

Abstract
The mammalian Y Chromosome sequence, critical for studying male fertility and dispersal, is enriched in repeats and palindromes, and thus, is the most difficult component of the genome to assemble. Previously, expensive and labor-intensive BAC-based techniques were used to sequence the Y for a handful of mammalian species. Here, we present a much faster and more affordable strategy for sequencing and assembling mammalian Y Chromosomes of sufficient quality for most comparative genomics analyses and for conservation genetics applications. The strategy combines flow sorting, short- and long-read genome and transcriptome sequencing, and droplet digital PCR with novel and existing computational methods. It can be used to reconstruct sex chromosomes in a heterogametic sex of any species. We applied our strategy to produce a draft of the gorilla Y sequence. The resulting assembly allowed us to refine gene content, evaluate copy number of ampliconic gene families, locate species-specific palindromes, examine the repetitive element content, and produce sequence alignments with human and chimpanzee Y Chromosomes. Our results inform the evolution of the hominine (human, chimpanzee, and gorilla) Y Chromosomes. Surprisingly, we found the gorilla Y Chromosome to be similar to the human Y Chromosome, but not to the chimpanzee Y Chromosome. Moreover, we have utilized the assembled gorilla Y Chromosome sequence to design genetic markers for studying the male-specific dispersal of this endangered species.
The sequence of the mammalian male-specific sex chromosome—the Y—is crucial for understanding male infertility disorders (Case and Teuscher 2015), population genetics of male-specific dispersal (Mendez et al. 2011; Karmin et al. 2015), and male mutation bias (Kuroki et al. 2006; Hughes et al. 2010, 2012a; Wilson Sayres et al. 2011; Li et al. 2013). Despite its importance, the sequence of the Y Chromosome has so far been determined only for a handful of mammals—human, chimpanzee, rhesus macaque, mouse, and pig (Skaletsky et al. 2003; Hughes et al. 2010, 2012a; Soh et al. 2014; Skinner et al. 2016), as well as partially sequenced for bull, dog, cat, marmoset, opossum, and rat (Chang et al. 2013; Li et al. 2013; Bellott et al. 2014).
The paucity of mammalian Y Chromosome assemblies is partially due to the haploid nature of this chromosome. Many mammalian genome projects have focused on females to obtain reliable X Chromosome sequences (Rozen et al. 2003; Graves 2010). Even when the Y is targeted, its unusual highly repetitive structure makes it the most challenging mammalian chromosome to sequence and assemble. Indeed, although the X has largely retained the ancestral autosomal structure and gene content (Graves 2010), the Y has undergone degradation via the accumulation of repeats and gene loss (Charlesworth and Charlesworth 2000; Skaletsky et al. 2003). The primate Y Chromosome, for example, is composed of pseudoautosomal regions (PAR), and X-degenerate, ampliconic, X-transposed, and heterochromatic regions (Skaletsky et al. 2003). The recombining PAR is present in both Y and X Chromosomes. The X-degenerate regions—the live remnants of the progenitor autosomes—harbor single-copy ubiquitously expressed genes with homologs on the X. The ampliconic regions are repetitive regions that contain palindromes (inverted repeats from several kilobases to several megabases long), whose arms are >99.9% identical (Rozen et al. 2003) and which harbor multicopy genes important for spermatogenesis (Bhowmick et al. 2007). The X-transposed region, detected so far only on the human Y, was created via a transposition from the X to the Y after the human–chimpanzee split (Skaletsky et al. 2003). The heterochromatic regions have high interspersed repetitive content and have not been entirely sequenced for any primate Y. These characteristics pose technical challenges requiring the development of specialized methods to sequence and assemble the mammalian Y Chromosome.
The main method has been single-haplotype iterative mapping and sequencing (SHIMS), which was used to sequence the human, chimpanzee, macaque, and mouse Y Chromosomes (Skaletsky et al. 2003; Hughes et al. 2010, 2012a; Soh et al. 2014). Although it is highly accurate, SHIMS remains expensive and tedious. Novel sequencing technologies have opened opportunities to make Y Chromosome sequencing faster and more affordable. One such approach sequences both male and female genomes and uses a differential analysis to identify Y-linked contigs (Carvalho and Clark 2013; Vicoso et al. 2013). However, this approach still requires substantial amounts of whole-genome sequencing.
In this study, we propose a cost-effective alternative method that integrates both existing and novel experimental and computational strategies. We first use flow sorting to significantly enrich the DNA for Y sequence and then apply both short- (Illumina) and long-read (Pacific Biosciences) technologies. We then combine existing assembly tools with a new algorithm, RecoverY, to efficiently identify Y-specific reads from the flow-sorted material. Finally, our assembly is augmented by testis transcriptome reconstruction, which is instrumental in building the Y Chromosome gene catalog, and by the estimation of the sizes of ampliconic gene families using droplet digital PCR (ddPCR) (Hindson et al. 2011).
We apply our strategy to produce a draft de novo assembly of the gorilla Y Chromosome. Gorilla diverged from the human–chimpanzee common ancestor 6–10 million years ago (Mya); however, only the genome of gorilla female has been so far sequenced (Scally et al. 2012). The sequence of the gorilla Y Chromosome is important for several specific applications. It is an endangered species, and the Y sequence can be used to design genetic markers to study male-specific dispersal patterns. It is also important to inform the evolutionary history of the hominine (human, chimpanzee, and gorilla) Y Chromosomes, two of which—human Y and chimpanzee Y—were recently found to be highly divergent from each other (Hughes et al. 2010). Importantly, our strategy can be applied to reconstruct the sex chromosomes present in the heterogametic sex (Y or W) of other species.
Results
We used an integrated strategy to sequence and assemble the gorilla Y Chromosome. In short, as the first step, the Y Chromosome was flow sorted. Next, Illumina paired-end (PE) and mate pair (MP), as well as Pacific Biosciences (PacBio), libraries were constructed. From the resulting Illumina reads, we extracted the Y Chromosome–specific reads using a novel algorithm developed in-house, RecoverY (see below). Next, such reads were assembled into contigs with SPAdes (Bankevich et al. 2012) and scaffolded with SSPACE (Boetzer et al. 2011). PacBio reads were used to further scaffold the assembly with SSPACE-LR (Boetzer and Pirovano 2014) and to close assembly gaps with PBJelly (English et al. 2012). The resulting assembly was additionally improved by creating super-scaffolds based on transcript information. To resolve the copy number of ampliconic genes, we utilized ddPCR (Hindson et al. 2011).
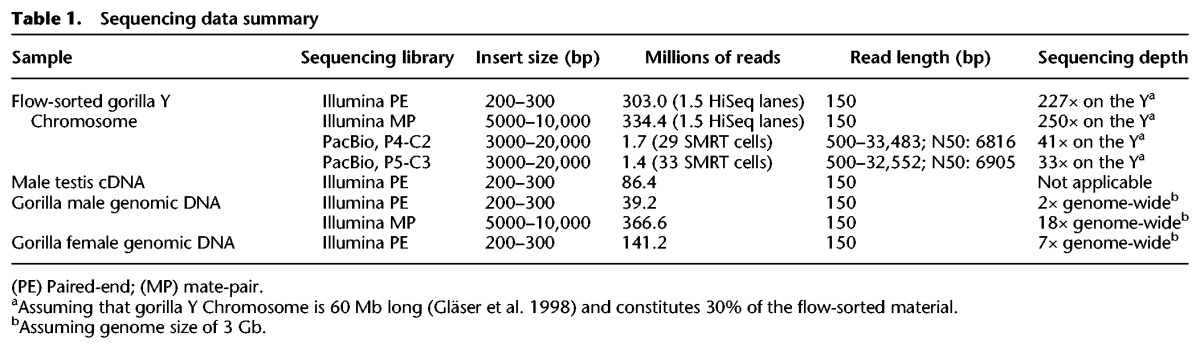
Flow sorting and sequencing
Approximately 12,000 copies of the Y Chromosome were flow sorted (Supplemental Fig. S1) from a fibroblast cell line of western lowland gorilla male. The flow-sorted DNA was used as a template for whole-genome amplification (WGA) (Supplemental Figs. S2–S3; Supplemental Table S1). The WGA DNA was utilized to construct three types of sequencing libraries, i.e., Illumina PE, Illumina MP, and PacBio, as specified in Table 1. Our analysis indicated that Chromosome Y constituted ∼30% of sequenced flow-sorted material (the rest might be debris from the other chromosomes; see below), in sharp contrast to sequencing gorilla male DNA, in which only 1%–2% of reads come from the Y (Supplemental Table S2). In this analysis, to remove the mapping bias caused by repetitive elements, we used the RepeatMasked human Y as reference and Bowtie 2 (Langmead and Salzberg 2012) as it offered a relatively unbiased mapping (Supplemental Table S2). Thus, sequencing of the Illumina (for the PE and MP libraries combined) and PacBio libraries resulted in depths of ∼477× and ∼74×, respectively, for gorilla Y (Table 1; Supplemental Fig. S4). Sequencing depth analysis suggested that WGA did not introduce any gross biases in the subsequent read distribution (Supplemental Fig. S5), although some sequences were potentially not amplified. Additionally, cDNA from gorilla testis was sequenced to assemble the transcriptome, and genomic (and not flow sorted) DNA of gorilla male and female was sequenced at low depth for validation (Table 1).
