

Abstract
Adeno-associated virus (AAV) has shown great promise as a gene therapy vector in multiple aspects of preclinical and clinical applications. Many developments including new serotypes as well as self-complementary vectors are now entering the clinic. With these ongoing vector developments, continued effort has been focused on scalable manufacturing processes that can efficiently generate high-titer, highly pure, and potent quantities of rAAV vectors. Utilizing the relatively simple and efficient transfection system of HEK293 cells as a starting point, we have successfully adapted an adherent HEK293 cell line from a qualified clinical master cell bank to grow in animal component-free suspension conditions in shaker flasks and WAVE bioreactors that allows for rapid and scalable rAAV production. Using the triple transfection method, the suspension HEK293 cell line generates greater than 1 × 105 vector genome containing particles (vg)/cell or greater than 1 × 1014 vg/l of cell culture when harvested 48 hours post-transfection. To achieve these yields, a number of variables were optimized such as selection of a compatible serum-free suspension media that supports both growth and transfection, selection of a transfection reagent, transfection conditions and cell density. A universal purification strategy, based on ion exchange chromatography methods, was also developed that results in high-purity vector preps of AAV serotypes 1–6, 8, 9 and various chimeric capsids tested. This user-friendly process can be completed within 1 week, results in high full to empty particle ratios (>90% full particles), provides postpurification yields (>1 × 1013 vg/l) and purity suitable for clinical applications and is universal with respect to all serotypes and chimeric particles. To date, this scalable manufacturing technology has been utilized to manufacture GMP phase 1 clinical AAV vectors for retinal neovascularization (AAV2), Hemophilia B (scAAV8), giant axonal neuropathy (scAAV9), and retinitis pigmentosa (AAV2), which have been administered into patients. In addition, we report a minimum of a fivefold increase in overall vector production by implementing a perfusion method that entails harvesting rAAV from the culture media at numerous time-points post-transfection.
Introduction
The benefits of using adeno-associated virus (AAV) for gene therapy include long-term gene expression, the inability to autonomously replicate without a helper virus, transduction of dividing and nondividing cells, and the lack of pathogenicity from wild-type infections. A number of phase 1 and phase 2 clinical trials utilizing AAV have been performed worldwide.1,2 However, many preclinical studies and clinical trials have demonstrated a number of challenges that will need to be addressed to sustain rAAV use for human gene therapy.2 One major challenge is establishing large-scale manufacturing technologies in accordance with current good manufacturing practices (cGMP) to yield the purified vector quantities needed for the expanding clinical need. The success of generating a scalable production technology relies heavily on understanding the basic biology of AAV in regard to generating reagents such as cell lines, plasmids, or recombinant viral vectors that when used together, will closely mimic wild-type (wt) AAV production.
The generation of rAAV vectors entails encapsidation of a therapeutic gene of interest utilizing the wt replication (rep) and capsid (cap) genes. rAAV production requires delivery of the proviral (vector DNA) construct, the rep and cap genes specific to the serotype of interest and Adenovirus (Ad) or Herpes Simplex Virus (HSV) helper virus functions.3,4
Adenovirus (Ad) and HSV have been shown to be necessary for rAAV replication. In regard to the Ad helper virus, the E1a, E1b, E2a, E4orf6, and VA RNA genes were determined to supply the helper functions necessary for rAAV production.5 Infection of Ad into producer cells to generate rAAV was effective in producing rAAV, but a consequence was that it also produced Ad particles. A significant improvement in the evolution of rAAV production was the introduction of the triple plasmid transfection method.5 This method used an AAV serotype-specific rep and cap plasmid as well as the vector DNA plasmid, but eliminated the use of Ad infection by supplying the essential Ad genes on a third plasmid (pXX6). Supplying the Ad helper genes on the pXX6 plasmid eliminated Ad production in the transfected cells yielding only rAAV vector. Multiplasmid transient transfection of adherent HEK293 cells remains the most widely used method for rAAV production.5,6,7
Advances and novel approaches in rAAV production have been made recently that have allowed some laboratories to move away from production using adherent HEK293 cells and move toward scalable technologies. These novel approaches use alternate helper viruses and nonmammalian cells. The dual HSV infection system has been shown to generate 5 × 104 to 1 × 105 vg/cell with low particle to infectivity ratios.8,9,10,11,12 rAAV production using the insect baculovirus Autographa californica nuclear polyhedrosis (baculovirus expression vector system (BEVS)) has also been investigated in Spodoptera frugiperda (Sf9) cells.13,14,15,16,17,18,19 Yields of up to 5 × 104 vg/cell have been reported using BEVS.1
A few recent studies have reported rAAV production from mammalian HEK293 cells adapted to grow in serum-free suspension media.20,21,22 Park et al. and Durocher et al. demonstrated that approximately 1.4 × 104 and 3 × 104 vg/cell respectively were generated using their optimized serum-free suspension HEK293 cell transient transfection production systems. Common with these studies is the fact that the yield of vector continues to be the impediment and significantly below the vg/cell generated via transfection of adherent HEK293 cells in the presence of serum and the rHSV and BEVS production systems.
