

Abstract
The regulation of miRNAs is critical to the definition of cell identity and behavior in normal physiology and disease. To date, the dynamics of miRNA degradation and the mechanisms involved in remain largely obscure, in particular, in higher organisms. Here, we developed a pulse-chase approach based on metabolic RNA labeling to calculate miRNA decay rates at genome-wide scale in mammalian cells. Our analysis revealed heterogeneous miRNA half-lives, with many species behaving as stable molecules (T1/2 > 24 h), while others, including passenger miRNAs and a number (25/129) of guide miRNAs, are quickly turned over (T1/2 = 4–14 h). Decay rates were coupled with other features, including genomic organization, transcription rates, structural heterogeneity (isomiRs), and target abundance, measured through quantitative experimental approaches. This comprehensive analysis highlighted functional mechanisms that mediate miRNA degradation, as well as the importance of decay dynamics in the regulation of the miRNA pool under both steady-state conditions and during cell transitions.
MicroRNAs (miRNAs) are an evolutionary conserved class of short, single-stranded noncoding RNAs (∼18–22 nt in length) that act in post-transcriptional regulation of gene expression in higher eukaryotes. Each miRNA can potentially target hundreds of genes, even simultaneously, causing their down-modulation through multiple mechanisms, including translational repression and/or mRNA destabilization (Eulalio et al. 2008; Bartel 2009; Jonas and Izaurralde 2015). Most miRNAs display a tightly regulated expression pattern that is often tissue specific or even cell type specific, emphasizing their importance in defining the space, time, and developmental stage of gene expression (Bushati and Cohen 2007; Gangaraju and Lin 2009; Lujambio and Lowe 2012). Hence, the regulation of the miRNA pool is a critical event in the definition of cell identity and behavior, both under steady-state conditions and during cell transitions.
Overall, the levels of miRNAs in the cell are the result of both biosynthetic and decay processes. The expression of miRNAs starts with the transcription of a primary RNA (called pri-miRNA), a long transcript that contains a stem–loop region for each encoded miRNA, and is dependent on the same transcriptional machinery and regulatory mechanisms as used by protein coding genes, e.g., transcription factors, enhancers, and epigenetic regulators (for review, see Ha and Kim 2014). This pri-miRNA is usually short-lived and generates mature miRNA molecules through two sequential processing events. The first processing event occurs in the nucleus and is mediated by the Microprocessor complex (DROSHA/DGCR8), which generates a pre-miRNA (70–100 nt long). The pre-miRNA is then exported to the cytosol, where the second cleavage event occurs, mediated by the DICER1/TRBP complex, producing a miRNA duplex (18–22 nt long). MiRNA processing is followed by loading of the miRNA duplex onto Argonaute proteins (AGO) and retention of just one of the two strands (the “guide” strand), resulting in the formation of the miRNA-induced silencing complex (miRISC) that eventually executes gene silencing (Krol et al. 2010b). Once loaded onto the AGO-based complexes, miRNAs are stabilized (Diederichs and Haber 2007) and have long half-lives (many hours or even days [van Rooij et al. 2007]). Yet, under physiological or pathological conditions, some miRNAs are rapidly down-regulated, even in the absence of cell division, such as upon growth factor or serum stimulation (Avraham et al. 2010; Rissland et al. 2011; Zhu et al. 2012). This observation poses an apparent paradox on how cells get rid of the pre-existing pool of such stable molecules.
Insights into the mechanisms and relevance of miRNA degradation in physiology are just beginning to emerge. Some reports suggested that slow turnover might not be a general feature of miRNAs and, in some instances, miRNA decay could be relatively fast (few hours), such as for miR-503 in fibroblasts (Rissland et al. 2011), miR-29b during mitosis (Hwang et al. 2007), and several neuronal miRNAs (Krol et al. 2010a). The degradation dynamics of miRNAs and the mechanisms involved remain largely obscure, in particular in mammals, and apparently involve enzymatic activities able to modify miRNA ends by adding (“tailing”) or removing (“trimming”) nucleotides (Ramachandran and Chen 2008; Chatterjee and Grosshans 2009; Ameres et al. 2010; Kim et al. 2010; Burns et al. 2011; Neilsen et al. 2012; Ruegger and Grosshans 2012; Lee et al. 2014). A target-dependent mechanism has also been identified, in which turnover is mediated by the interaction of miRNAs with their mRNA targets, promoting miRNA unloading from AGO and degradation (Baccarini et al. 2011; Ruegger and Grosshans 2012; De et al. 2013; de la Mata et al. 2015). One main reason for this lack of knowledge is the lack of experimental approaches that allow the analysis of miRNA half-lives at the global level without perturbing gene transcription or miRNA biogenesis (Bail et al. 2010; Gantier et al. 2011; Guo et al. 2015). To overcome such issues, we exploited metabolic pulse labeling, the gold standard method used to determine RNA and protein half-lives, and developed a “pulse-chase” approach optimized for the analysis of miRNAs. Variants of this approach based on nonradioactive labels, such as 4-thiouridine (4sU), which is incorporated into nascent RNA in place of uridine, have been successfully developed and used to study RNA dynamics in different species (Cleary et al. 2005; Dolken et al. 2008; Friedel et al. 2009; Schwanhausser et al. 2011). By combining the 4sU-based “pulse-chase” approach with high-throughput RNA sequencing, we have now measured miRNA decay rates and revealed how turnover affects the regulation and the activity of miRNAs in mammalian cells.
Results
Development of an optimized pulse-chase methodology to determine miRNA degradation rates in mammalian cells
To determine the half-lives of miRNAs, with minimal perturbation to cell physiology, we cultivated 3T9 mouse fibroblasts in medium supplemented with 4sU; this ribonucleoside analog is incorporated into nascent RNA, allowing thiol-specific biotinylation and selective pull-down of newly synthesized RNA from total RNA (Schwanhausser et al. 2011). Initially, we examined the incorporation dynamics of continuous 4sU labeling and its effects on cell physiology (Supplemental Fig. S1A–F; Dolken et al. 2008; Burger et al. 2013). Administration of 4sU (300 μM) for up to 3 h did not (1) affect cell proliferation (Supplemental Fig. S1D), (2) induce nucleolar stress or rRNA degradation (Supplemental Fig. S1E), or (3) alter global miRNA expression (Supplemental Fig. S1F).
We next checked the labeling of a number of mRNA species (N = 4), pri-miRNAs (N = 7), and mature miRNAs (N = 18) by RT-qPCR (Supplemental Fig. S1G–I). As expected, the incorporation rates (4sU-labeled/total RNA) correlated with the stability of the RNA species, with unstable mRNAs (Myc and Ccne2) incorporating 4sU more efficiently than stable mRNAs (Gapdh and Rplp0) (Supplemental Fig. S1G). Accordingly, pri-miRNAs, which are short-lived, were efficiently labeled at early time points (15–30 min), reaching a maximal incorporation rate (∼100%) after 1–2 h (Supplemental Fig. S1H), while mature miRNAs, which are stable molecules, were poorly labeled even after 2 h (<8%) (Supplemental Fig. S1I). We reasoned that a 2- to 3-h pulse might be compatible with the complete biogenetic process of miRNAs, including the loading onto AGO2-based complexes, where silencing occurs. Exploiting a lentiviral inducible expression system (pSLIK-neo), we performed a time-course induction of two miRNAs: i.e., miR-34a, which has a low incorporation rate, and miR-182, which has a high incorporation rate. Mature miRNAs were significantly induced after 3 h of doxycycline treatment (Supplemental Fig. S1L). Concomitantly, we observed an increase in the quantity of miR-34a loaded onto AGO2-based complexes, isolated by RNA immunoprecipitation (AGO2-RIP, Supplemental Fig. S1M,N). Based on these results, we established that 3 h of continuous 4sU treatment (hereafter referred to as the “pulse labeling” method) was sufficient to label mature miRNA molecules, which are immediately loaded on AGO2.