Table 1.
Sequencing data summary
RecoverY: extracting Y Chromosome–specific reads
Flow sorting greatly enriches the content of the Y Chromosome; however, as any other enrichment technique, it is not 100% efficient. For instance, as the Y Chromosome is small, it might flow sort together with debris from other chromosomes (Supplemental Fig. S6). To further increase the contribution of Y-specific reads to our assembly, we developed the RecoverY algorithm that separates Y and non-Y reads based on differential sequencing depth. RecoverY plots the distribution of the number of occurrences (the abundance) of k-mers from the flow-sorted read data. We used the k-mer size of 25 that was selected after testing RecoverY on a range of k values from 15 to 25 using simulated data from the human genome with Y enrichment ranging from 10% to 50%. We found that the maximum number of Y-specific k-mers was recovered at k = 25. The distribution reveals two categories of k-mers (Fig. 1A): low-abundance k-mers from sequencing errors, autosomes and Chromosome X, versus high-abundance k-mers from the Y and from transposable and other repetitive elements. RecoverY applies an abundance threshold to classify the k-mers and then filters out reads in which more than half of the constituent k-mers have an abundance lower than the chosen threshold. This strategy is designed to retain reads from the Y and from transposable and other repetitive elements, along with PAR found on both X and Y Chromosomes, while filtering out reads from the X and the autosomes. We note that RecoverY has the potential to be applied more generally to reads from any flow-sorted, or otherwise enriched (e.g., microdissected), chromosome.
Figure 1.
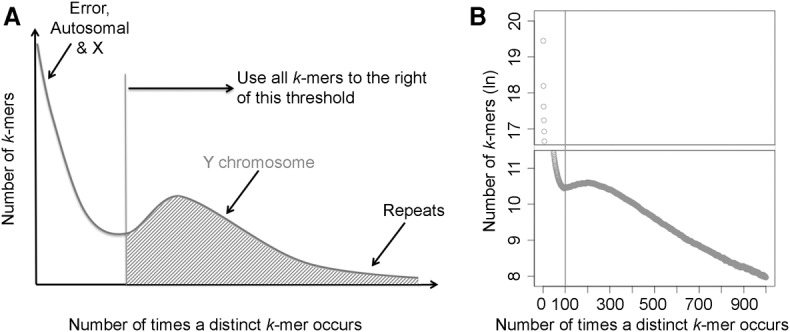
RecoverY—a novel algorithm for extracting Y Chromosome–specific reads from sequences of flow-sorted material. (A) The expected distribution of k-mer abundances. (B) The abundance of k-mers from paired-end flow-sorted gorilla Y sequencing data. The k-mers with an abundance greater than 100 are considered to be Y-specific or repetitive.
Assembly
We explored combinations of different sequencing technologies (Illumina only versus Illumina combined with PacBio versus PacBio only), data processing approaches (e.g., the use of RecoverY), and assembly tools (Fig. 2A; Supplemental Table S3; see below). Specifically, we evaluated the performance of these approaches and tools in terms of the total length assembled, N50 (Fig. 2A), NG50 (Supplemental Table S3), and the number of genes and palindromes recovered (Fig. 2B,C).
Figure 2.

(A) The global workflow applied for the Y Chromosome assembly (see text for details). Four assemblies in the dotted frame are nested within each other. The best assembly is framed in red. (Orange) Illumina data; (blue) PacBio data. All assemblies were filtered against the reference female genome. The total (including Ns) and unambiguous (non-N, shown in parentheses) lengths are shown. N50 is the contig/scaffold length for which all contigs/scaffolds of that length or longer contain half of the assembly length. (B) Gene and (C) palindrome recovery. The heatmaps show how sequences homologous to 25 human genes, eight human palindromes, and 12 chimpanzee-specific palindromes were recovered in the assemblies (see Methods). Genes lost on the chimpanzee Y are marked with an asterisk.
Our best assembly had a total length of 25.4 Mb with a scaffold N50 of 97.45 kb and an NG50 of 99.19 kb (assuming that the size of the euchromatic portion of gorilla Y Chromosome is the same as that for human Y, i.e., ∼25 Mb) (Skaletsky et al. 2003). This assembly was generated using both short reads (Illumina data) and long reads (PacBio data), applying a succession of assembly and scaffolding tools that led to assembly improvement at each step (Fig. 2A). First, we applied the RecoverY algorithm to the Illumina PE and MP reads, reducing the number of reads by ∼35% (Fig. 1B; Supplemental Fig. S7). The remaining reads were further subsampled using in silico normalization to reduce downstream memory and computational requirements (Haas et al. 2013). As a result, we obtained 12.5 and 20 million pairs of reads for PE and MP data sets, respectively, that were used in subsequent steps. Next, the PE reads were assembled into contigs using the SPAdes genome assembler (Bankevich et al. 2012). The initial development of SPAdes focused on single-cell sequencing data. As a result, SPAdes does not make any assumptions about coverage, which is advantageous for the assembly of flow-sorted data having unique coverage patterns. The use of RecoverY resulted in fewer and larger contigs and in a smaller number of non-Y contigs, as compared to not using RecoverY (Supplemental Table S4). These contigs were then scaffolded with SSPACE (Boetzer et al. 2011) using MP reads.
In the next step, PacBio reads longer than 12 kb were error corrected with HGAP (Chin et al. 2013) using the full PacBio data set, resulting in 153,310 error-corrected long reads (a total of 666 Mb of sequence). Next, we ran SSPACE-LR (Boetzer and Pirovano 2014), which utilizes these error-corrected long reads to improve the Illumina assembly by merging scaffolds and filling in gaps between contigs. Smaller read length thresholds for HGAP yielded a larger number of corrected sequences with a smaller average length, but did not improve the results of SSPACE-LR (Supplemental Table S5). Finally, we applied PBJelly (English et al. 2012) to align uncorrected PacBio subreads longer than 10 kb to minimize the gaps in the assembly (Supplemental Note S1). The combination of SSPACE-LR and PBJelly produced a 42% increase in assembly size and a 270% improvement in scaffold N50 over the Illumina-only assembly (Fig. 2A).
To improve the Y-Chromosome specificity of our assembly, we aligned the resulting scaffolds to the gorilla reference female genome using the long-read aligner BLASR (Chaisson and Tesler 2012). The scaffolds mapping with a best hit of >70% identity to gorilla autosomes or non-PAR gorilla X Chromosome sequences were discarded. The number of scaffolds was thus reduced by 30%; however, the assembly length decreased by only 13%, indicating that the effect of non-Y reads that were undetected by RecoverY was limited to the formation of very short non-Y scaffolds. We named the resulting assembly as “the best assembly.” The insert size distribution of MP data mapping to the best assembly is presented in Supplemental Figure S8A. This assembly was evaluated with an independent scaffolder, BESST (Sahlin et al. 2014), which resulted in a similar insert size distribution of MP data (Supplemental Fig. S8B,C). We also evaluated the best assembly with REAPR (Hunt et al. 2013), which reported a relatively low proportion of assembly errors (Supplemental Table S6). A total of 55 scaffolds in the best assembly aligned to gorilla PAR (Supplemental Table S7).
