongly represses DCL1 mRNA abundance by cleavage of an miRNA precursor in intron 14 (Xie et al. 2003; Rajagopalan et al. 2006). In contrast, in Chlamydomonas, the id-miRNAs do not affect the abundance of the corresponding mRNA (Supplemental Table S3) or the miRNA-induced phenotype (Fig. 4) and so, even when higher plants have some id-miRNAs, there are major differences from Chlamydomonas miRNA features and mechanisms.
A second nonplant feature associated with the miRNA-related mechanisms of C. reinhardtii is with the UTR miRNAs. The mRNAs with miRNA structures in the UTR overaccumulated in the dcl3 mutants, indicating that they are targeted for degradation by DCL3 in wild-type cells (Supplemental Table S3). An equivalent mechanism occurs with the mammalian FSTL1 mRNA that is destabilized by Drosha during hs-miR198 biogenesis (Sundaram et al. 2013). Similarly the DGCR8 mRNA is destabilized by Drosha cleavage via cleavage of a hairpin-like structure at the 3′ UTR, although there is no miRNA produced (Han et al. 2009).
There are 13 mRNAs in C. reinhardtii with miRNA in their UTRs (Table 1) of which eight accumulate at higher level in dcl3 mutants (Supplemental Table S3). In addition, there are 26 mRNAs with siRNA precursors in their UTRs (Supplemental Table S3) and nine mRNAs with hairpin structures without associated sRNAs that are up-regulated in dcl3 (Supplemental Table S3). It is likely, therefore, that there are at least 43 mRNAs in C. reinhardtii that may be subject to direct cleavage by DCL3. Remarkably, six of 13 UTR miRNAs bind AGO3 (Voshall et al. 2015), one of the three AGO proteins in C. reinhardtii. These observations prompt us to suggest that C. reinhardtii DCL3, like animal Drosha, has a dual role in mRNA regulation: It is first a ribonuclease that controls the levels of certain mRNAs by direct cleavage; and second, it is involved in biogenesis of sRNAs that act in trans to influence either mRNA accumulation or translation (Ma et al. 2013; Yamasaki et al. 2013; Voshall et al. 2015).
Finally, a third nonplant feature associated with C. reinhardtii miRNAs concerns the complementarity requirement for miRNAs to produce an effective down-regulation of their targets. Effective miRNA silencing in higher plants depends on near complete complementarity of the miRNA and its target (Liu et al. 2014) whereas, in C. reinhardtii, pairing in the miRNA seed region is sufficient to induce down-regulation (Yamasaki et al. 2013).
Evolution of miRNA silencing in C. reinhardtii
Animal and plant miRNA pathways are very different and it is likely that they evolved separately from an ancestral RNA silencing pathway with Dicer proteins and small RNAs with 5′ phosphate and 3′ hydroxyl groups that bind to AGO proteins (Ghildiyal and Zamore 2009; Axtell et al. 2011). The algal/Chlamydomonas miRNA pathway is also distinct from that of higher plants, as discussed above, and we can envision either of two evolutionary scenarios to explain those differences. The first of these is that animal, higher plant, and algal miRNA pathways all evolved independently of each other. A second scenario is that an animal-like miRNA pathway evolved early and persisted in lower plant lineages, including the green algae and C. reinhardtii, although it was not retained in higher plants.
Our data are consistent with the second scenario because C. reinhardtii and animal miRNA pathways share the presence of a Drosha-like structure (absence of PAZ and presence of P-rich domain) of the miRNA processing enzyme (Supplemental Fig. S3), Drosha-like dual function exerted by the miRNA processing enzyme (Fig. 6; Supplemental Table S3), and miRNA association with introns or exons of RNA coding sequences (Table 1). In addition, as mentioned above, the animal and C. reinhardtii miRNA systems depend only on seed region complementarity (Yamasaki et al. 2013), and they both use VIG and TSN1 (Voshall et al. 2015; Ibrahim 2009).
At present there are insufficient data to resolve these two alternative scenarios, although the further characterization of additional class I –IV mutants (Supplemental Table S1) may shed more light on the evolutionary origin of miRNAs in C. reinhardtii.