Usually, pulse labeling of mRNA allows the indirect estimation of the half-life (T1/2) by calculating newly transcribed–over–total RNA ratios (Dolken et al. 2008). However, the biogenesis of mature miRNAs is more complex and depends on the transcription of the pri-miRNA plus several maturation steps, including DROSHA/DICER1 processing, nuclear export, and AGO loading (for review, see Krol et al. 2010b). Thus, we decided to follow directly the decay of labeled mature miRNAs, minimizing the contribution of transcription/maturation. We employed a “pulse-chase” protocol, where 4sU is removed from the medium after the labeling period (3 h pulse), and labeled RNA is purified at different time points (chasing time) (Fig. 1A) to measure the decay of labeled molecules. 4sU-RNA could be isolated with high purity (>97%) (Supplemental Material), and its levels decreased linearly up to 16 h of chasing (Fig. 1B), reflecting the degradation of total RNA. We measured the half-lives (T1/2) of several types of RNA species (mRNAs, small RNAs, pri-miRNAs, mature miRNAs) during the chase period (Fig. 1C–E). For mRNAs and pri-miRNAs, the experimental half-lives were of few hours, which is compatible with previous observations (Yang et al. 2003) and remarkably in line with those determined with the pulse labeling method (R2 = 0.98) (Fig. 1F). Conversely, mature miRNAs showed longer half-lives ranging from 3 to 24 h (Fig. 1E), with a poor correlation with those calculated with the “pulse” method (R2 = 0.10) (Fig. 1F). For instance, only with the “pulse-chase” approach was it possible to confirm the short half-life of miR-503 (T1/2 = 3.2 h), which was previously described as highly unstable in growing fibroblasts (T1/2 = 3.6 h) (Rissland et al. 2011). Hence, we concluded that only by using the “pulse-chase” approach it is possible to accurately determine miRNA decay rates.
Figure 1.

4sU pulse-chase RNA labeling and miRNA decay rates. (A) The scheme illustrates the experimental strategy of the 4sU pulse-chase approach used to follow miRNA decay dynamics. (B) The yield of 4sU-RNA at the indicated chasing times is reported. Forty micrograms of labeled total RNA was used at each time point. The dashed line marks the background level (yield of the negative control). (C) The half-life of Myc was measured by RT-qPCR using the pulse-chase protocol according to a one-phase exponential fitting (N = 2). Data (log2 scale) were normalized on the levels of Actb, a highly stable gene (Yang et al. 2003). (D,E) Half-lives of mRNAs, pri-miRNAs, small RNAs (D), and mature miRNAs (E) were estimated as in C. Shown is the average and the SEM of two independent experiments. The miRNA data were normalized to the levels of Snord72, a highly stable short RNA. (F) Half-lives calculated using the pulse-chase approach (shown in D,E) were compared with those indirectly determined by 4sU pulse labeling, as described previously (Dolken et al. 2008). A linear correlation was calculated for mRNAs/pri-miRNAs (red dashed line) or miRNAs (yellow dashed line). Shown also is the coefficient of determination (R2) of the linear fitting. (G) Canonical miRNA species analyzed in the “decay data sets,” distinguished as guide or passenger miRNAs according to miRBase and TargetScan databases. (H) Distribution of miRNA half-lives. The red and blue lines mark boundaries of fast- and slow-decaying species. (I) Half-lives (mean with confidence interval) are plotted against miRNA expression (copies per cell [CPCs]; log10 scale). (J) Degradation dynamics of fast (N = 25, T1/2 < 14 h) and slow (N = 32, T1/2 > 24 h) guide miRNAs are shown along the time courses. Data were normalized to miR-92a-3p and reported as log2 fold-change over the 0-h time point. (K) The frequency of passenger versus guide miRNAs within the decay classes has been analyzed by contingency (χ2 = 6.24, P = 0.042). Ambiguous species were not included. (L) Box plot shows the distribution of half-lives for matched guide and passenger species. P-value (Wilcoxon test) is shown.
Genome-wide analysis of miRNA degradation rates in growing fibroblasts
To obtain a global picture of miRNA decay, we performed high-throughput sequencing (Illumina) of labeled miRNAs along with the “pulse-chase” experiment. In total, 21 libraries were produced from 4sU-labeled RNA isolated at seven time points in three independent biological repeats. We obtained 125 million reads that constitute the “decay data set,” which was analyzed with the IsomiRage tool, a bioinformatics pipeline for the characterization and analysis of miRNAs and their variants in next-generation sequencing data sets (Muller et al. 2014). After data filtering and normalization (Supplemental Fig. S2A–D; Supplemental Material), we retrieved data for 564 species with a threshold of expression along the entire time course (more than 10 reads), including 186 canonical miRNAs (Fig. 1G). Importantly, no major biases in miRNA length or base composition, including the uridine number (Duffy et al. 2015), were observed as a consequence of the labeling and purification procedure (see Supplemental Fig. S2A; Supplemental Table S1). The half-lives of miRNAs were modeled assuming a one-phase exponential decay, fitting to the average value at each time point of three independent biological replicas (T1/2ave) with 95% confidence intervals (T1/2min and T1/2max) for the best fit (Supplemental Table S1). A simple one-phase exponential decay model fitted the data better than more complex models, such as a two-phase decay, arguing against the existence of multiple pools of the same miRNA with different stabilities in the cell. A consistent fraction of miRNAs (57/186, 30.6%) displayed half-lives >1 d, consistent with previous reports (Fig. 1H,I; Supplemental Table S1; Ruegger and Grosshans 2012). Nonetheless, a number of miRNAs displayed short half-lives (T1/2 < 12 h; N = 61, 32.8%), with some of them disappearing relatively quickly (T1/2 < 8 h; N = 28). Mature miRNAs are usually distinguished as “guide” or “passenger” miRNAs, according to whether they are preferentially retained in the miRISC complex after loading or they are released into the cytosol and quickly degraded, respectively (Krol et al. 2010b). As expected, “passenger” miRNAs (N = 50) were short lived and accumulated to levels that were significantly lower than their paired “guide” miRNAs (P-value < 0.001) (Fig. 1J,K). A number of “guide” miRNAs also decayed rapidly (“fast,” N = 25) in a reproducible manner (R2 > 0.9), with half-lives ranging from 4–14 h, as opposed to “slow” decaying miRNAs (N = 32) with half-lives of >24 h (Fig. 1J–L; Supplemental Table S1).
Next, we explored the relationship between miRNA decay rates and their genomic organization. The half-lives of miRNAs were not influenced by the location of their genes in intergenic or intronic regions or by their chromosomal location (Fig. 2A). In most instances, miRNAs belonging to the same transcriptional cluster displayed similar half-lives (25/33 coherent clusters) (Fig. 2B). Moreover, clusters often contained miRNAs belonging to the same family, which shared sequence and functional similarities, as well as similar decay rates (10/17 coherent families) (Fig. 2C). As expected, fast turnover strongly correlated with 4sU incorporation (P < 0.0001) (Fig. 2D,E). Although we could not precisely calculate miRNA half-lives based on 4sU incorporation (see Fig. 1F), a reciprocal relationship between half-life and incorporation was observed (Fig. 2E). MiRNAs displaying a very high incorporation rate (first quartile of the distribution) (Supplemental Table S1) were strongly enriched in “fast” species, while those with a very low incorporation rate (last quartile of the distribution; <3% 4sU) were enriched for “slow” miRNAs (P < 0.0001) (Fig. 2E,F; Supplemental Table S1). Hence, incorporation rates (3 h, 4sU pulse) could serve as a proxy to qualitatively infer miRNA turnover rates in a given biological system, as shown by others (Duffy et al. 2015). We measured the incorporation rate of miRNAs in two other human cell lines: BJ (foreskin fibroblasts) and HeLa (cervical carcinoma epithelial cells) (Fig. 2G). Despite the very different origins of the two human cell lines, they displayed a good correlation in their 4sU incorporation rates (Fig. 2H) and a strong overlap in the class of miRNAs with the highest incorporation rates (P < 0.0001), which included several members of the let-7 family (Supplemental Table S1). In addition, the 4sU incorporation rates of miRNAs were remarkably similar between mouse and human cells (Fig. 2I), suggesting that decay rates might be maintained across different cell types and species. We verified this hypothesis by directly measuring the half-lives of a number of miRNAs by pulse-chase in human BJ fibroblasts. We confirmed the fast decay (T1/2 < 14 h) of seven out of eight miRNAs and the slow decay of one miRNA (Fig. 2J).