In this report, we demonstrate that it is possible to adapt a HEK293 cell line in animal component-free conditions to generate greater than 1 × 105 vector genome containing particles per cell and yield greater than 1 × 1013 purified rAAV from 1 l of transiently transfected cell culture. The adaptation and optimization spanned over a 2-year period. However, the resulting cell clone and conditions have remained stable over thousands of production runs, which include both preclinical and clinical manufacturing. The manufacturing system can be used to produce and purify numerous rAAV serotypes and chimeric capsids (e.g., 1–9) with significantly reduced empty particles. The system has been successfully adapted to WAVE bioreactors and small scale stir tank bioreactors demonstrating a seamless transition from shaker flasks and establishing that the transient transfection manufacturing technology is scalable. Furthermore, we have shown that it is possible to continuously harvest nonheparin binding rAAV serotypes from the suspension culture media over a 6-day period.
Results
Development of a suspension HEK293 cell line
The purpose of this work was to generate a scalable manufacturing technology to produce high-titer and highly pure rAAV using transient transfection technology and mammalian HEK293 cells. To begin, an adherent HEK293 cell line from our GMP facility's qualified master cell bank (derived from a clone previously selected for high transfection efficiency and rAAV production) was adapted to grow in serum-free suspension conditions as described in the methods section. After adaptation to growth in animal component-free and antibiotic-free suspension conditions and selection of a compatible serum-free suspension medium, the suspension HEK293 cell line maintained its ability for high transfection efficiency and rAAV production.
Optimization of transfection conditions using polyethyleneimine Max
Previously, our laboratory utilized polyethyleneimine (PEI) (linear 25-kDa) for transfection of adherent HEK293 cells.23 PEI was selected for use in this study because it is well-tolerated by HEK293 cells, able to transfect HEK293 cells very efficiently, does not require a media change after transfection, and is very inexpensive compared to commercially available lipid transfection reagents. It was recently shown that removal of the residual N-acyl moieties from commercial linear 25-kDa PEI enhanced its plasmid DNA delivery efficiency 21 times in vitro, as well as 10,000 times in mice with a 1,500-fold enhancement in lung specificity.24 Although the fully deacylated forms of PEI that Thomas et al. generated are not manufactured and available commercially, Polysciences supplies PEI Max which is built from the same linear 25-kDA polymer, but contains more than 11% additional free nitrogens. Tested side by side and consistent with data from Thomas et al., PEI Max was superior in transfection efficiency than PEI on our adherent and suspension HEK293 cells (data not shown). Therefore, all transfection optimization studies on the suspension HEK293 cells were conducted using PEI Max.
Two of the major requirements for production of rAAV using transient transfection are determining the optimal plasmid ratios of the three plasmids and the transfection reagent to total DNA ratio. The suspension HEK293 cells were initially grown in SFM4Transfx-293 media and were seeded at 1 × 106 viable cells/ml in a 30 ml volume in 125 ml shaker flasks on the day of transfection. Various ratios of XX680:pXR2:TReGFP were tested with PEI Max to DNA ratios of 2:1 and 4:1 with 1 µg DNA/ml of cells and incubated at room temperature for 10–15 minutes. As shown in Figure 1a, the plasmid molar ratio of 2:1.5:1 with a PEI Max to DNA ratio of 2:1 generated the most vector genomes (vg)/cell. Vg/cell from cell lysates were determined using Quantitative PCR (qPCR) methods as described in the methods section. It was evident that the PEI Max to DNA ratio of 4:1 generated the least amount of vg/cell at all plasmid ratios tested. This was most likely due to a lower cell viability post-transfection, detected by the Beckman ViCell XR Viability analyzer, suggesting a level of toxicity was reached with PEI Max. In addition, the 4:1 PEI Max to DNA ratio led to larger cell aggregates after transfection. This occurrence was most likely due to an increased concentration of PEI Max/DNA complexes in the cell culture, which have a net positive charge leading to cell to cell interaction. Experiments described from this point on will be utilizing the plasmid ratio of 2:1.5:1 along with PEI Max to DNA ratio of 2:1.