The role of sRNAs in C. reinhardtii
To explain the absence of physiological phenotype in our dcl3 mutants in normal laboratory conditions as described here and in a description of another unrelated RNA silencing mutant of Chlamydomonas (Voshall et al. 2015) we propose that DCL3 has a role at certain stages of the life cycle or under conditions that have not yet been tested. A role under starvation of sulphate and/or phosphate is possible because these conditions affect sRNA profiles in Chlamydomonas (Shu and Hu 2012; Zheng et al. 2015). The DCL3-dependent silencing might also act redundantly with other silencing systems as indicated by the loss of transposon silencing in C. reinhardtii that was dependent on loss of function at both DCL1 and of a histone methyltransferase (Casas-Mollano et al. 2008). The availability of DCL3 mutants will now allow us to test these possibilities. We cannot, however, rule out the possibility that at least some of the C. reinhardtii sRNAs have a silencing-independent role.
Methods
Strain, culture conditions, and transformation
The C. reinhardtii cell-wall deficient strain CC-1883 (cw15, NIA, NIT2, mt−) was used in this study as wild-type background. It was obtained from the Chlamydomonas Resource Center (University of Minnesota) and grown in either solid or liquid 2-amino-2-(hydroxymethyl)-1,3-propanediol (TRIS)-acetate-phosphate (TAP) media (Harris 2009) at 25°C under continuous illumination. When indicated, cells were grown in nitrate TAP (TAP medium in which ammonium was replaced by the equivalent amount of nitrate).
For transformation, the indicated DNA cassettes were excised from their backbones, and ∼100 ng purified fragments were used for each transformation experiment. Transformations of mid-log-phase cells were done by electroporation following a previously described method (Shimogawara et al. 1998) in a Gene Pulser Xcell apparatus (Bio-Rad) with exponential electric pulses (2250 kV/cm, 10 µF). After recovery, cells were plated on solid media in the presence of starch.
DNA oligonucleotides
DNA sequence of primers used in this study are listed in Supplemental Table S4.
Plasmids
The nitrate-inducible amiRNA construct (ni-amiRNA) was generated from pMS539 (Schmollinger et al. 2010) by subcloning a XbaI/DraI excised fragment that contains the Nit1 promoter/5′ UTR, amiRNA precursor, and terminator into XbaI/SmaI digested pSI103-1 (a derivative of pSI103) (Sizova et al. 2001). Unlike the original pMS539, the resulting plasmid confers resistance to paromomycin once integrated into the C. reinhardtii genome.
The amiRNA that targets PSY (Cre02.g095092) mRNA was designed using the Web MicroRNA Designer (http://wmd3.weigelworld.org/). The 21-nt amiRNA 5′-UGAUUUUGGAAGCGUUCGGCC-3′ was introduced in ni-amiRNA as a 90-nt double- stranded DNA (obtained by in vitro annealing of amiFor-PSY and amiRev-PSY primers) in its unique SpeI restriction site, following a previously described method (Molnár et al. 2009), to generate the ni-amiRNA-PSY.
The intron-derived cre-miR1157 precursor that lacks the cre-miR1157 stem–loop was amplified by PCR from ni-amiRNA with primers miR1157-Prec-For and miR1157-Prec-Rev, which carry tails in order to reconstitute the whole intron 22 from Cre12.g537671 and add PmlI and PvuII restriction sites at the 5′ and 3′ ends of the PCR product, respectively. This PCR product was cloned into pGEM-T Easy (Promega) to create pGEMT-miR1157. The gene splicing via overlap extension method (Horton et al. 1989) was used to generate the Spect/intron, a plasmid based on pALM32 (Meslet-Cladière and Vallon 2011) that carries the whole intron-derived cre-miR1157 (without the stem–loop precursor) in the middle of the aadA gene. A mix of three different DNA fragments was used as template for the overlapping PCR: (1) a PCR product obtained by amplification from pALM32 using the primers RBSC2_Pro-For and Spect+intron1157-Rev; (2) a PCR product obtained by amplification from pALM32 using the primers Spect+intron1157-For and RBSC2_3′UTR-Rev; and (3) a PmlI/PvuII digested fragment from pGEMT-miR1157. The resulting PCR fragment was digested with KpnI and cloned into SmaI/KpnI digested pALM32 to generate the Spect/intron plasmid. Spect/intron(mi) was generated by cloning an amiRNA (5′-UAUGUACACAAUGCACUUCAG-3′), which targets the tryptophan synthase beta-subunit mRNA, into the Spect/intron plasmid by following the procedure described above. Site-directed mutagenesis of the splicing donor site in Spect/intron(mi) was carried out by two PCR steps, as previously described (Herlitze and Koenen 1990). The first round PCRs were done by using Spect/intron(mi) as template plus primer pair HSP70-For and Intron_Donor-Mut-Rev primers, or primers Intron_Donor-Mut- For and SpeI-Rev. These two PCR products were then used as template for the second round PCR with primers HSP70-For and SpeI-Rev. The resulting PCR product was digested with AatII/SpeI and cloned by triple ligation with AatII/HindIII and SpeI/HindIII digested fragments from Spect/intron(mi) to generate Spect/Δintron(mi).