Figure 2.

Correlation of decay dynamics with genomic and functional properties of miRNAs. (A, left) The distribution of half-lives for intergenic versus intronic guide miRNAs (N = 129) is shown (P = 0.7488, Wilcoxon test). (Right) miRNA half-lives are reported according to chromosomal location of miRNA loci. (B,C) miRNAs belonging to the same transcriptional unit (cluster) or sequence similarity group (family), according to miRBase 21, were analyzed. Clusters and families with coherent behavior (differences in T1/2 < 12 h) are shown. A color code indicates miRNA decay class (red, “fast”; blue, “slow”). (D–F) 4sU incorporation in miRNAs was correlated with decay. (D) The histogram shows the distribution of the incorporation rates of guide miRNAs, while the box-plot compares fast versus slow decaying miRNAs (P < 0.0001, Wilcoxon test). (E) miRNA half-lives plotted against 4sU incorporation rates. The curved line highlights the reciprocal relationship between decay and incorporation. The dashed curves mark 95% confidence intervals. Coefficient of determination (R2) is shown. (F) Guide miRNAs were distinguished into four classes (quartiles of 4sU incorporation rates). Shown is the distribution of “fast” and “slow” miRNAs among the four classes by contingency (χ2 = 44.8, P < 0.0001). (G) The box plots show the distribution of 4sU incorporation rates in the different cell lines. Purple and yellow lines mark areas of high and low incorporating miRNA species, respectively. (75-PCTL) More than 75th percentile; (25-PCTL) less than 25th percentile. (H) The scatter plot compares the 4sU incorporation rates in HeLa and BJ human cells. The dashed line highlights the linear correlation, and the coefficient of determination (R2) is shown. (I) The scatter plot compares 4sU incorporation rates in human (BJ) and mouse (3T9) fibroblasts. Only miRNAs that are perfectly conserved between human and mouse were considered in the analysis (N = 72). (J) Half-lives of nine miRNAs were measured in human BJ cells by the pulse-chase method and compared with those from mouse 3T9 cells. The dashed line indicates the cut-off (T1/2 < 14 h) previously used for classifying miRNAs as fast decaying in 3T9 cells. The Pearson correlation coefficient (r) is shown.
Transcription and decay contribute to the regulation of the miRNA pool
MiRNA levels are dictated by both biosynthetic and decay processes, which can be represented by three events—transcription, maturation, and degradation—and by dilution due to cell division (α; Fig. 3A). At steady state, the generation and degradation of miRNAs are at equilibrium and, assuming that maturation is much faster than cell division (k2 >> α), miRNA amount (µ) can be calculated using just three parameters: the transcription rate (k1), the degradation rate (k3, which is ln(2)/T1/2 − α), and the dilution by cell division (α, which is 1/TD − cell doubling time). We calculated absolute values of miRNA amounts in growing fibroblasts (steady state) by combining digital PCR and sequencing (for details, see Supplemental Material). Then, we correlated miRNA abundance expressed as copies per cell (CPC) (Fig. 3B) with decay rates. We observed a trend (P = 0.0599), for “fast” miRNAs being less abundant than “slow” miRNAs (Fig. 3B,C), implying that decay, by itself, contributes partially to miRNA abundance.
Figure 3.
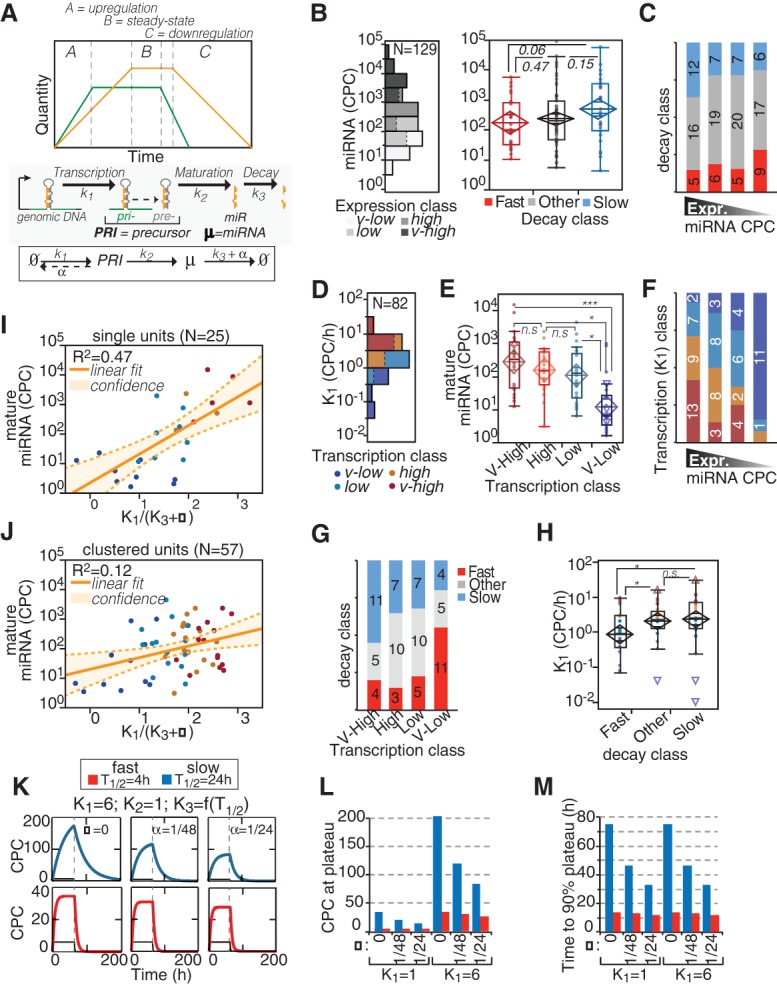
Regulation of miRNA expression by transcription and decay. (A) The scheme describes the variations of miRNA (yellow) expression in time according to changes in transcription of pri-miRNA (green). The regulation of miRNA expression (µ) is dependent on transcription, maturation, and decay rates (k1, k2, and k3, respectively) and on dilution by cell division (α). Half-life (T1/2) and k3 are related: [T1/2 = ln(2)/(k3 + α)]. (B,C) The absolute expression of miRNAs in cells (log10 scale) was correlated with miRNA decay. (B) According to the distribution of miRNA expression in 3T9 cells (histogram on the left), four expression classes (quartiles, highlighted in gray scale) could be defined. The box-plot (right) shows the abundance of miRNAs by decay class. P-values were calculated by Wilcoxon test. (C) The distribution of miRNA decay rates along the expression classes was analyzed by contingency (χ2 = 4.7, P = 0.585). (D–F) The expression of miRNAs in cells was correlated with miRNA transcription, expressed as copies of pri-miRNA per cell (CPCs) (which is k1; log10 scale). (D) Distribution of transcription rates of 82 miRNA loci. The color code defines the four transcription classes (quartiles) in the distribution. (E) The abundance of miRNAs (CPCs; log10 scale) was correlated with transcription classes as in B. (*) P < 0.05; (***) P < 0.001; Wilcoxon test. (F) The distribution of miRNA transcription rates among the expression classes was analyzed by contingency (χ2 = 38.07, P < 0.0001). (G,H) The transcription of miRNAs (CPC/h, log10 scale) was correlated with the decay of mature miRNAs by contingency test (χ2 = 13.06, P = 0.0421; G) and by one-way analysis (H). Asterisks mark significant values (P < 0.05). (I,J) The expression of miRNAs at steady state (“B zone”; A) was inferred using the kinetic parameters (k1 and k3), which were measured by “pulse labeling” and “pulse-chase” experiments, respectively. Scatter plots show the fitting of experimental (CPCs, log scale) and theoretical (k1/[k3 + α], log scale) copies, for miRNAs transcribed as single units (I) or clustered units (J). Straight and dashed lines mark linear correlation and 95% confidence intervals, respectively. (K) Mathematical modeling of accumulation and degradation dynamics of miRNAs (thick colored line) and pri-miRNAs (thin black line) by defined kinetic parameters and according to different proliferation states (α = 1/TD, doubling time). The dashed vertical line marks when synthesis was shut off. The blue and red lines highlight the behavior of slow and fast decaying miRNAs, respectively. (L,M) Bar graphs show the theoretical values of miRNA abundance at plateau (CPCs; L) or the time required to reach 90% of the plateau (in hours; M) inferred by modeling. Values are reported according to indicated k1 and T1/2 in cells, further distinguished by cell proliferation rates (α, as in K).