We also performed two additional assemblies using only PacBio reads and the HGAP (Chin et al. 2013) and MHAP (Berlin et al. 2015) software, but found the results to be inferior to our best assembly (Fig. 2; Supplemental Note S2; Supplemental Table S3). The improvement in PacBio data analysis tools (which are undergoing active development) could make PacBio-only assemblies a more attractive option in the future.
Human and chimpanzee sequence alignments
We next aligned (see Methods) the best assembly to the sequences of human and chimpanzee Y Chromosomes. Because the gorilla lineage diverged prior to the human–chimpanzee split (Scally et al. 2012), we expected a similar sequence identity for the gorilla–human and gorilla–chimpanzee pairwise alignments. Indeed, at the nucleotide level, we observed highly similar sequence identities for gorilla–human (97.09%) and gorilla–chimpanzee (97.10%). Contrary to the expectation, though, different proportions of the gorilla Y aligned to the human Y and chimpanzee Y (83.4% and only 70.3%, respectively).
Experimental validations and genome rearrangements
To validate the best gorilla Y assembly experimentally, we first designed primers for 32 randomly selected regions (Supplemental Table S8) that aligned to human and/or chimpanzee Y Chromosome. Of these, 25 (78%) were validated (PCR products were obtained), one (3%) amplified from the WGA Y but not from male genomic DNA, and six (19%) could not be amplified or resulted in nonspecific PCR products.
After aligning the scaffolds of the best assembly to the human and chimpanzee Y Chromosomes, we identified alignment breakpoints (Methods; Supplemental Note S3). We found 162 putative gorilla-specific breakpoints that had PacBio read support in the gorilla but were present in neither the human nor the chimpanzee Y Chromosomes. We attempted to validate 42 of them with PCR using gorilla male DNA as a template (Supplemental Table S8). Of these, 32 (76%) were validated as gorilla-specific rearrangements (PCR products were obtained), four (10%) amplified from the WGA Y but not from male genomic DNA, and six (14%) could not be amplified or represented nonspecific PCR products.
Gene repertoire
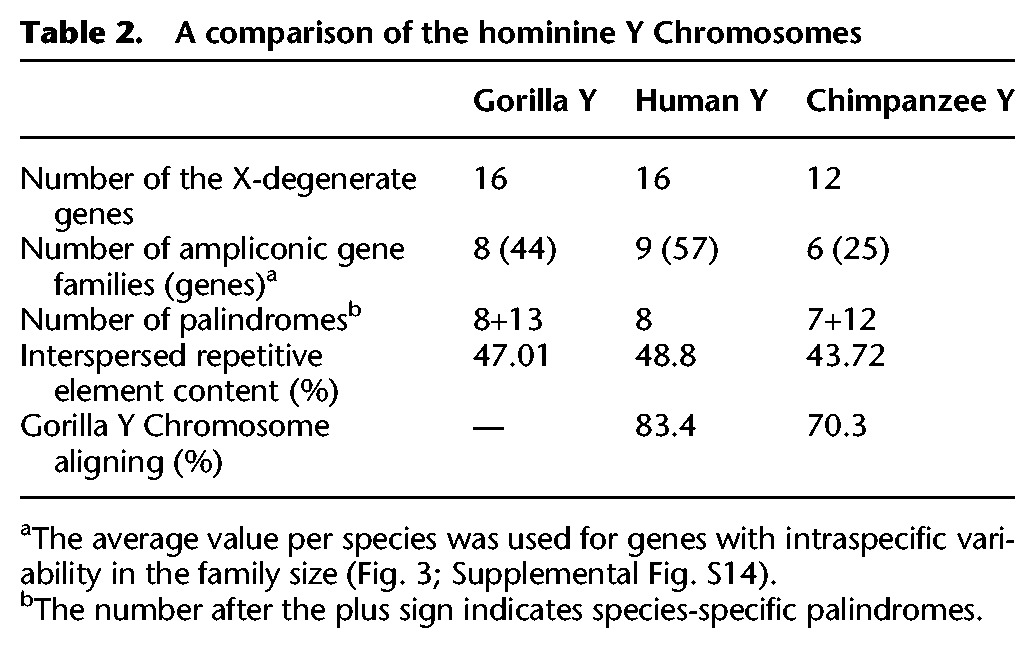
The gorilla Y gene repertoire we recovered from the best assembly was validated by the analysis of gorilla testis transcriptome and appears to be remarkably similar to that on the human Y but different from that on the chimpanzee Y. We detected in the best assembly (via alignment) (see Methods; Figs. 2B, 3) the homologs of all 16 human X-degenerate genes and eight of the nine human ampliconic gene families (Skaletsky et al. 2003). The VCY gene family was not detected, which we confirmed experimentally (see below). In contrast, the gorilla Y shares only 12 X-degenerate genes and five ampliconic gene families with the chimpanzee Y (Figs. 2B, 3; Table 2); TXLNGY, TBL1Y, TMSB4Y, USP9Y, and XKRY were pseudogenized, whereas HSFY and PRY families were lost, on the chimpanzee Y (Fig. 3; Hughes et al. 2010).
Figure 3.

A comparison of the gene content among the hominine Y Chromosomes. (A) X-degenerate genes. (B) Ampliconic genes.
Table 2.
A comparison of the hominine Y Chromosomes
We assembled the RNA-seq data from gorilla testis (Supplemental Table S9) and used various filtering strategies and additional gorilla and human data to reconstruct the gorilla Y-Chromosome genes (Methods; Supplemental File S1). This was performed independently of the Y Chromosome reference to validate our best assembly (a genome-guided transcriptome reconstruction including the Y Chromosome assembly as a reference led to inferior results). The best assembly accurately recovered the 24 gorilla Y protein-coding genes. For the 21 genes found in single copy (all but CDY, DAZ, and RBMY), 94.7% of exonic sequences were retrieved, and the position and orientation of >95% of their exons were consistent with the transcript data.
Using 11 X-degenerate genes whose exons were spread among multiple scaffolds, we merged 43 scaffolds into 11 super-scaffolds (Supplemental Fig. S9). TBL1Y alone guided the joining of four scaffolds, generating an ∼779-kb super-scaffold. No novel full-length protein-coding genes were found on the gorilla Y (Supplemental Note S4), but we found 59 noncoding transcripts (of which 13 were reported previously) (Cortez et al. 2014) and 166 expressed pseudogenes (out of 193 previously reported) (Cortez et al. 2014; Supplemental Table S10; Supplemental File S2).
Ampliconic genes
We found substantial intra- and interspecific variability in the sizes of ampliconic gene families (i.e., in the number of duplicate gene copies per family). The size of each of the families was estimated experimentally with ddPCR (Supplemental Table S11; Hindson et al. 2011). The approach was initially validated for nine ampliconic gene families in two human males (Fig. 4). We obtained copy numbers consistent with that in the reference human genome for all but two gene families (Supplemental Table S12)—RBMY and TSPY. For these two families, intraspecific variability in family size was noted previously (Tyler-Smith et al. 1988; Giachini et al. 2009; Case et al. 2015). Next, we examined the size of ampliconic gene families for 14 wild-born gorillas. The intraspecific size variation was observed for the RBMY and TSPY gene families (similar to human), but also for the CDY and HSFY gene families (Fig. 4; Supplemental Table S12). We found that ddPCR is a more reliable method for measuring gene family size than a computational analysis of the assembly (see Methods). Because these gene families tend to lie in the hard-to-assemble (with our strategy) palindromes, their family size is often underestimated in the assembly (Supplemental Table S13).
Figure 4.