BAC clones 29A6 (A6) and 29M20 (M20) carrying the C. reinhardtii DCL3 genomic sequence were identified in a BAC library generated by Paul Lefebvre (University of Minnesota) by using the JGI v4 browser (http://genome.jgi-psf.org/Chlre4/Chlre4.info.html). The whole library, named CRCCBa, was obtained from the Clemson University Genomics Institute, and the indicated clones were isolated from E. coli glycerol stocks by using the QIAGEN Large-Construct Kit (Qiagen) following the manufacturer's instructions.
Plasmids pSI103-1 (J Moy, M LaVoie, C Silflow, unpubl.), pHyg3 (Berthold et al. 2002), and pALM32 (Meslet-Cladière and Vallon 2011) were obtained from the Chlamydomonas Resource Center (University of Minnesota).
Mutant screen and mapping of mutagen integration sites
Two independent transgenic lines (here called A4-1 and E9-3 parental lines) carrying a functional ni-amiRNA cassette were grown on liquid TAP until mid-log-phase (this medium carries ammonium as the only nitrogen source, which represses the Nit1 promoter). Random insertional mutants were obtained by transformation of A4-1 and E9-3 with the corresponding resistance cassettes from pALM32 and pHyg3, respectively, following the transformation protocol described above. Mutant lines potentially affected in the miRNA silencing pathway were observed at 5–7 d after plating the cells in solid amiRNA induction media (TAP medium in which the ammonium was replaced for an equivalent amount of nitrate) at high light intensity (200 µmol photons m−2sec−1). Insertions were mapped in the C. reinhardtii genome by RESDA-PCR as previously described (González-Ballester et al. 2005). Primer pairs annealing across the insertion sites were used for easy genotyping of dcl3 mutant lines by direct PCR, from a tiny amount of cells, by using the Phire Plant Direct PCR Kit (Thermo Scientific) according to the manufacturer's instructions.
RNA extraction and analyses
RNA isolation and small RNA detection by Northern blot were carried out as previously described (Molnár et al. 2007). A detailed protocol can be found at http://www.plantsci.cam.ac.uk/research/davidbaulcombe/methods/downloads/smallrna.pdf/view. DNA primers corresponding to the reverse complementary sequence of the indicated miRNAs (amiRPSY-det, miR1151b-det, miR1157-det, miR1162-det, siRgypsy-det, miR_Cre07.g341100-det, miR_ Cre07.g344260-det, miR_Cre14.g623850-det, and miR_Cre16.g670000-det) (Supplemental Table S4) were radiolabeled with γ32P-ATP by the action of polynucleotide kinase and used to probe membranes with immobilized RNA samples. Radioactive signals were further detected with a phosphorimager.
The accumulation level of PSY mRNA was estimated by qRT-PCR from 5 µg DNA-free RNA. Briefly, RT was primed with random hexamers and SuperScript III (Invitrogen) following the manufacturer's guidelines. The PCR amplification step was carried out with primers PSY-qPCR-For and PSY-qPCR-Rev in the presence of the dsDNA-specific dye SYBR Green (Sigma) and monitored with a Chromo4 qPCR machine (Bio-Rad). The RACK1 gene (Cre06.g278222) was used as an internal control for normalization. The delta-delta Ct method was used to calculate the differences in mRNA abundance.