Next, we sought to determine transcription rates (k1) of pri-miRNAs by combining nascent RNA sequencing data obtained by short “pulse labeling” (10′ 4sU) with the INSPEcT bioinformatic tool (de Pretis et al. 2015). We manually defined genomic boundaries of pri-miRNAs in growing 3T9 fibroblasts according to a combination of 4sU-RNA-seq and ChIP-seq (POL2 and H3K4me3) tracks, which mark the transcriptional unit and the promoter region, respectively (Supplemental Fig. S3A,B; Supplemental Material). The synthesis rate (k1), expressed as reads per kilobase per million mapped in an hour (RPKM/h), was converted into an absolute value (CPC/h) by measuring different pri-miRNAs (N = 6) with digital PCR (Supplemental Fig. S3C). Overall, we could calculate the transcription rate for approximately 100 different miRNA loci (including 82 of the 129 guide miRNAs mentioned in this study), with values ranging from 0.1–30 CPC/h (Fig. 3D; Supplemental Table S2). As expected, miRNA transcription highly correlated with miRNA abundance (Fig. 3E,F). Transcription and decay appeared as coupled, as previously observed for clustered miRNAs (Fig. 2B). MiRNAs with low synthesis rates (low k1) were enriched for “fast” decaying species (11/20), and vice versa, miRNAs that are actively transcribed (high k1) were enriched for “slow” miRNAs (11/20) (Fig. 3G,H; Supplemental Table S2). We calculated the theoretical miRNA abundance in cells with experimentally measured k1 and k3. The fitting with miRNA absolute expression was good (R2 = 0.47), but only for miRNA loci that are organized as single (nonclustered) transcriptional units (N = 25) (Fig. 3I). In contrast, miRNAs that belong to clusters (N = 57) could not be modeled properly (Fig. 3J, R2 = 0.12), suggesting that the assumption made about the maturation step (k2 >> α) is not verified and that processing has to be taken into account to infer absolute values of miRNA expression. By use of kinetic parameters (k1, k2, k3, α) in the range of what was measured experimentally in 3T9 cells, we evaluated the impact of transcription and decay on the dynamic regulation of miRNAs (Fig. 3K; Supplemental Table S2; Supplemental Fig. S3D). While transcription (k1) primarily regulated the magnitude of miRNA expression changes (copies at plateau), the decay influenced the dynamics of miRNA regulation (Fig. 3L,M; Supplemental Table S2). In particular, only fast decaying miRNAs could give rise to sharp and quick changes in miRNA levels (i.e., the time to shift from two different steady levels upon a change in k1).
Fast decaying miRNAs are associated with high stoichiometric target:miRNA ratios
MiRNAs function by silencing the expression of target genes (TGs) through various mechanisms (Carthew and Sontheimer 2009; Jonas and Izaurralde 2015). Conversely, high-affinity TGs can influence miRNA stability by promoting their unloading from AGO2 (De et al. 2013) or the generation of miRNA isoforms (3′-end “tailing” and “trimming”) that are associated with miRNA degradation (Ameres et al. 2010; Baccarini et al. 2011; Xie et al. 2012; de la Mata et al. 2015). Collectively, these mechanisms are referred to the so-called “target-induced (or target-directed) miRNA decay” (TIMD or TDMD). We addressed the interaction of miRNAs with their target pool by identifying high-confidence targets in 3T9 cells (Fig. 4A). We computed the total abundance of high-affinity binding sites (context score, CS < −0.2; targets with “seed”) and estimated the target per miRNA ratio (TPM) (Supplemental Table S3), under the simplest hypothesis that no competition occurs for the same binding site between different miRNAs. Since the presence of additional complementary sequences between targets and the 3′-end of miRNAs has been shown to promote unloading and degradation of miRNAs (Ameres et al. 2010; Xie et al. 2012; de la Mata et al. 2015), we also stratified targets according to the presence of 3′ compensatory regions (CS < −0.1 plus 3′-end pairing < −0.03 or < −0.05, defined as 3C.03 or 3C.05, respectively) (Fig. 4B). TPM_seed ranged from a 1:1 ratio to thousands of binding sites for a single miRNA molecule, while TPM_3C.03 and TPM_3C.05 reached a maximum of hundreds or tens of binding sites, respectively (Fig. 4C; Supplemental Table S3). Of note, “fast” miRNAs displayed higher TPM with respect to “slow” miRNAs (P < 0.01) (Fig. 4C–E), and conversely, miRNAs with very high TPM (High; top 20% distribution) (Supplemental Fig. S4A) had a significant enrichment of “fast” species and a shorter half-life compared with those with very low TPM (Low; bottom 20% distribution) (Fig. 4F). This result was far more significant than the association of either miRNA abundance (miRNA CPC) or target abundance (TG CPC) with half-life (Supplemental Fig. S4B–D). To corroborate this observation, we also analyzed the ratio of targets and miRNA copies predicted by the synthesis rate of pri-miRNAs (“virtual copies”). In this way, we could verify that high TPM correlates with faster decay per se and not as a result of reduced miRNA expression due to target-induced miRNA degradation. MiRNAs with high “virtual TPM” still displayed shorter half-lives (Fig. 4G), with a much stronger association compared with the previously observed association between synthesis and half-life (see Fig. 3G,H; Supplemental Fig. S4E). In most cases, we observed a much stronger association between TPM and miRNA half-lives when considering targets with 3′ complementarity (Fig. 4D–F). This observation further supports a model in which unfavorable stoichiometric ratios between miRNAs and targets with extensive complementarity promote miRNA degradation (Fig. 4H).
Figure 4.