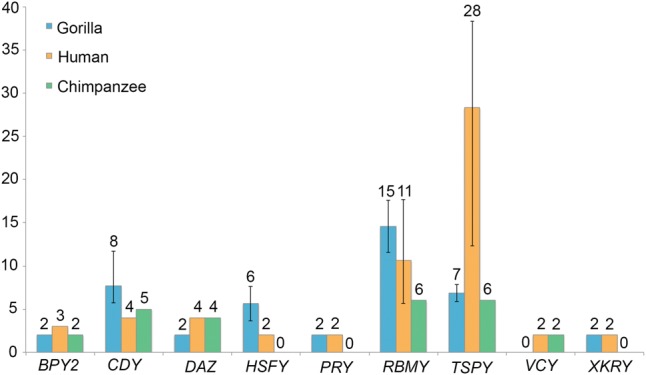
Sizes of ampliconic gene families on the hominine Y Chromosome. The number of functional genes was evaluated for 14 gorilla males using ddPCR (blue), evaluated for two human males using ddPCR and retrieved from the reference human genome sequence (orange), and retrieved from the chimpanzee reference genome sequence (green). For families with intraspecific size variation (Supplemental Table S12), size averages (numbers above bars) and ranges (error bars) are shown.
Palindromes
Our assembly contained sequences homologous to all eight palindromes present on the human Y (Fig. 2C; Methods). The sequences of P1, P2, and P8 were present only partially (Supplemental Table S14). In the case of P2, this was likely because of its highly repetitive structure (Supplemental Fig. S10); in the case of P8, this was confirmed by the absence of the VCY gene family, corroborating our analyses above (Figs. 2B, 3). The assembly also harbored complete or partial sequences homologous to nine of 12 chimpanzee-specific palindromes (Fig. 2C; Supplemental Note S5). Our analysis suggests that most homologs to human and chimpanzee palindromes have high read depth and thus likely also form palindromes in gorilla (Supplemental Fig. S11A–D). However, our ability to fully reconstruct the sequence of palindromes on the gorilla Y might be limited due to potential palindrome collapses in our assembly.
Additionally, the intra-scaffold sequence similarity analysis (see Methods) identified 13 novel, very short (6–16 kb long), gorilla-specific palindromes (Supplemental Fig. S12). The length of palindromes is limited by the length of our scaffolds, and thus their shortness is not indicative of the size distribution of gorilla-specific palindromes in general. The sequences homologous to gorilla-specific palindromes were present on the human Y and with one exception also on the chimpanzee Y, but did not exhibit the palindrome structure in these species (Supplemental Fig. S12).
Repetitive element content
The interspersed repetitive element content on the gorilla Y (47.0%) was similar to that on the human Y (48.8%), but higher than that on the chimpanzee Y (43.7%) (Supplemental Table S15; see Methods). The low repetitive element content on the chimpanzee Y is due to the relatively low LTR and SINE element content (Supplemental Table S15), the latter being consistent with the Alu insertion slowdown in the chimpanzee–bonobo common ancestor (Hormozdiari et al. 2013). The interspersed repetitive element content on the gorilla Y was different than that for gorilla autosomes (43.3%) and gorilla Chromosome X (52.2%). Similar to the human Y (Skaletsky et al. 2003), the gorilla Y ampliconic regions (defined here as scaffolds containing ampliconic genes) had a lower interspersed repetitive element content (42.5%) than the X-degenerate regions (51.0%). Note that we used the primate library for masking the repetitive elements and thus might have missed some gorilla-specific repeats.
Designing a panel of gorilla Y-specific microsatellite markers
To enable future studies of gorilla male-specific dispersal and migration (Douadi et al. 2007), we designed a panel of Y-specific microsatellite markers based on the assembly generated in this study (see Methods). This panel includes seven novel fluorescently labeled tri- and tetranucleotide microsatellite markers that can be assayed in a single run on ABI3700 and are polymorphic when tested in 14 wild-born gorillas (Supplemental Table S16).
Discussion
The strategy for sequencing sex chromosomes
Our proposed strategy for sequencing and assembling the Y Chromosome provides a more accessible alternative to existing approaches and can make sex chromosome reconstruction more widespread in the future while catalyzing novel biological discoveries. Compared with SHIMS (Skaletsky et al. 2003; Hughes et al. 2010, 2012a), the proposed strategy is several orders of magnitude cheaper and faster, making it affordable for many more laboratories. The strategy presented here is also more targeted and is thus more affordable than a strategy based on sequencing with the equivalent technologies of both male and female genomes for the same species (Carvalho and Clark 2013; Vicoso and Bachtrog 2013). Although we expect the overall quality of the assemblies to be comparable between the two approaches, we estimate that our targeted approach can decrease the cost of sequencing needed to achieve the same depth by at least 10 times (Supplemental Table S17). In particular, PacBio sequencing is still expensive, and obtaining enough coverage of the Y Chromosome through genome-wide sequencing of the male genome remains prohibitive.
We utilized flow sorting based on chromosome size and GC-content to enrich for the Y-Chromosome DNA, which contributed to the increased coverage of this small chromosome. This approach might be challenging to apply for very young sex chromosomes that might have not diverged enough in size and GC-content from each other. However, individual chromosomes can also be enriched by microdissection (Zhou and Hu 2007) or laser capture dissection (Keinath et al. 2015), and the computational techniques developed here also have the potential to be utilized in such situations, thus increasing the applicability of the method. Therefore, our approach provides a timely opportunity to generate data needed for the studies on sex chromosome evolution and sex-bias in dispersal across populations. Such data can provide significant new insights and can find immediate applications, e.g., to conservation genetics of endangered species.
The power of our strategy is in combining the unique strengths of orthogonal experimental and computational approaches to reconstruct a detailed picture of the Y Chromosome. By combining flow sorting with a novel computational method RecoverY, we are able to enrich our read data sets for the Y Chromosome sequence and make sequencing faster and more cost effective. By combining short- and long-read technologies, we are able to obtain high coverage while also increasing assembly contiguity. Compared with the use of short reads only, this resulted in fewer scaffolds and an almost threefold increase of N50 (Fig. 2A). By sequencing the testis transcriptome, we can build an improved gene catalog and detect novel transcripts. By demonstrating how ddPCR can be used to measure the size of ampliconic gene families, we can enable future studies of male fertility (Nickkholgh et al. 2010). This is particularly important because of an association of the sizes of some ampliconic families with fertility in men (Elliott 2000; de Vries et al. 2002; Writzl et al. 2005; Nickkholgh et al. 2010).
Our study provides a workflow for future sequencing projects of sex chromosomes present in the heterogametic sex—Y and W (Fig. 5). By generating an abundance of data, we were able to determine levels of coverage beyond which the benefits became incremental. A preliminary sequencing run (e.g., on MiSeq) can test for chromosome enrichment in the flow-sorted DNA. If the enrichment is satisfactory, we recommend sequencing to the chromosome-specific depth of ∼50× for each of the Illumina PE and MP data (Supplemental Note S6). Whereas we generated an amount of PacBio data that may not be cost effective for other projects, we show that ∼5–10× sequencing depth already provides a substantial improvement of Illumina assemblies for Y Chromosomal data (Supplemental Fig. S13A,B). To achieve the highest N50, the most accurate PacBio chemistry should be used, and fewer PacBio long reads are preferred to many shorter reads (Supplemental Fig. S13C).
Figure 5.
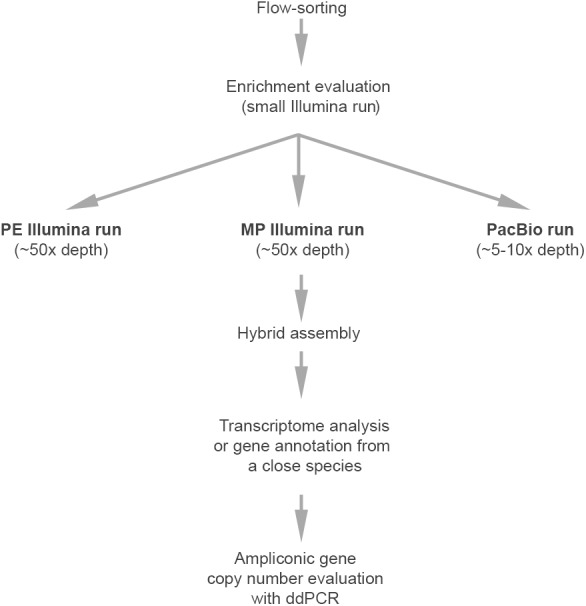
The workflow for sequencing mammalian Y Chromosomes.