RT-PCR was used to confirm splicing of the artificially generated, intron-derived, miRNA precursor. To do that, RT reaction was carried out as described above, whereas a normal PCR amplification step with primers Spect-For and Spect-Rev, which flank both sides of the intron, was done using the RT reaction as template.
5′ RNA ligase-mediated RACE was done as described (Llave et al. 2002) with the GeneRacer kit (Invitrogen). First PCR round was done with distal primers (PSY-Rev, OMT2-Rev and CPLD52-Rev), while nested primers were used for the second round PCRs (PSY_nested-Rev, OMT2_nested-Rev, CPLD52_nested-Rev). The final PCR fragments were gel purified using MinElute gel extraction kit (Qiagen) and cloned into pCRII vector (Invitrogen). Positive clones were further analyzed by DNA sequencing to map exact miRNA cleavage sites.
Preparation of RNA libraries
Prior to preparing the sRNA libraries, samples carrying 10 µg total RNA were subjected to the FDF-PAGE method as previously described (Harris et al. 2015). The sRNA libraries were further prepared according to the TruSeq small RNA cloning protocol (Illumina) and run in an Illumina HiSeq 2000 (BGI HongKong).
Libraries for RNA-seq were prepared from poly(A) RNAs, which were purified from 50 µg total RNA by using the MicroPoly(A)Purist Kit (Ambion), following the manufacturer's instructions. Poly(A) RNA was used as starting material for the ScriptSeq v2 RNA-seq Library Preparation Kit (Illumina). Libraries were prepared following the manufacturer's protocol and run in an Illumina HiSeq 2000 (BGI HongKong).
Analysis of sRNA high-throughput sequencing data
Illumina sRNA libraries were preprocessed using the ADDAPTS pipeline and tracking system (http://www.plantsci.cam.ac.uk/bioinformatics/addapts). After 3′ adaptor removal, all sequences <15 nt in length are discarded, and the remaining sequences are aligned against C. reinhardtii genome v5.0 using PatMaN (Prüfer et al. 2008). Only sequences with at least one perfect match are included. The initial sequencing data for each library and the number of reads obtained after each step are indicated in Supplemental Table S5. For the definition of sRNA producing loci, segmentSeq_2.2.1 (Hardcastle et al. 2012), available as part of Bioconductor, was used. This package takes the density of matches of sRNAs to the genome to determine regions corresponding to sRNA producing transcripts, taking into account replicate data. Segments with a higher than 0.9 posterior probability of being loci were used. Loci were subjected to differential expression analysis using baySeq 2.2.0 (Hardcastle and Kelly 2010). This package uses the negative binomial distribution for the count data produced by high-throughput sequencing and estimates its parameters using empirical Bayes, with the number of iterations determined by the parameter “sample size.” Models for different patterns of differential expression (including no differential expression) among the samples are specified, and the model with the highest posterior probability is used. The library scaling factor (surrogates for library size) has to be specified for each sample, and they were calculated by using the previously described quantile normalization (Bullard et al. 2010). This method sums all counts in each sample for which the value below the qth quantile of nonzero counts for that particular sample. Only those loci with a likelihood ≥0.9 of being differentially expressed in the specified model were considered.
A Python (v2.7.9) script was developed to count the number of overlaps between genomic annotations (Phytozome v5.5), repeat masker annotations (Phytozome v10.3), inverted repeats, tandem repeats, and miRNA precursors (these three last features were predicted as explained below) with the sets of differentially and nondifferentially expressed loci. Inverted repeats and tandem repeats were predicted by Inverted Repeat Finder v3.0.7 and Tandem Repeat Finder v4.0.7b, respectively (Benson 1999; Warburton et al. 2004).