miRNA targets and decay. (A,B) Scheme of strategy used for the target:miRNA ratio calculation. Targets of miRNAs were further distinguished according to their complementarity to the miRNA 3′ end. (TG_seed) Seed interaction; (TG_3C) seed plus 3′ compensatory site with moderate (3C.03) or extensive (3C.05) interaction. Examples are reported in panel B. (C,D) The bar graph (C) and the table (D) summarize the TPM, the number of targets [TG (N)], and their level of expression in “fast” versus “slow” miRNA classes. (*) P < 0.05; (**) P < 0.01; Wilcoxon test. (E) Box-plots show the distribution of the target:miRNA ratios (TPM, distinguished according to target complementarity as shown in B) in “fast” versus “slow” miRNAs. (F,G) miRNA half-life (T1/2) distribution within low (<20th Pctl) or high (>80th Pctl) TPM (F) or virtual TPM (G) miRNA classes. Virtual TPMs were calculated using transcription rate (k1) and cell doubling time (α) data. (H) The scheme summarizes the mutual relationship between targets, miRNA synthesis, miRNA expression levels and miRNA turnover rates. (E–G) P-values were calculated by Wilcoxon test.
These results suggest that the ratio between high-affinity targets and miRNA abundance plays a role in the rate of miRNA decay, and supports TIMD as a general mechanism that promotes miRNA degradation (Baccarini et al. 2011; De et al. 2013; de la Mata et al. 2015).
Fast decaying miRNAs are enriched in tailed and trimmed isoforms
MiRNA sequences are quite heterogeneous, resulting in the existence of multiple isoforms (Neilsen et al. 2012), called “isomiRs” (schematized in Fig. 5A), which appear to be associated with miRNA degradation and TIMD (Ameres et al. 2010; Kim et al. 2010; Baccarini et al. 2011; Wyman et al. 2011; de la Mata et al. 2015). By exploiting the IsomiRage tool (Muller et al. 2014), we analyzed the expression of miRNA isoforms in the decay data set (Supplemental Table S3). Globally, tailing (3′-NT) and trimming isoforms were significantly represented (>25%) (Supplemental Fig. S5A). Among 3′-NT isomiRs, adenylation and urydilation were prevalent (Supplemental Fig. S5B). On average, tailing (3′-NT) and trimming isoforms accounted for 11.9% and 15.1% of all guide miRNAs, respectively (Fig. 5B, “all guide”). These proportions were maintained in the slow decaying miRNA subset. Conversely, the “fast” miRNA subset was significantly enriched in both tailing and trimming isoforms (Fig. 5B; P < 0.001), supporting the hypothesis that these isoforms represent degradation intermediates. Most, but not all, fast decaying miRNAs were represented by tailing isoforms (P = 0.0061) (Fig. 5C, Supplemental Fig. S5C–D), with 19/25 fast miRNAs falling in the top 50% tailing distribution (P = 0.0235) (Fig. 5D). Among 3′-NT isomiRs, adenylation was the most enriched modification in fast decaying species (Supplemental Fig. S5E). Very similar observations were obtained for trimming isoforms, with high levels of trimmed isoforms (P = 0.0065) (Fig. 5E; Supplemental Fig. S5F,G) and a high trimming frequency (P = 0.0069) (Fig. 5F; Supplemental Fig. S5G) in the “fast” miRNA subset. Co-occurrence of tailing and trimming isoforms was significantly high (P = 0.0270) (Fig. 5G), suggesting that the two phenomena could be associated. In this respect, 15/25 (60%) “fast” miRNAs, but only eight of 32 (25%) “slow” miRNAs, displayed both high tailing and high trimming (P = 0.005) (Fig. 5G).
Figure 5.

miRNA isoforms (isomiRs) and decay. (A) The scheme illustrates the type of isomiRs that are generated starting from mouse mir-92-1, which is used as the model. Dashed lines mark the two cuts (by DROSHA and DICER1) that generate the “canonical” mature miR-92a-1-3p. Alternative cuts originate “templated” (T) isoforms, while tailing by terminal transferases produces nontemplated (NT) species, which are further distinguished according to the nucleotide added. (B) The pie charts show the frequency of isomiRs for all the guide miRNAs included in the decay data set or just those of the “fast” and “slow” subsets. Data refer to the average of three independent sequencing experiments (shown in Supplemental Fig. S3A,B). Asterisks mark significant values (P < 0.001, paired t-test). (C,E) Distribution of 3′-NT isoforms (C) or trimmed isoforms (“Trim-2,” which includes only species shortened by at least two bases) (E) between “fast” and nonfast (other) miRNA subsets. (**) P < 0.01, Wilcoxon test. (D,F) Guide miRNAs were distinguished into classes (quartiles) according to the amount of 3′-NT (D) or Trim-2 (F) modifications and were correlated with “fast” decay by contingency analysis (3′-NT χ2 = 9.6, P = 0.0235; Trim-2 χ2 = 12.1, P = 0.0069). (G) Co-occurrence of 3′-NT and Trim-2 modifications determined by contingency analysis (χ2 = 5.35, P = 0.0207). The two classes with concordant “high” or “low” tailing/trimming are highlighted and further distinguished by decay classes. (*) P < 0.01, contingency test. (H) Frequency of 3′-NT or Trim-2 modifications in miRNAs distinguished by arm type (3p or 5p). 3′-NT forms were further distinguished in uridylated (U) or adenylated (A) variants. (I) The pie charts show the frequency of isomiRs in the AGO2-RIP (“AGO2”) experiments compared with the total RNA (“Input”) used as input. (*) P < 0.001, paired t-test. (J) Abundance (top; [RPM] reads per million, log10 scale) and loading efficiency (bottom; AGO2/input ratio) of “fast” and “slow” miRNAs associated with AGO2, as measured by AGO2-RIP. Data are the average of three independent biological experiments. (H,J) P-values were calculated by the Wilcoxon test.
Mature miRNAs could derive from any of the two arms of the hairpin, called the 3p- and 5p- arms (Fig. 5A), implying that the 3′-end is generated either in the nucleus by the Microprocessor (3p miRNAs) or in the cytosol by DICER1 (5p miRNAs). We did not observe differences in the frequency of trimming between the 5p and 3p miRNAs (Fig. 5H), suggesting that this modification likely occurs in the cytosol on mature molecules, as previously shown (Krol et al. 2010b). In contrast, 3′-NT modifications were more frequent among 3p miRNAs (P = 0.0012) (Fig. 5H), implying that some tailing events might also occur in the nucleus at the level of the pre-miRNAs. This correlation was almost exclusively restricted to uridylation [“3′NT (U)”; P = 0.0144] (Fig. 5H) and might, therefore, be ascribed to the nuclear terminal uridylyl transferases (TUTases) (Krol et al. 2010b; Lin and Gregory 2015). Conversely, adenylation, the most frequent tailing event, was similar among 5p and 3p miRNAs and likely occurs in the cytosol, as previously suggested (Lee et al. 2014). Indeed, target-induced nontemplated nucleotide addition (tailing) is believed to occur in the cytosol, while miRNAs are bound to AGO2 (De et al. 2013; de la Mata et al. 2015). To corroborate these findings, we analyzed the abundance and enrichment of miRNA isoforms at the level of the RISC complex by performing small RNA sequencing on AGO2-RIP experiments. Overall, the abundance of miRNAs on AGO2 closely resembled the total pool of cellular miRNAs (“input”; R2 = 0.91) (Supplemental Fig. S5H), whereas some specific miRNAs could be differentially loaded, as recently reported (Flores et al. 2014). IsomiR distribution on AGO2 was very similar to the distribution of total RNA (input), confirming the enrichment of trimmed and 3′-NT species among “fast” miRNAs (Fig. 5I) and ruling out the possibility that these modifications occur after the unloading of miRNAs from AGO2. Of note, “fast” miRNAs did not display a different abundance on AGO2 and were not differently loaded (AGO2/input ratio) compared with other decay groups (Fig. 5J; Supplemental Fig. S5I), suggesting that the fast decay is not due to poor loading on the RISC complex.