Figure 1.

Optimization of triple transfection plasmid ratios and transfection cocktail volumes. (a) Varying plasmid ratios of XX680, pXR2 helper, and TReGFP plasmid were explored to determine optimal plasmid ratio for rAAV vector production. (b) Varying transfection cocktail volumes were tested to determine optimal transfection conditions with PEI Max to DNA ratio of 2:1 and an incubation time of 10–14 minutes. Percent volume of transfection cocktail is in reference to percent of final cell culture volume. These parameters were optimized using the SFM4Transfx-293 media (HyClone).
A number of lipid-based transfection protocols describe the importance of the transfection cocktail volume. To determine the impact of transfection cocktail volume on transfection efficiency and vector production using PEI Max, transfection cocktail volumes of 2, 5, and 10% were selected. The % volume is based on the final cell culture volume in the shaker flask. As illustrated in Figure 1b, a 5% transfection cocktail volume was shown to have the better transfection efficiency (based on green fluorescent protein (GFP) flow cytometry) and generated the most vg/cell.
Optimization of cell density and plasmid DNA concentrations
Prior to initiating these studies, we began using the FreeStyle F17 serum-free suspension media from Invitrogen that supported better growth of our cell clone, increased transfection efficiency and increased vector production/cell. The suspension HEK293 cells were diluted in the FreeStyle F17 media to 1 × 106 and 2 × 106 viable cells/ml to achieve a final cell culture volume of 30 ml. 1, 1.5, and 2 µg/ml of plasmid DNA were utilized in this set of experiments. Based on the data shown in Figure 2a, it was evident that the rAAV vector production was optimal when the cell density was at 1 × 106 viable cells/ml at the time of transfection using 1.5 µg/ml of plasmid DNA.
Figure 2.

Cell density transfection optimization experiments and impact of cell culture age on vector production. (a) 1 × 106 and 2 × 106 viable cells/ml were transfected with 1, 1.5, and 2 μg of plasmid DNA (2:1.5:1 XX680:pXR:TR plasmid). (b) Low, medium, and high passage cells were transfected using optimized parameters to determine if cell culture age impacted transfection efficiency and rAAV production.
Over the course of experimentation, the suspension HEK293 cells were passaged a number of times and when low, medium and high passage cells were available, they were transfected to determine if cell culture age impacted transfection efficiency and rAAV production. As shown in Figure 2b, transfection efficiency is not impacted by cell culture age, but rAAV production per cell decreases over time suggesting that the suspension HEK293 cell clone that was developed should be used a maximum of 30–40 passages.
Purification of rAAV serotypes using ion exchange chromatography
In conjunction with the optimization of rAAV production in suspension HEK293 cells, we focused on AAV purification. We set out to develop a universal purification strategy for all AAV serotypes and chimeric capsids using ion exchange chromatography. Previous studies reported that purification of different serotypes could be accomplished through the use of various ion exchange chromatography methods and resins.25,26,27 We rationalized that ion exchange chromatography, using a single anionic or cationic resin, could potentially be used to purify all serotypes through modulation of specific parameters during the binding and elution steps. It was previously reported that efficient clarification of rAAV cell lysates can be accomplished through the use of discontinuous iodixanol gradient purification and that the iodixanol did not impact rAAV interaction with the ion exchange resin.28 We thus employed the use of a modified discontinuous iodixanol gradient to clarify rAAV cell lysates prior to ion exchange chromatography. Buffers and pH chosen for the iodixanol gradient were modified to be consistent with a single downstream ion exchange chromatography step. Figure 3a illustrates the initial experimental results using the modified discontinuos iodixanol gradient in conjunction with ion exchange chromatography. The silver stain was loaded with 1 × 1010 vector genome containing particles of rAAV2 cytomegalovirus transcriptoinal promoter (CMV)-eGFP peak elution fractions. It is evident that the latter two elution peak fractions contain a small circular contaminant ranging in size between 5 and 10 microns (arrows in the transmission electron microscopy (TEM) negative stain image) with an individual/subunit molecular weight ranging between 20–25 kDa. This contaminant was present in all rAAV serotype preparations and therefore not common to only AAV2. Through the use of mass spectrometry (Applied Bioimics, Hayward, CA), the contaminant was identified as ferritin. Through further manipulation of the ion exchange chromatography step, we were able to physically separate the rAAV from the ferritin. Figure 3b depicts the physical separation of rAAV from the ferritin based on silver stain and negative stain TEM analyses of peak elution fractions and downstream fractions. The modified versions of the discontinuous iodixanol gradient and ion exchange chromatography are the downstream steps in the current purification process used to purify all rAAV serotype and chimeric capsid vectors in this report. The purification procedure is considered universal because a single set of parameters (components, reagent/buffer concentrations, pH, chromatography resin, and chromatography protocol) are used to purify all AAV serotypes and chimeric capsids.