Notwithstanding its advantages, our strategy remains less accurate and produces a more fragmented assembly than SHIMS. WGA, unavoidable when working with limited material, might introduce artificial junctions (Lasken and Stockwell 2007). Although we demonstrate that such artifacts are rare (Supplemental Table S8), the WGA step should be omitted if the material is more abundant. Also, as the longest PacBio reads (Table 1) are shorter than most palindrome arms (Supplemental Table S14), we cannot resolve all palindromes. In situations in which palindrome reconstruction is critical, SHIMS (Skaletsky et al. 2003) could be used.
A comparison of the hominine Y Chromosomes
We have demonstrated that the assemblies produced by our strategy are highly informative, despite these potential limitations. In applying this strategy to the gorilla Y Chromosome, we refined its gene repertoire, identified several lineage-specific palindromes, determined the interspersed repetitive element content, and generated its alignments with human and chimpanzee Y Chromosomes, which allowed us to compare hominine Y Chromosomes.
Our analysis of the sequence alignments indicated that the Y Chromosome gene tree among hominines studied was congruous with the species tree. At the nucleotide level, we observed a greater sequence identity between human and chimpanzee (97.99%) than between either of those and gorilla, consistent with chimpanzee and human sharing a more recent common ancestor (Scally et al. 2012). Moreover, the gorilla–human and gorilla–chimpanzee identities were highly similar (97.09% and 97.10%). These results are inconsistent with incomplete lineage sorting that would lead to higher gorilla–human than gorilla–chimpanzee sequence identity or vice versa. Note that the use of different sequencing data and assembly approaches for the gorilla Y Chromosome could have affected sequence identity values in alignments, including this chromosome.
We found the gorilla Y to be more similar to the human than to the chimpanzee Y in terms of shared palindrome sequences, the percentage of aligned sequence, the interspersed repetitive element content, and gene repertoire (Table 2). Although the gorilla and human Y Chromosomes share all but one (VCY) protein-coding gene family (Table 2), the chimpanzee Y lost one-quarter of X-degenerate (Goto et al. 2009) and one-third of ampliconic gene families compared to the gorilla or human Y (Table 2; Fig. 3). The smaller number of gene families on the chimpanzee Y compared with human Y is consistent with a high rate of gene loss on chimpanzee autosomes and Chromosome X (Demuth et al. 2006). In fact, the proportion of Y gene families among all gene families is not significantly different between chimpanzee and human (18/9,711 versus 25/10,374, P = 0.395, Z-test). However, the overall number of genes on the chimpanzee Y Chromosome is one-half that on the human or gorilla Y (37 versus 73 or 60, respectively) (Table 2), and the proportion of Y among all genes is significantly lower for chimpanzee than human (37/20,984 versus 73/22,836, P = 0.002, Z-test), suggesting additional forces acting on the chimpanzee Y.
We hypothesize that one such force could be selection potentially elevated in the chimpanzee lineage due to polyandrous mating and resulting sperm competition (Møller 1988; Dixson 2012). In agreement with this hypothesis, the DAZ ampliconic gene family evolves under positive selection in the chimpanzee lineage (Hughes et al. 2012b), and we found significantly higher nonsynonymous-to-synonymous rate ratios on the chimpanzee than on the human or gorilla Y Chromosome for five X-degenerate (DDX3Y, EIF1AY, PRKY, KDM5D , and SRY) (Supplemental Table S18) as well as for one ampliconic gene (CDY) (Supplemental Table S19); none of the ratios was significantly greater than one (Supplemental Note S7). Selection was likely accompanied by genetic hitchhiking, particularly strong on the Y because of no recombination (Charlesworth and Charlesworth 2000; Bachtrog 2008), and increasing the presence of nonadaptive mutations that could include gene and palindrome loss on the chimpanzee Y (Hughes et al. 2010). When the data on the Y Chromosome structure and gene sequences in other primates with different levels of sperm competition become available, one will be able to more explicitly test a hypothesis about sperm competition shaping Y Chromosome evolution. Currently the only other primate (non-hominine) Y Chromosome deciphered is the macaque Y (Hughes et al. 2012a) that is remarkably similar in gene content to the human and gorilla Y Chromosomes (Supplemental Fig. S14). This similarity is despite the presence of sperm competition in macaque, which, according to our hypothesis, would lead to a disparate gene content on the macaque Y (Møller 1988; Dorus et al. 2004; Dixson 2012).
Only five of nine ampliconic gene families were shared by all hominine species compared (Fig. 3) and four also with macaque (Supplemental Fig. S14B). Some gene family losses might be random; however, as most ampliconic genes are expressed in testis (Skaletsky et al. 2003), such rapid alteration in their content might be associated with changes in sperm production among species (Bhowmick et al. 2007). Our results indicate a remarkable level of variability in the ampliconic gene family size among hominines and within gorilla—with six and four of nine gene families displaying inter- and intraspecific variability, respectively (Fig. 4). All but one (TSPY) of the studied ampliconic gene families are located within palindromes (Bhowmick et al. 2007). Mechanistically, such organization facilitates recombination within individual and among homologous palindromes and results in frequent gene gain and loss within families, as well as in gene conversion (Rozen et al. 2003) that counteracts the degeneration of the Y by efficiently removing deleterious mutations (Connallon and Clark 2010).
Applications for conservation genetics
The novel fluorescent Y-Chromosome microsatellite multiplex assay presented here can be used to investigate gorilla male dispersal patterns determining the genetic diversity of gorilla populations in the wild (Douadi et al. 2007). Such patterns influence population structure; thus, their analyses will ultimately aid conservation efforts on behalf of this endangered species that faces numerous threats, including habitat loss and disease impacts (Genton et al. 2014). The designation of protected areas that encompass the largest possible extent of the species genetic diversity is thus strategic for long-term conservation of wild gorilla populations.
Methods
Samples, flow sorting, WGA, and sequencing
Gorilla Y Chromosome was flow sorted from a fibroblast cell line of western lowland gorilla male (ID KB3781). Flow sorting of the gorilla Y was performed as described (Yang et al. 1995). Laser-based flow cytometry consists of applying an electrical charge to droplets containing chromosomes of interest. Chromosomes to be sorted are classified according to the size and A/T to G/C base pair ratio. The Y-Chromosomal markers were used to confirm this chromosome-sorted specificity. Flow-sorted Y DNA was subsequently used as a template for WGA performed with REPLI-g Single Cell Kit (Qiagen). Male genomic DNA was extracted from the same sample. Gorilla female genomic DNA (ID 2000-0150) was isolated from liver with the DNeasy Blood and Tissue kit (Qiagen). RNA from gorilla testis (ID 2006-0091) was extracted with the RNeasy Mini kit (Qiagen). Additional DNA samples from 13 wild-born western lowland gorilla males (Supplemental Table S12) were provided by the San Diego Zoological Society. PE, MP, and stranded RNA-seq libraries were constructed with the TruSeq DNA Sample Preparation Kit (Illumina), Nextera Mate Pair Library Preparation Kit (Illumina), and TruSeq RNA Sample Prep Kit (Illumina), respectively. These libraries were sequenced on the HiSeq 2500 (Rapid mode). Prior to constructing PacBio libraries, we performed debranching (Zhang et al. 2006).
Preprocessing raw read data
We trimmed adapters in raw reads using Trimmomatic version 0.32 (http://www.usadellab.org/cms/index.php?page=trimmomatic) with the following settings: ILLUMINACLIP: ${adapter_file}:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:30. PE reads were trimmed in “palindrome” mode, and MP reads were trimmed in “simple” mode. All FASTQ files were checked with fastQValidator (http://github.com/statgen/fastQValidator). We additionally preprocessed the MP reads using NxTrim (http://github.com/sequencing/NxTrim), which classified reads into three categories based on the location of biotinylated adapters, e.g., MP (long fragment size), PE (short fragment size), and unknown (adapter absent, but validation suggests mostly long fragment size). The “PE contamination” was eliminated, retaining approximately two-thirds of the MP data.