Taken together, these results suggest that enzymatic activities able to modify miRNA 3′-ends are in place and mediate miRNA degradation.
miRNA decay rate confers flexibility to miRNA regulation particularly in nondividing cells
We sought to verify the interplay between miRNA decay and transcription, target abundance, and 3′-end modification during the dynamic regulation of the miRNA pool in physiological processes, such as serum stimulation of quiescent fibroblasts (Supplemental Fig. S6A). Indeed, in this condition, a consistent change in gene and miRNA expression occurs without cell division; thus, miRNA dilution is negligible (α = 0). To minimize the effects of cell density on miRNA processing (Hwang et al. 2009; Mori et al. 2014), quiescent and serum-stimulated cells were kept at low density (<70%). We followed miRNA expression by small RNA sequencing for up to 12 h post serum stimulation, when DNA synthesis starts (Supplemental Fig. S6B,C). We isolated a group of serum-regulated miRNAs (N = 52) (Supplemental Table S5), consistent with previous observations (Bueno et al. 2010; Rissland et al. 2011), composed of a module of serum-induced miRNAs (“UP”; N = 39) (Fig. 6A; Supplemental Fig. S6D) and of serum-repressed miRNAs (“DOWN”; N = 13), whose down-regulation was observed even when data were normalized for the increase in total RNA levels induced by serum stimulation. Overall, the “DOWN” group was completely depleted of “slow” miRNAs and significantly enriched for “fast” miRNAs (Fig. 6B). This group also displayed substantially shorter half-lives (Fig. 6C, P < 0.01), compatible with the kinetics of cell cycle re-entry. For some serum-regulated miRNAs, we also measured decay rates in quiescence and observed values similar to those determined in growing cells (Supplemental Fig. S6E,F), suggesting that, in this limited number of cases, decay was not affected.
Figure 6.

Regulation of miRNAs by decay dynamics following serum stimulation. (A) The heat-map shows fold-change (log2) of serum-regulated miRNAs (n = 52) over the 12-h time course following serum treatment. Data are the average of two independent biological replicas (shown in Supplemental Fig. S6A). “Fast” and “slow” decay miRNAs are indicated by red and blue, respectively. (B) The distribution of serum-regulated miRNAs among decay classes was analyzed by contingency (χ2 = 9.3, P = 0.0096). (C) Distribution of miRNAs half-lives (T1/2) within classes of serum-regulated miRNAs. The “ALL” class (N = 119) contains all the common miRNAs of the serum-regulation and decay data sets. (D) Heat-maps show the variation of tailing (3′-NT/TOT) and trimming (TRIM-2/TOT) frequencies over the 12-h time course, as in A. (E) Number of miRNAs with changes in tailing (χ2 = 12.0, P = 0.0025) and trimming (χ2 = 15.2, P = 0.0005) within classes of serum-regulated miRNAs. (F–I) Representative examples of “DOWN” (F,H) and “UP” (G,I) serum-regulated miRNAs are shown. For each miRNA, the regulation of templated (T), tailed (3′-NT/TOT), and trimmed (TRIM-2/TOT) forms is reported, along with the changes in the primary transcript (below; in red and measured as CPC/h). The bar plot summarizes the frequency of each miRNA isoform. (J) Mean target abundance (with SEM) of down-regulated (in blue), up-regulated (in red), or all miRNAs (in black) over the time course of serum stimulation. (K) Correlation between target:miRNA ratio (TPM) in growing versus serum-depleted fibroblasts. Straight lines with shaded confidence intervals highlight the linear correlation. Targets were further distinguished according to their complementarity to the miRNA 3′ end, as described in Figure 4, A and B.
As miRNA degradation and “fast” decay are associated with the accumulation of certain miRNA isoforms, we analyzed isomiRs upon serum stimulation. We found a dynamic regulation of tailing and trimming during the time course for serum-regulated miRNAs, with a relative increase of both tailing and trimming for “DOWN” miRNAs and a decrease in trimming among “UP” miRNAs (Fig. 6D,E; Supplemental Fig. S6J). In many cases, the fluctuations of tailed and trimmed forms were the mirror image of miRNA regulation (templated forms) either for down- or up-regulated species (Fig. 6F–I).
Our previous observations (see Fig. 3; Supplemental Fig. S3) suggest that transcription plus decay mainly regulates changes in the miRNA pool. We verified this hypothesis by calculating the synthesis rate of pri-miRNAs along the time course by integrating RNA-seq data on total and nascent RNA (with a short 4sU pulse of 10 min). Indeed, most of the changes in the miRNA pool could be ascribed to concomitant coherent variations in the synthesis rate of pri-miRNAs (Supplemental Fig. S6G,H; Supplemental Table S5), although in some cases (five of 10), we observed down-regulation with no evidence of a significant reduction in the transcription rate (see also miR-30b in Fig. 6F). In the case of miR-155, an “UP” miRNA, we observed a strong peak of transcriptional activation at 2 h post stimulation (Fig. 6G, bottom panel); afterward, miR-155 levels decreased according to its intrinsic stability. Thus, we could model miR-155 decay from 4 to 12 h, at the end of its transcriptional activation, calculating a T1/2 of 10.5 h, in very good agreement with the half-life measured by 4sU pulse-chase (12.3 h) (Supplemental Table S1). Of interest, miR-155 is a “fast” decaying miRNA, which our modeling (Fig. 3K; Supplemental Fig. S3D) predicted would be involved in sharp and quickly regulated mechanisms, such as the early serum response.
Finally, we analyzed the expression of miRNA TGs over the time course by exploiting RNA-seq data. We observed a general increase of targets with seed along the time course, which is independent of the regulation of miRNAs by serum (“seed”) (Fig. 6J; Supplemental Fig. S6I). However, when considering targets with 3′ compensatory binding, we observed an increase just for targets of “DOWN” miRNAs (“3C.03” and “3C.05”) (Fig. 6J; Supplemental Fig. S6I). This was particularly evident for miR-30b and miR-30c (two “DOWN” miRNAs), leading to a sudden increase in TPM_3C soon after serum stimulation (Fig. 6K). These two miRNAs are not transcriptionally repressed (Fig. 6F), supporting the hypothesis that induction of high-complementary targets might promote the clearance of certain miRNAs by TIMD.
Discussion
miRNA degradation dynamics are heterogeneous
Exploiting an approach that combines the metabolic labeling of RNA with high-throughput sequencing, we measured the decay rates of approximately 200 miRNAs in mammalian cells under physiological conditions. While most miRNAs are, indeed, stable molecules, with half-lives >24 h (“slow” miRNAs), as previously reported (Krol et al. 2010b; Ruegger and Grosshans 2012), a consistent number of miRNAs display shorter half-lives (<12 h; “fast” miRNAs, N = 61). “Fast” miRNAs include not only “passenger” miRNAs, which are poorly loaded onto AGO and thus quickly degraded (Guo et al. 2015), but also many “guide” miRNAs (N = 25), including the few (i.e., miR-503) that were previously described as having a rapid turnover (Krol et al. 2010a; Rissland et al. 2011). Previous attempts to measure globally the degradation rates of miRNAs relied on the use of inhibitors of miRNA transcription or processing, which represent nonphysiological conditions under which the assessment of “fast” turnover rates is arduous. More specifically, the block/loss of miRNA processing enzymes (i.e., DICER1) requires up to 3 d to be effective, a time frame in which “fast” decaying miRNAs are cleared and just the “slow” species are retained (Gantier et al. 2011). Conversely, the block of transcription by the use of RNA Polymerase II inhibitors can be maintained for only a few hours before inducing toxic effects and, thus, identifies just those miRNAs that are rapidly disappearing (the “passenger” miRNAs) (Guo et al. 2015). Nonetheless, among the “unstable” miRNA species found by such an approach, we found enrichment of the “fast” species identified here (seven in the top 10% of unstable species) (shown in Supplemental Table S1). Recently, an improved chemistry for 4sU labeling and isolation has been developed and used to analyze RNA stability (including miRNAs) by pulse labeling in HEK293T cells (Duffy et al. 2015). Of note, two different behaviors of miRNA decay also emerged by this approach (fast vs. slow), although the precise determination of miRNA degradation rates still requires more tailored methodologies (i.e., pulse-chase) to minimize the effects of processing on labeling efficiency (see Fig. 1E).