MiRNA prediction
The identification of miRNA precursors was performed by a multistep process, which first uses a combination of three different miRNA prediction algorithms: miRDP, miRDeep2 (with minimum score of 5), and miRCat (Mackowiak 2011; Yang and Li 2011; Stocks et al. 2012). These results were then combined to remove duplicate predictions, precursors with mature miRNAs with sizes outside 20–22 nt, and/or precursors with less than 100 sRNA reads. Finally, the last automated step was performed by removing those predicted miRNA precursors that did not overlap with those sRNA loci that had been previously identified by segmentSeq (see above). The resulting precursors were manually curated for the presence of miRNA* and defined miRNA stacks in an attempt to follow the standards of high confidence recommended by miRBase (Kozomara and Griffiths-Jones 2014). The number of identified miRNA precursors at each stage of the multistep process is shown in Supplemental Table S6. Detailed information about predicted miRNA precursors (exact location in the genome and nucleotide sequences of the corresponding mature miRNAs and miRNAs*) is shown in Supplemental Table S7.
Analysis of RNA-seq high-throughput sequencing data
RNA-seq libraries were first analyzed with FastQC v0.11.2 for quality control (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic 0.32 (Bolger et al. 2014) was then used for adaptor removal and trimming of bases with a quality score lower than 20. Reads shorter than 40 nt were discarded, and the remaining reads were subsequently aligned to rRNA, ncRNA, cpDNA, and mtDNA from C. reinhardtii using Bowtie 2 (Langmead and Salzberg 2012). Positive matches were discarded. Finally, the remaining reads were aligned with Bowtie 2 against the C. reinhardtii transcriptome (Phytozome v5.5). The initial sequencing data for each library and the number of reads obtained after each filtering step are indicated in Supplemental Table S5. Quantification of transcript abundance was performed using express 1.5.1 (Roberts and Pachter 2013). The est_counts and eff_length from express were then passed as input to baySeq (Hardcastle and Kelly 2010) for the differential expression analysis. Transcripts for which a likelihood ≥0.9 in the specified model were considered as differentially expressed.
Data access
Small RNA-seq and RNA-seq data sets generated during this study have been submitted to the ArrayExpress database (EMBL-EBI; https://www.ebi.ac.uk/arrayexpress/) under accession numbers E-MTAB-3851 and E-MTAB-3852, respectively. Plasmids and strains generated in this study are available at the Chlamydomonas Resource Center (University of Minnesota).
Supplementary Material
Acknowledgments
We thank James Barlow for technical assistance and media preparation, Michael Schroda and Stefan Schmollinger for providing pMS539, and Olivier Vallon for his useful advice about BACs and DNA sequencing data. We thank Witold Filipowicz and Ian Henderson for critical reading of the manuscript. Work in the Baulcombe laboratory is supported by the Balzan Prize award and the European Research Council Advanced Investigator Grant ERC-2013-AdG 340642 TRIBE. A.A.V. was supported by a Marie-Curie fellowship (PIEF-GA-2010-276037). B.Y.C. was supported by an EMBL long-term postdoctoral fellowship and a Sir Henry Wellcome Fellowship (096082). D.C.B. is the Royal Society Edward Penley Abraham Research Professor.
Author contributions: A.A.V. and D.C.B. designed the research. A.A.V. performed most of the experiments. B.A.C.M.S. analyzed high-throughput sequencing data. S.H. mapped the mutation in the dcl3-3 allele. A.R.B., A.M., and B.Y.C. designed and carried out experiments leading to the identification of a mutant affecting miRNA and siRNA pathways. A.A.V. and D.C.B. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.199703.115.
Freely available online through the Genome Research Open Access option.