RecoverY
RecoverY (version 1.0) is composed of the following steps: (a) Run the DSK (version 2.0.2) (Rizk et al. 2013) k-mer counter on the quality-controlled reads and construct an abundance histogram depicting the count of each distinct k-mer (k = 25, in our case); (b) choose a threshold to separate putative Y Chromosome k-mers from other erroneous, autosomal, or X Chromosome k-mers; (c) store the putative Y k-mers in a hash table for efficient retrieval; and (d) flag a read to be Y-Chromosomal if the majority of its constituent k-mers are present in the hash table. We used conservative abundance thresholds of 100× for PE and 50× and 10× for the two MP libraries.
Assemblies
The remaining post-RecoverY reads were subsampled using in silico normalization in Trinity (version 20140717) (Grabherr et al. 2011), with a target coverage of 220× of the Y Chromosome. The target coverage was chosen to be sufficiently high to ensure that reads at a lower coverage would be retained by Trinity normalization, whereas reads at a much higher coverage would be mostly eliminated. The PE and MP reads were normalized independently. The PE reads were provided to SPAdes (version 3.1.1) (Bankevich et al. 2012) with parameters --only-assembler --careful -t 32. The contigs produced were scaffolded using MP reads by SSPACE (version 3.0) (Boetzer and Pirovano 2014). SSPACE was run with default parameters, which required either a minimum of five mate pair links or 15-bp overlaps to merge contigs into a scaffold. As an alternative to SPAdes and SSPACE, we used ALLPATHS-LG (release 47875) (Gnerre et al. 2011); however, this resulted in more scaffolds that were also shorter and in fewer genes and palindromes recovered (Supplemental Table S3). Therefore, we proceeded with the SPAdes/SSPACE assembly. Using >12-kb PacBio reads error-corrected with HGAP (Chin et al. 2013), we ran SSPACE-LR (version 1-1) (Boetzer and Pirovano 2014) to improve the scaffolding. We applied PBJelly (PBSuite_14.9.9) (English et al. 2012) to align uncorrected >10-kb PacBio subreads to close gaps in the resulting assembly and filter it against the gorilla female genome (see next section). We used the following BLASR parameters for the PBJelly (PBSuite_14.9.9) protocol: <blasr>-minMatch 8 -minPctIdentity 85 -bestn 1 -nCandidates 20 -maxScore -500 -nproc 60 –noSplitSubreads </blasr>. For PacBio-only assemblies, see Supplemental Note S2.
Autosomal and X Chromosome contamination filtering
We concatenated the gorilla female genome (gorGor3), the human Y Chromosome (GRCh38), and the chimpanzee Y Chromosome (gi|326910934|gb|DP000054.2) into a single reference. Accounting for the divergence of gorilla from the chimpanzee–human ancestor, and the error rate of PacBio data, we performed mapping of scaffolds to this reference using BLASR (Chaisson and Tesler 2012) with a 70% minimum percentage identity required for a match. We then filtered out scaffolds whose best mapping was to the non-PAR region of the gorilla female. We also filtered out short (<1 kb) scaffolds.
Repeat masking
Repeats on the gorilla Y were identified with RepeatMasker version open-4.0.5 (http://www.repeatmasker.org), search engine NCBI/RMBLAST [2.2.27+] and database RepeatMaskerLib.embl (20140131) with parameters “-species Primates -s.” The repetitive element content for the ampliconic and X-degenerate regions was computed from scaffolds containing exons of the ampliconic and X-degenerate genes, respectively. In order to make the comparison of repeat content consistent between different genomes, we re-ran RepeatMasker on human Y Chromosome, chimpanzee Y Chromosome, and gorilla female genome using the same parameters as for the gorilla Y.
Chromosome sequence alignments
Scaffolds from the gorilla assembly were aligned to the latest human and chimpanzee Chromosome Y sequences using LASTZ version 1.03.66 (Harris 2007). Masking was disabled to allow the reporting of alignments for duplicated elements. We set substitution scores identical to those used for LASTZ alignments of primates generated by the UCSC (Miller et al. 2007) but used more relaxed gap scores. The exact LASTZ command line was: lastz human.chrY gorilla.contigN.chrY W=12 O=500 E=30 K=3000 L=4500 X=900 Y=15000 Q=human_primate.scores. The identity distribution was estimated by attributing to each location in gorilla the highest identity of any alignment crossing that location. We discarded short alignments (less than 30 alignment columns) and any alignments with lower than 94% identity, following Hughes et al. (2010). To validate our alignment procedure, we first aligned the human and chimpanzee Y Chromosomes. The resulting nucleotide identity (97.99%) was similar to that reported previously (Hughes et al. 2010). Alignments with lower than 94% identity contributed to the proportion of gorilla aligned to human or chimpanzee, reported as the ratio of aligned bases to non-N bases.
Retrieval of genes and palindromes in the assembly
To test for gene and palindrome presence, we mapped human gene and gorilla or human palindrome sequences to the best assembly, using BWA (version 0.7.5a-r428) (Li and Durbin 2009) with seed length = 5 to increase sensitivity. This procedure captured the presence of at least one copy and evaluated neither possible fragmentation nor copy number.
Transcriptome analysis
Testis RNA-seq reads were mapped to the gorilla female reference genome with TopHat2 (v.2.0.10) (Kim et al. 2013), and the unmapped reads (enriched for male-specific transcripts) were assembled with Trinity (version 20140717) (Haas et al. 2013) and SOAPdenovo-Trans (Luo et al. 2012) with k-mer size of 25 bp. The generated contigs were aligned to the gorilla female reference genome with BLAT (Kent 2002), and contigs that aligned at >90% of their length with 100% identity were removed from subsequent steps. Additionally, we removed contigs that were covered at >90% of their length by mapped female gorilla RNA-seq reads from another study (Brawand et al. 2011). The contigs were then repeat-masked (RepeatMasker open-3.3.0, Repbase library with parameters -s -species ‘mammal’) (http://www.repeatmasker.org) and combined to generate gene consensus sequences with TGICL (Pertea et al. 2003). We then scaffolded the TGICL contigs using SSPACE (version 3.0) (Boetzer et al. 2011). We next mapped male and female genomic reads to the gene scaffolds with Bowtie 2 (v.2.1.0) (Langmead and Salzberg 2012) and retained only male-specific gene scaffolds (with at least 80% of the sequence covered by male-specific reads and no more than 20% of the sequence covered by female-specific reads). Following Brawand et al. (2011), we utilized a threshold of <20% (we also tested 10%) of a transcript covered by female genomic reads to retain the Y-specific transcripts as some regions of the Y Chromosome are almost identical to the X Chromosome, particularly the gametologous genes. The mapping of RNA-seq and genomic reads was performed with the local alignment, and the read threshold was equal to 1. Lastly, the RNA-seq reads were mapped back to the final transcripts to evaluate coverage and gene sequence reconstruction. Annotation of the final transcripts was performed using nucleotide and protein databases. The transcripts generated here, the gorilla transcripts from Cortez et al. (2014) and the gorilla X-degenerate genes from Goto et al. (2009), as well as human Y genes/cDNAs (http://www.biomart.org), were aligned to the best assembly with BLAT (Kent 2002). Focusing on the matches with identities >95% for gorilla and >90% for human, we determined the level of completeness for each gene in the best assembly and performed additional ordering of the scaffolds based on exon connectivity of certain genes that spanned several scaffolds, resulting in super-scaffolds.