A more general question is whether miRNA half-lives are an intrinsic feature or whether they can be regulated according to the cellular context. We observed limited variations in miRNA half-lives between quiescent versus proliferating cells and between human versus murine fibroblasts. In addition, our decay rates were similar to those measured by others in different systems (Krol et al. 2010a; Rissland et al. 2011). Nonetheless, we cannot exclude significant changes in miRNA stability in specific physiopathological contexts, as observed for maternal miRNAs during embryogenesis (Lee et al. 2014).
Impact of different biogenetic steps on the miRNA pool: the role of transcription and decay
By use of multiple layers of genome-wide analysis, in particular, ChIP-seq of histone marks and nascent RNA sequencing, we were able to accurately define the genomic boundaries of miRNA transcriptional units, an approach that could be applied to other cells and conditions. Overall, we calculated the synthesis rates of approximately 100 miRNA genes. The dynamic range of transcription rates was wide (up to 500-fold) and correlated nicely with miRNA levels, supporting the hypothesis that miRNA expression is primarily controlled at the transcription step. By considering the synthesis and decay rates, we could reliably predict the abundance of miRNAs in steady-state conditions. Our theoretical calculation assumes that processing is much faster than cell division (k2 >> α), a simplification that did not hold for complex miRNA clusters, where processing efficiency plays an important role in dictating the levels of each miRNA contained in the cluster. A paradigmatic example is the miR-17-92 cluster, comprising six individual miRNAs processed with different efficiencies, despite being cotranscribed due to a particular tertiary structure of the pri-miRNA (Chakraborty et al. 2012). We have to underline that low efficiency maturation also affects half-life calculations by metabolic labeling. Indeed, we supposed a negligible production of mature miRNAs from labeled precursors during the chase period, an assumption that might not be true for miRNAs with very low processing rates. In such cases, the “apparent” half-life increases, and hence, the actual miRNA decay is faster than measured. More sophisticated experimental approaches are needed if we are to quantitatively and globally measure miRNA maturation in mammalian cells.
Nonetheless, by modeling the kinetic parameters (k1, k2, k3, and α) in a range compatible with experimental observations, we highlighted the impact of different biosynthesis and degradation rates on the regulation of miRNA within the cell. While transcription primarily regulates the magnitude of miRNA expression, the decay influences the dynamics of miRNA regulation: “Slow” decaying miRNAs can accumulate to high levels and are subjected to fewer fluctuations; conversely, miRNAs with “fast” decay rates can give rise to sharp and quick changes in miRNA levels. This contention implies that miRNAs with “fast” turnover should be involved in specific physiological processes that require a rapid change in miRNA levels. In our study, we showed that several of the miRNAs that are rapidly repressed (<12 h) upon serum stimulation of quiescent fibroblasts have a “fast” turnover. In addition, miR-155 (another “fast” miRNA) displayed a spike of expression during the early serum response. Other examples of biological processes that require a relatively fast regulation of miRNAs can also be found in the literature, with “fast” miRNAs implicated, such as during developmental transitions (let-7b/c, miR-99, miR-100) (Thornton et al. 2014), growth factor response (miR-320, miR-155) (Avraham et al. 2010), and epithelial-mesenchymal transition (miR-335, miR-205) (Pencheva and Tavazoie 2013).
High complementary targets and enzymatic activities are determinants of miRNA decay
Our analyses suggest that the ratio between high-affinity targets and miRNA abundance (TPM) is an important determinant in miRNA decay, in line with evidence from other groups claiming a target-mediated mechanism that promotes miRNA degradation (TIMD) (Baccarini et al. 2011; De et al. 2013; de la Mata et al. 2015). Such a mechanism has been associated with two conditions: an extensive complementarity between the target and miRNA, which promotes miRNA unloading (De et al. 2013), and the induction of 3′-end nontemplated miRNA modifications (Ameres et al. 2010; Baccarini et al. 2011; de la Mata et al. 2015). Both of these conditions were found to be associated with “fast” decaying miRNAs. “Fast” miRNAs were enriched in tailed and trimmed variants, in particular, in adenylation and uridylation modified variants (Fig. 5), and when the targets were selected on the basis of 3′ complementarity (3C.03 or 3C.05), the association of TPM with half-lives became much stronger (Fig. 4). In addition, “fast” and “slow” miRNAs displayed a similar number of seed-containing targets (and target copies) in 3T9 cells, but different values for targets with 3′ complementarity (Fig. 4D). Although these correlations are strong, more direct evidence will be required to demonstrate that the behavior of “fast” miRNAs depends on the level of their 3′ complementary targets. Of note, several studies pointed out the existence of a threshold in target repression that depends on miRNA expression and target abundance (Mukherji et al. 2011; Mullokandov et al. 2012; Bosson et al. 2014), suggesting that the activity of a miRNA on its target decreases sharply at high TPM ratios.
A relevant implication of our study is the theoretical possibility to switch from “high” to “low” TPM and thus modify the activity and the decay of miRNAs. However, the number of miRNA targets is typically very high, and only a few of them are expressed at hundreds of CPCs. Thus, it is very unlikely that TPM can be consistently affected by altered expression of one or two TGs. Hence, a better understanding of the role of competing endogenous RNAs (ceRNAs) in miRNA mechanisms is urgently needed. We predict such a phenomenon might take place only at high TPM, given the preferential interaction of a miRNA with high-affinity targets and the reduction of the effective target pool, as previously observed (Bosson et al. 2014).
Overall, our study provides an integrated framework for the quantitative analysis of miRNA biology, with ad hoc tools to assess pri-miRNA transcription rates, miRNA turnover, structural heterogeneity (isomiRs), and activity on targets (TPM). By monitoring simultaneously and quantitatively these intertwined features, we obtained important insights into the dynamics of miRNA function and regulation. We now need to extend this kind of approach to other (physiological and pathological) systems and to include missing regulatory mechanisms (such as processing, subcellular compartmentalization, enzymatic activities). We expect that with such an integrated and quantitative framework, we will achieve a deeper understanding of the regulatory circuitries controlling miRNA concentration and, more importantly, activity.
Methods
4sU labeling
4sU (Sigma catalog no. T4509) was added to culture medium at a final concentration of 300 μM. Cells were pulsed for 3 h and subsequently chased with 1 mM uridine (Sigma catalog no U3003). For continuous labeling experiments, cells were treated with 4sU (300 μM) for increasing lengths of time (15 min, 30 min, 45 min, 60 min, 120 min, 180 min). Total RNA was extracted at each time point by the miRNeasy mini kit (Qiagen catalog no. 217004), according to the manufacturer's instructions, including the recommended DNase I digestion. Separation of 4sU-labeled newly synthesized RNA from nonlabeled pre-existing RNA was performed as previously described (Rabani et al. 2011) with minor modifications (see Supplemental Material).