References
- Axtell MJ, Westholm JO, Lai EC. 2011. Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulcombe D. 2004. RNA silencing in plants. Nature 431: 356–363. [DOI] [PubMed] [Google Scholar]
- Baumgarten S, Bayer T, Aranda M, Liew YJ, Carr A, Micklem G, Voolstra CR. 2013. Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC Genomics 14: 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold P, Schmitt R, Mages W. 2002. An engineered Streptomyces hygroscopicus aph 7′′ gene mediates dominant resistance against hygromycin B in Chlamydomonas reinhardtii. Protist 153: 401–412. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun L, Cannella D, Ortet P, Barakat M, Sautel CF, Kieffer S, Garin J, Bastien O, Voinnet O, Hakimi MA. 2010. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog 6: e1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P, Voinnet O. 2009. Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol 10: 141–148. [DOI] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136: 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Mollano JA, Rohr J, Kim EJ, Balassa E, Van Dijk K, Cerutti H. 2008. Diversification of the core RNA interference machinery in Chlamydomonas reinhardtii and the role of DCL1 in transposon silencing. Genetics 179: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. 2013. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14: 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XS, Collins LJ, Biggs PJ, Penny D. 2009. High throughput genome-wide survey of small RNAs from the parasitic protists Giardia intestinalis and Trichomonas vaginalis. Genome Biol Evol 1: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. 2009. Small silencing RNAs: an expanding universe. Nat Rev Genet 10: 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Ballester D, De Montaigu A, Galván A, Fernández E. 2005. Restriction enzyme site-directed amplification PCR: a tool to identify regions flanking a marker DNA. Anal Biochem 340: 330–335. [DOI] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Han J, Pedersen JS, Kwon SC, Belair CD, Kim YK, Yeom KH, Yang WY, Haussler D, Blelloch R, Kim VN. 2009. Posttranscriptional crossregulation between Drosha and DGCR8. Cell 136: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardcastle TJ, Kelly KA. 2010. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics 11: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardcastle TJ, Kelly KA, Baulcombe DC. 2012. Identifying small interfering RNA loci from high-throughput sequencing data. Bioinformatics 28: 457–463. [DOI] [PubMed] [Google Scholar]
- Harris EH. 2009. The Chlamydomonas sourcebook: introduction to Chlamydomonas and its laboratory use. Academic Press, New York. [Google Scholar]
- Harris CJ, Molnar A, Müller SY, Baulcombe DC. 2015. FDF-PAGE: a powerful technique revealing previously undetected small RNAs sequestered by complementary transcripts. Nucleic Acids Res 43: 7590–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Koenen M. 1990. A general and rapid mutagenesis method using polymerase chain reaction. Gene 91: 143–147. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61–68. [DOI] [PubMed] [Google Scholar]
- Huang PJ, Lin WC, Chen SC, Lin YH, Sun CH, Lyu PC, Tang P. 2012. Identification of putative miRNAs from the deep-branching unicellular flagellates. Genomics 99: 101–107. [DOI] [PubMed] [Google Scholar]
- Ibrahim F. 2009. Molecular characterization of RNA interference components in Chlamydomonas: a novel nucleotidyltransferase (MUT68) and a Vasa intronic gene homolog (MUT70). University of Nebraska, Lincoln, NE. [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H. 2015. PIWI-interacting RNA: its biogenesis and functions. Annu Rev Biochem 84: 405–433. [DOI] [PubMed] [Google Scholar]
- Joshi PK, Gupta D, Nandal UK, Khan Y, Mukherjee SK, Sanan-Mishra N. 2012. Identification of mirtrons in rice using MirtronPred: a tool for predicting plant mirtrons. Genomics 99: 370–375. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wang F, Axtell MJ. 2014. Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 26: 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. 2002. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297: 2053–2056. [DOI] [PubMed] [Google Scholar]
- Ma X, Kim E-J, Kook I, Ma F, Voshall A, Moriyama E, Cerutti H. 2013. Small interfering RNA–mediated translation repression alters ribosome sensitivity to inhibition by cycloheximide in Chlamydomonas reinhardtii. Plant Cell 25: 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackowiak SD. 2011. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr Protoc Bioinformatics Chapter 12: Unit 12.10. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Zhou K, Doudna JA. 2007. Structural determinants of RNA recognition and cleavage by Dicer. Nat Struct Mol Biol 14: 934–940. [DOI] [PubMed] [Google Scholar]
- Mallick B, Ghosh Z, Chakrabarti J. 2008. MicroRNA switches in Trypanosoma brucei. Biochem Biophys Res Commun 372: 459–463. [DOI] [PubMed] [Google Scholar]