Ampliconic gene number estimation with ddPCR
Primers for the ddPCR assays (Supplemental Table S11) were designed with Primer3Plus (v2.3.6) using parameters recommended in the Droplet Digital PCR Applications Guide (Bio-Rad). General parameter settings were: product size range of 60–150 bp; primer size of 15–30 nt with an optimum of 22 nt; melting temperature (Tm) range of 58°C–65°C with an optimum of 62°C; GC content range of 50%–60% with an optimum of 55%; 50 mM monovalent cations; 50 nM annealing primer; 3.8 mM divalent cations; 0.8 mM dNTPs; and the human mispriming/repeat library. Advanced parameter settings were default except that GC clamp was turned on, the maximum end GCs was three, the maximum end stability was ≤3.0, and sometimes the maximum hairpin was 30. Primers were first designed for the human Y by targeting only a section of the known functional ampliconic genes not found in any pseudogenes according to the latest annotation of the human Y Chromosome (GCF_000001405.26 GRCh38/hg38). This approach enabled primer targeting of all the known functional ampliconic genes, but none of the known pseudogenes on the human Y (Supplemental Table S11). The exception was TSPY in which a section of the known functional ampliconic genes was also present in some of the pseudogenes, so the section that hits the least number of pseudogenes was used. Once it was demonstrated with ddPCR that a primer pair captures the in silico determined ampliconic gene copy number on the human Y, the gorilla Y Chromosome best assembly was searched with the human ddPCR amplicon to locate scaffolds containing this amplicon with high identity (≥95%) and 100% coverage. These gorilla scaffolds were also examined to determine that the amplicon is located within a gene annotated as the respective ampliconic gene. None to small manual changes in the human primers enabled targeting the homologous gorilla amplicon (Supplemental Table S11). The sequence specificity of primers used was determined via BLASTn against the appropriate species-specific databases in GenBank (i.e., nucleotide collection, reference genomic sequences, NCBI genomes, reference RNA sequences, and transcriptome shotgun assembly). All ddPCR primers and amplicons were confirmed through forward and reverse Sanger sequencing of a PCR product consisting of the amplicon ± 200 bp, and then alignment of these sequences to each other and the respective primers, amplicon, and the best assembly.
Quantification of gene copy number was performed by ddPCR using a Bio-Rad QX200 Droplet Digital PCR system (Hindson et al. 2011; McDermott et al. 2013). Simplex sample PCR reaction mixtures (20 µL) contained the final concentration of the following components: 1× EvaGreen Supermix (Bio-Rad), 100 nM of each primer, 0.1 unit/µL of HindIII, and template DNA at 0.5, 1.0, or 2.0 ng/µL (i.e., 10, 20, or 40 ng/reaction) depending on the expected gene copy number. Formation of droplet emulsions was performed by mixing 20 µL of PCR reaction and 70 µL of EvaGreen droplet generation oil with the Automatic Droplet Generator (Bio-Rad). These emulsions of about 20,000 droplets contained in a 96-well plate were cycled to amplicon saturation using a C1000 Thermal Cycler (Bio-Rad) operating at the following conditions: for 5 min at 95°C, 45 cycles of 30 sec at 94°C and for 1 min at 55°C–65°C, for 5 min at 4°C, for 5 min at 90°C, and a 4°C hold. Amplitude of fluorescence by amplicons in each cycled droplet was measured using flow cytometry on a QX200 Droplet Reader (Bio-Rad) set on the EVA channel. The QuantaSoft droplet reader software (v1.4.0.99; Bio-Rad) was used to cluster droplets into distinct positive and negative fluorescent groups and fit the fraction of positive droplets to a Poisson algorithm to determine the starting concentration (copies/µL) of the input target DNA molecule ([Miotke et al. 2014], Droplet Digital Applications Guide). Copy number was determined by calculating the ratio of the target (unknown) concentration to the reference concentration and then multiplying this ratio by the number of copies the reference gene has in the genome ([Miotke et al. 2014], Droplet Digital Applications Guide). Two reference genes used simultaneously were the single-copy Y-Chromosomal SRY and the two-copy (diploid) autosomal RPP30. Mean copy number are reported with the Poisson 95% confidence interval calculated by QuantaSoft (Supplemental Tables S11, S12).
Detection of microsatellites and development of a fluorescent assay
Microsatellites in the best assembly were detected with PHOBOS v3.3.12 (http://www.rub.de/spezzoo/cm/cm_phobos.htm). We searched for uninterrupted tri- and tetranucleotide microsatellites because they are easier to score than dinucleotide microsatellites (Eckert et al. 2002; Ananda et al. 2013). BLAST analyses of the designed microsatellites have been performed against the NCBI nucleotide database to check for their gorilla Y specificity. Subsequently, seven gorilla Y-specific microsatellite amplifications were performed in each of the 14 wild-born gorilla males in GeneAmp PCR system 9700 (Applied Biosystems) using the following thermal conditions: initial denaturation at 94°C, 30 or 35 cycles of: denaturation for 1 min at 94°C, annealing at primer specific temperature for 45 sec, extension for 45 sec at 72°C; followed by a final extension for 5 min at 72°C. The forward primer of each of the seven primer pairs was labeled with one of the fluorescent dyes compatible with Applied Biosystems 3730XL sequencer: FAM, HEX, NED, or ROX (Supplemental Table S16). PCR reaction mixtures consisted of 20 ng DNA, 1 unit of ChoiceTaq DNA polymerase (Denville Scientific), 10× PCR buffer, 1.5 µM MgCl2 (Denville Scientific), 500 µM dNTPs (Roche), 1.25 µM of each primer, 1.25 µL DMSO (Sigma), and water to a final volume of 25 µL. All fluorescently labeled PCR products were electrophoresed for each gorilla male in one assay on an Applied Biosystems 3730XL Sequencer. The collected data were analyzed by the Peak Scanner Software v1.0 (Life Technologies).
Software, assembly, and alignment availability
All scripts are available in Supplemental File S4, which represents the content of the code repository (http://github.com/makovalab-psu/GorillaY_project/) as of January 20, 2016. Readers are encouraged to download the latest versions of the scripts directly from the GitHub repository. The gorilla Y assembly and alignments are available at https://usegalaxy.org/u/rsharris/p/gor-hum-chi-y.
Data access
Sequencing data and assembly for the gorilla Y Chromosome from this study have been submitted to the NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA293447. RNA-seq data from this study have been submitted to BioProject under accession number PRJNA304995.
Supplementary Material
Acknowledgments
We thank Michael DeGiorgio and Raquel Assis for useful discussions of the results and the San Diego Zoological Society and Smithsonian Institution for providing us with samples used in this study. Kristoffer Sahlin provided assistance with running BESST. All HiSeq and Sanger sequencing were performed at Pennsylvania State Genomics Core Facility, University Park, Pennsylvania; PacBio sequencing was performed at Johns Hopkins University Deep Sequencing and Microarray Core Facility. This study was funded by National Science Foundation (NSF) awards DBI-ABI 0965596 (to K.D.M.), DBI-1356529 (to P.M.), IIS-1453527, IIS-1421908, and CCF-1439057 (to P.M.). Additionally, this study was supported by the funds made available through the Penn State Clinical and Translational Sciences Institute, and through the Pennsylvania Department of Health (Tobacco Settlement Funds). M.C. was supported by the National Institutes of Health (NIH)-PSU funded Computation, Bioinformatics and Statistics (CBIOS) Predoctoral Training Program (1T32GM102057-0A1). O.A.R. acknowledges financial support from the John and Beverly Stauffer foundation and the Alice B. Tyler Charitable Trust. M.A.F.-S. thanks the Leverhulme Trust for the Emeritus Fellowship award, which provided support for his contribution to the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.199448.115.
Freely available online through the Genome Research Open Access option.