Determination of miRNA half-lives
For mRNA and pri-miRNAs, half-lives (T1/2) were calculated indirectly by the “pulse labeling” method, measuring the ratio (R) of newly transcribed RNA (labeled) over total RNA (Dolken et al. 2008), according to the following equation:
For miRNAs, half-lives (T1/2) were calculated by pulse-chase, measuring the decay of labeled RNA species during the chasing time. For genome-wide analysis of miRNA half-life, three independent pulse-chase experiments were used (decay data set). The decay data set (normalized and background subtracted) (Supplemental Material) was imported in GraphPad Prism 6, and half-lives were modeled by a one-phase exponential decay, with a plateau fixed to a constant value of zero and a fitting method based on least squares. In the fitting, the replicate values at each time point (N = 3) were averaged. The simple one-phase decay was compared with a two-phase decay, which would imply the existence of different miRNA pools with specific decay rates (e.g., miRNA unloaded and loaded on AGO2). The extra sum-of-squares F-test was used to select the best model. We preferred the simpler model unless the P-value was <0.05. “Fast” and “slow” classes were defined as follows: “fast,” T1/2ave < 14 h with T1/2max < 20 h; “slow,” T1/2ave > 24 h with T1/2min > 15 h. Half-lives and confidence intervals are reported in Supplemental Table S1.
Statistical analysis
Analyses (Oneway, Scatter Plot, Contingency) and statistics were produced using JMP 10 (SAS) software. Microsoft Excel was used to generate bar graphs with average and SD of repeated experiments. The number of replicates and the statistical tests used are indicated in the figure legends. Heat-maps were generated by Java TreeView software (http://jtreeview.sourceforge.net) for Mac OSX.
Data access
The small-RNA-seq data sets from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72656 for the “decay data set” and under accession number GSE72655 for the “serum-stimulation data set.”
Supplementary Material
Acknowledgments
We thank the Genomic Unit (Luca Rotta, Salvatore Bianchi, Thelma Capra) for sequencing runs. We thank Stefano Campaner and Gioacchino Natoli for helpful discussions and critical suggestions and Rosalind Gunby for manuscript editing. This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to F.N. (IG14085), B.A. (IG13182), and M.J.M. (TRIDEO 15904) and the European Research Council (268671), European Commission (622934 and 259743), and Ministero della Salute (RF-2011-02346976) to B.A.
Author contributions: M.J.M. and F.G. performed 4sU labeling and 4sU pulse-chase experiments. H.M. and B.C. performed bioinformatics analyses. M.J.M., F.G., M.M.G., and C.G. prepared samples and libraries related to the serum-stimulation data set. T.K., M.M.G., and B.A. produced nascent RNA-seq data. F.G. and P.B. performed experiments with inducible systems and miRNA reporters. B.C. developed mathematical modeling of miRNA abundance and dynamic changes. M.J.M. and S.d.P. calculated miRNA synthesis rates. M.J.M. and F.N. performed data analyses. M.J.M. and F.N. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.198788.115.
Freely available online through the Genome Research Open Access option.
References
- Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD. 2010. Target RNA-directed trimming and tailing of small silencing RNAs. Science 328: 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham R, Sas-Chen A, Manor O, Steinfeld I, Shalgi R, Tarcic G, Bossel N, Zeisel A, Amit I, Zwang Y, et al. 2010. EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors. Sci Signal 3: ra43. [DOI] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, Brown BD. 2011. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr Biol 21: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, Kiledjian M. 2010. Differential regulation of microRNA stability. RNA 16: 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA. 2014. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno MJ, Gomez de Cedron M, Laresgoiti U, Fernandez-Piqueras J, Zubiaga AM, Malumbres M. 2010. Multiple E2F-induced microRNAs prevent replicative stress in response to mitogenic signaling. Mol Cell Biol 30: 2983–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K, Muhl B, Kellner M, Rohrmoser M, Gruber-Eber A, Windhager L, Friedel CC, Dolken L, Eick D. 2013. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol 10: 1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DM, D'Ambrogio A, Nottrott S, Richter JD. 2011. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. 2007. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136: 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Mehtab S, Patwardhan A, Krishnan Y. 2012. Pri-miR-17-92a transcript folds into a tertiary structure and autoregulates its processing. RNA 18: 1014–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Grosshans H. 2009. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 461: 546–549. [DOI] [PubMed] [Google Scholar]
- Cleary MD, Meiering CD, Jan E, Guymon R, Boothroyd JC. 2005. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat Biotechnol 23: 232–237. [DOI] [PubMed] [Google Scholar]
- De N, Young L, Lau PW, Meisner NC, Morrissey DV, MacRae IJ. 2013. Highly complementary target RNAs promote release of guide RNAs from human Argonaute2. Mol Cell 50: 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P, Filipowicz W, Grosshans H. 2015. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep 16: 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pretis S, Kress T, Morelli MJ, Melloni GE, Riva L, Amati B, Pelizzola M. 2015. INSPEcT: a computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA- and 4sU-seq time course experiments. Bioinformatics 31: 2829–2835. [DOI] [PubMed] [Google Scholar]
- Diederichs S, Haber DA. 2007. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131: 1097–1108. [DOI] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD. 2015. Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol Cell 59: 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E. 2008. Getting to the root of miRNA-mediated gene silencing. Cell 132: 9–14. [DOI] [PubMed] [Google Scholar]
- Flores O, Kennedy EM, Skalsky RL, Cullen BR. 2014. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res 42: 4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Dolken L, Ruzsics Z, Koszinowski UH, Zimmer R. 2009. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res 37: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangaraju VK, Lin H. 2009. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol 10: 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, et al. 2011. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 39: 5692–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Liu J, Elfenbein SJ, Ma Y, Zhong M, Qiu C, Ding Y, Lu J. 2015. Characterization of the mammalian miRNA turnover landscape. Nucleic Acids Res 43: 2326–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. 2007. A hexanucleotide element directs microRNA nuclear import. Science 315: 97–100. [DOI] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT. 2009. Cell–cell contact globally activates microRNA biogenesis. Proc Natl Acad Sci 106: 7016–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E. 2015. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet 16: 421–433. [DOI] [PubMed] [Google Scholar]
- Kim YK, Heo I, Kim VN. 2010. Modifications of small RNAs and their associated proteins. Cell 143: 703–709. [DOI] [PubMed] [Google Scholar]
- Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J, Duebel J, Bicker S, Fehling HJ, Schubeler D, et al. 2010a. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 141: 618–631. [DOI] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. 2010b. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11: 597–610. [DOI] [PubMed] [Google Scholar]
- Lee M, Choi Y, Kim K, Jin H, Lim J, Nguyen TA, Yang J, Jeong M, Giraldez AJ, Yang H, et al. 2014. Adenylation of maternally inherited microRNAs by Wispy. Mol Cell 56: 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Gregory RI. 2015. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 15: 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Lowe SW. 2012. The microcosmos of cancer. Nature 482: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. 2014. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 156: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Marzi MJ, Nicassio F. 2014. IsomiRage: from functional classification to differential expression of miRNA isoforms. Front Bioeng Biotechnol 2: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. 2012. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9: 840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen CT, Goodall GJ, Bracken CP. 2012. IsomiRs—the overlooked repertoire in the dynamic microRNAome. Trends Genet 28: 544–549. [DOI] [PubMed] [Google Scholar]
- Pencheva N, Tavazoie SF. 2013. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol 15: 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. 2011. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29: 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V, Chen X. 2008. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321: 1490–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Hong SJ, Bartel DP. 2011. MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 43: 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegger S, Grosshans H. 2012. MicroRNA turnover: when, how, and why. Trends Biochem Sci 37: 436–446. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342. [DOI] [PubMed] [Google Scholar]
- Thornton JE, Du P, Jing L, Sjekloca L, Lin S, Grossi E, Sliz P, Zon LI, Gregory RI. 2014. Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res 42: 11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. 2007. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316: 575–579. [DOI] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, Tewari M. 2011. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res 21: 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, et al. 2012. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods 9: 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE Jr. 2003. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res 13: 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Lu Y, Zhang Q, Liu JJ, Li TJ, Yang JR, Zeng C, Zhuang SM. 2012. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res 40: 4615–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.