- Mar-Aguilar F, Trevino V, Salinas-Hernández JE, Taméz-Guerrero MM, Barrón-González MP, Morales-Rubio E, Treviño-Neávez J, Verduzco-Martínez JA, Morales-Vallarta MR, Reséndez-Pérez D. 2013. Identification and characterization of microRNAs from Entamoeba histolytica HM1-IMSS. PLoS One 8: e68202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SS, Kobayashi MC, Niyogi KK. 2004. White mutants of Chlamydomonas reinhardtii are defective in phytoene synthase. Genetics 168: 1249–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G. 2013. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459. [DOI] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L, et al. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meslet-Cladière L, Vallon O. 2011. Novel shuttle markers for nuclear transformation of the green alga Chlamydomonas reinhardtii. Eukaryot Cell 10: 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. 2007. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447: 1126–1129. [DOI] [PubMed] [Google Scholar]
- Molnár A, Bassett A, Thuenemann E, Schwach F, Karkare S, Ossowski S, Weigel D, Baulcombe D. 2009. Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant J 58: 165–174. [DOI] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M. 2012. Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4: 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Stenzel U, Dannemann M, Green RE, Lachmann M, Kelso J. 2008. PatMaN: rapid alignment of short sequences to large databases. Bioinformatics 24: 1530–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. 2006. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20: 3407–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Pachter L. 2013. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10: 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr J, Sarkar N, Balenger S, Jeong BR, Cerutti H. 2004. Tandem inverted repeat system for selection of effective transgenic RNAi strains in Chlamydomonas. Plant J 40: 611–621. [DOI] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. 2007. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiya AA, Wang CC. 2008. snoRNA, a novel precursor of microRNA in Giardia lamblia. PLoS Pathog 4: e1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmollinger S, Strenkert D, Schroda M. 2010. An inducible artificial microRNA system for Chlamydomonas reinhardtii confirms a key role for heat shock factor 1 in regulating thermotolerance. Curr Genet 56: 383–389. [DOI] [PubMed] [Google Scholar]
- Shimogawara K, Fujiwara S, Grossman A, Usuda H. 1998. High-efficiency transformation of Chlamydomonas reinhardtii by electroporation. Genetics 148: 1821–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L, Hu Z. 2012. Characterization and differential expression of microRNAs elicited by sulfur deprivation in Chlamydomonas reinhardtii. BMC Genomics 13: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova I, Fuhrmann M, Hegemann P. 2001. A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 277: 221–229. [DOI] [PubMed] [Google Scholar]
- Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, Schwach F, Dalmay T, Moulton V. 2012. The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28: 2059–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram GM, Common JE, Gopal FE, Srikanta S, Lakshman K, Lunny DP, Lim TC, Tanavde V, Lane EB, Sampath P. 2013. ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing. Nature 495: 103–106. [DOI] [PubMed] [Google Scholar]
- Taylor RS, Tarver JE, Hiscock SJ, Donoghue PCJ. 2014. Evolutionary history of plant microRNAs. Trends Plant Sci 19: 175–182. [DOI] [PubMed] [Google Scholar]
- Voshall A, Kim EJ, Ma X, Moriyama EN, Cerutti H. 2015. Identification of AGO3-associated miRNAs and computational prediction of their targets in the green alga Chlamydomonas reinhardtii. Genetics 200: 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G. 2004. Inverted repeat structure of the human genome: The X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res 14: 1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Kasschau KD, Carrington JC. 2003. Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr Biol 13: 784–789. [DOI] [PubMed] [Google Scholar]
- Yamasaki T, Voshall A, Kim EJ, Moriyama E, Cerutti H, Ohama T. 2013. Complementarity to an miRNA seed region is sufficient to induce moderate repression of a target transcript in the unicellular green alga Chlamydomonas reinhardtii. Plant J 76: 1045–1056. [DOI] [PubMed] [Google Scholar]
- Yang X, Li L. 2011. miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27: 2614–2615. [DOI] [PubMed] [Google Scholar]
- Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D. 2004. A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol 21: 809–818. [DOI] [PubMed] [Google Scholar]
- Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang XJ, Qi Y. 2007. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev 21: 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wang Y, Wu J, Ding B, Fei Z. 2015. A dynamic evolutionary and functional landscape of plant phased small interfering RNAs. BMC Biol 13: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu QH, Spriggs A, Matthew L, Fan L, Kennedy G, Gubler F, Helliwell C. 2008. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res 18: 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.