References
- Ananda G, Walsh E, Jacob KD, Krasilnikova M, Eckert KA, Chiaromonte F, Makova KD. 2013. Distinct mutational behaviors differentiate short tandem repeats from microsatellites in the human genome. Genome Biol Evol 5: 606–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. 2008. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics 179: 1513–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. 2014. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K, Koren S, Chin CS, Drake JP, Landolin JM, Phillippy AM. 2015. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 33: 623–630. [DOI] [PubMed] [Google Scholar]
- Bhowmick BK, Satta Y, Takahata N. 2007. The origin and evolution of human ampliconic gene families and ampliconic structure. Genome Res 17: 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. 2014. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG. 2013. Efficient identification of Y chromosome sequences in the human and Drosophila genomes. Genome Res 23: 1894–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LK, Teuscher C. 2015. Y genetic variation and phenotypic diversity in health and disease. Biol Sex Differ 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LK, Wall EH, Osmanski EE, Dragon JA, Saligrama N, Zachary JF, Lemos B, Blankenhorn EP, Teuscher C. 2015. Copy number variation in Y chromosome multicopy genes is linked to a paternal parent-of-origin effect on CNS autoimmune disease in female offspring. Genome Biol 16: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Yang Y, Retzel EF, Liu WS. 2013. Male-specific region of the bovine Y chromosome is gene rich with a high transcriptomic activity in testis development. Proc Natl Acad Sci 110: 12373–12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. 2000. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci 355: 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Connallon T, Clark AG. 2010. Sex linkage, sex-specific selection, and the role of recombination in the evolution of sexually dimorphic gene expression. Evolution 64: 3417–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, Grützner F, Kaessmann H. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508: 488–493. [DOI] [PubMed] [Google Scholar]
- de Vries JW, Hoffer MJ, Repping S, Hoovers JM, Leschot NJ, van der Veen F. 2002. Reduced copy number of DAZ genes in subfertile and infertile men. Fertil Steril 77: 68–75. [DOI] [PubMed] [Google Scholar]
- Demuth JP, De Bie T, Stajich JE, Cristianini N, Hahn MW. 2006. The evolution of mammalian gene families. PLoS One 1: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixson AF. 2012. Primate sexuality. Oxford University Press, New York. [Google Scholar]
- Dorus S, Evans PD, Wyckoff GJ, Choi SS, Lahn BT. 2004. Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat Genet 36: 1326–1329. [DOI] [PubMed] [Google Scholar]
- Douadi MI, GATTI S, Levrero F, Duhamel G, Bermejo M, Vallet D, Menard N, Petit EJ. 2007. Sex-biased dispersal in western lowland gorillas (Gorilla gorilla gorilla). Mol Ecol 16: 2247–2259. [DOI] [PubMed] [Google Scholar]
- Eckert KA, Yan G, Hile SE. 2002. Mutation rate and specificity analysis of tetranucleotide microsatellite DNA alleles in somatic human cells. Mol Carcinog 34: 140–150. [DOI] [PubMed] [Google Scholar]
- Elliott DJ. 2000. RBMY genes and AZFb deletions. J Endocrinol Invest 23: 652–658. [DOI] [PubMed] [Google Scholar]
- English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, et al. 2012. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7: e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genton C, Pierre A, Cristescu R, Lévréro F, Gatti S, Pierre JS, Ménard N, Le Gouar P. 2014. How Ebola impacts social dynamics in gorillas: a multistate modelling approach. J Anim Ecol 84: 166–176. [DOI] [PubMed] [Google Scholar]
- Giachini C, Nuti F, Turner DJ, Laface I, Xue Y, Daguin F, Forti G, Tyler-Smith C, Krausz C. 2009. TSPY1 copy number variation influences spermatogenesis and shows differences among Y lineages. J Clin Endocrinol Metab 94: 4016–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gläser B, Grützner F, Willmann U, Stanyon R, Arnold N, Taylor K, Rietschel W, Zeitler S, Toder R, Schempp W. 1998. Simian Y Chromosomes: species-specific rearrangements of DAZ, RBM, and TSPY versus contiguity of PAR and SRY. Mamm Genome 9: 226–231. [DOI] [PubMed] [Google Scholar]
- Gnerre S, MacCallum I, Przybylski D. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Peng L, Makova KD. 2009. Evolution of X-degenerate Y chromosome genes in greater apes: conservation of gene content in human and gorilla, but not chimpanzee. J Mol Evol 68: 134–144. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JA. 2010. Review: sex chromosome evolution and the expression of sex-specific genes in the placenta. Placenta 31(Suppl: S27–S32. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University, College Park, PA, 1–84. [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. 2011. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83: 8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Konkel MK, Prado-Martinez J, Chiatante G, Herraez IH, Walker JA, Nelson B, Alkan C, Sudmant PH, Huddleston J, et al. 2013. Rates and patterns of great ape retrotransposition. Proc Natl Acad Sci 110: 13457–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SK, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463: 536–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. 2012a. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 482: 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Page DC. 2012b. Sequencing of rhesus macaque Y chromosome clarifies origins and evolution of the DAZ (Deleted in AZoospermia) genes. Bioessays 34: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. 2013. REAPR: a universal tool for genome assembly evaluation. Genome Biol 14: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmin M, Saag L, Vicente M, Wilson Sayres MA, Järve M, Talas UG, Rootsi S, Ilumäe AM, Mägi R, Mitt M, et al. 2015. A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Res 25: 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinath MC, Timoshevskiy VA, Timoshevskaya NY, Tsonis PA, Voss SR, Smith JJ. 2015. Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci Rep 5: 16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki Y, Toyoda A, Noguchi H, Taylor TD, Itoh T, Kim DS, Kim DW, Choi SH, Kim IC, Choi HH, et al. 2006. Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway. Nat Genet 38: 158–167. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken RS, Stockwell TB. 2007. Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol 7: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Davis BW, Raudsepp T, Pearks Wilkerson AJ, Mason VC, Ferguson-Smith M, OBrien PC, Waters PD, Murphy WJ. 2013. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res 23: 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott GP, Do D, Litterst CM, Maar D, Hindson CM, Steenblock ER, Legler TC, Jouvenot Y, Marrs SH, Bemis A, et al. 2013. Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal Chem 85: 11619–11627. [DOI] [PubMed] [Google Scholar]
- Mendez FL, Karafet TM, Krahn T, Ostrer H, Soodyall H, Hammer MF. 2011. Increased resolution of Y chromosome haplogroup T defines relationships among populations of the Near East, Europe, and Africa. Hum Biol 83: 39–53. [DOI] [PubMed] [Google Scholar]
- Miller W, Rosenbloom K, Hardison RC, Hou M, Taylor J, Raney B, Burhans R, King DC, Baertsch R, Blankenberg D, et al. 2007. 28-way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome Res 17: 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotke L, Lau BT, Rumma RT, Ji HP. 2014. High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR. Anal Chem 86: 2618–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller AP. 1988. Ejaculate quality, testes size and sperm competition in primates. J Hum Evol 17: 479–488. [Google Scholar]
- Nickkholgh B, Noordam MJ, Hovingh SE, van Pelt AM, van der Veen F, Repping S. 2010. Y chromosome TSPY copy numbers and semen quality. Fertil Steril 94: 1744–1747. [DOI] [PubMed] [Google Scholar]
- Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, et al. 2003. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- Rizk G, Lavenier D, Chikhi R. 2013. DSK: k-mer counting with very low memory usage. Bioinformatics 29: 652–653. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC. 2003. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423: 873–876. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Vezzi F, Nystedt B, Lundeberg J, Arvestad L. 2014. BESST - efficient scaffolding of large fragmented assemblies. BMC Bioinformatics 15: 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, et al. 2012. Insights into hominid evolution from the gorilla genome sequence. Nature 483: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837. [DOI] [PubMed] [Google Scholar]
- Skinner BM, Sargent CA, Churcher C, Hunt T, Herrero J, Loveland JE, Dunn M, Louzada S, Fu B, Chow W, et al. 2016. The pig X and Y Chromosomes: structure, sequence, and evolution. Genome Res 26: 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh YQ, Alföldi J, Pyntikova T, Brown LG, Graves T, Minx PJ, Fulton RS, Kremitzki C, Koutseva N, Mueller JL, et al. 2014. Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 159: 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-Smith C, Taylor L, Müller U. 1988. Structure of a hypervariable tandemly repeated DNA sequence on the short arm of the human Y chromosome. J Mol Biol 203: 837–848. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. 2013. Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature 499: 332–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Emerson JJ, Zektser Y, Mahajan S, Bachtrog D. 2013. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol 11: e1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson Sayres MA, Venditti C, Pagel M, Makova KD. 2011. Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 65: 2800–2815. [DOI] [PubMed] [Google Scholar]
- Writzl K, Zorn B, Peterlin B. 2005. Copy number of DAZ genes in infertile men. Fertil Steril 84: 1522–1525. [DOI] [PubMed] [Google Scholar]
- Yang F, Carter NP, Shi L, Ferguson-Smith MA. 1995. A comparative study of karyotypes of muntjacs by chromosome painting. Chromosoma 103: 642–652. [DOI] [PubMed] [Google Scholar]
- Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, Church GM. 2006. Sequencing genomes from single cells by polymerase cloning. Nat Biotechnol 24: 680–686. [DOI] [PubMed] [Google Scholar]
- Zhou RN, Hu ZM. 2007. The development of chromosome microdissection and microcloning technique and its applications in genomic research. Curr Genomics 8: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.