Figure 3.

Characterization of elution peak fractions of adeno-associated virus (AAV) after ion exchange chromatography. (a) Silver stain and negative stain transmission electron microscopy (TEM) images of elution peak fractions of AAV using nonoptimized elution conditions for rAAV. Arrows represent the major contaminant (Ferritin) on the silver stain gel and the TEM image. (b) Silver stain and negative stain TEM images of elution peak fractions of AAV using optimized elution conditions for rAAV. Silver stain and TEM images illustrate physical separation of the virions and the ferritin contaminants.
Production and purification of single-stranded and self-complementary rAAV serotypes 1–6, 8, and 9
As described in the methods section, 1 l culture volumes or 1 × 109 viable cells were transfected using the optimized transfection parameters to generate single-stranded and self-complementary rAAV serotypes 1–6, 8, and 9 packaging the CMV-eGFP transgene cassette. As shown in Table 1, the transfection efficiency (based on GFP flow cytometry) was greater than 70% and nearly equivalent among all transfections. All of the serotypes packaging a single-stranded genome generated around and above 1 × 105 vg/cell. Excluding rAAV serotype 4 (one of the lowest yielding serotypes), all single-stranded rAAV serotypes tested generated 8.2 × 1012 to 3.3 × 1013 total rAAV from one liter of cell culture postpurification and were relatively free of any proteins other than VP1, VP2, and VP3 as depicted in the silver stain in Figure 4a. Generally, self-complementary vector preps yield less total AAV vector than their single-stranded counterparts and are usually comprised of varying concentrations of both self-complementary (dimer) and single-stranded (monomer) genomes.29 The self-complementary rAAV serotypes tested generated 4.0 × 1012 to 1.3 × 1013 total rAAV from 1 l of cell culture postpurification. As shown in the alkaline gel Southern blot in Figure 4b, the rAAV particles efficiently packaged a high percentage of self-complementary genomes. When additional self-complementary rAAV vectors were produced with various transgene cassettes, a majority of the genomes packaged were self-complementary indicating that these results are not exclusive to self-complementary eGFP cassettes (data not shown).
Table 1. Production and purification of single-stranded and self-complementary rAAV serotypes 1–6, 8, and 9.

Figure 4.

Production and purification of single-stranded and self-complementary recombinant adeno-associated virus (rAAV) serotypes 1–6, 8, and 9 using optimized production and purification conditions. (a) Silver stain image of single-stranded serotypes 1–6, 8, and 9 postpurification. (b) Southern blot of alkaline agarose gel of self-complementary rAAV1–6, 8, and 9. (c) Negative stain transmission electron microscopy (TEM) images of rAAV serotype 1 (left image), serotype 2 (middle image), and empty rAAV2 capsids (right image). TEM images were also taken of serotypes 3–6, 8, and 9 (images not shown). Empty particles (indicated by arrows) can be distinguished based on the electron dense center within the particle.
A universal cell line that is permissive to most AAV serotypes has not been identified. However, a modified HeLa cell line called HeLaRC-32 (ref. 30) can be transduced in various degrees by all serotypes tested. The cell line contains an integrated AAV2 rep and cap gene and upon infection with Adenovirus, any vector cassette flanked by AAV2 ITRs will be replicated. The increased eGFP genome copy number will elicit higher expression of eGFP, which can then be visualized and quantitated using fluorescence microscopy. As expected, a range of vg:TU ratios (Table 1) was reported between serotypes upon infection of HeLaRC32 cells. Since cell lines and infectivity assays have not been established or standardized for AAV, transduction assays such as or similar to this can only be used as a guide or measuring tool to qualitatively determine whether a rAAV serotype batch falls within a specific acceptance criteria set by the investigator. As discussed thoroughly in Aucoin et al., comparison of transduction or infectivity data between publications should be done with vigilance due to the variety of assays utilized to quantitate infectivity.1
Negative stain TEM analysis of rAAV serotypes
To assess overall purity and ratio of genome-containing (full) to empty particles of the single-stranded and self-complementary rAAV, we imaged each serotype using negative stain TEM. It is well known that empty particles are generated when producing rAAV in any of the current production technologies. Discovering purification procedures that will effectively remove the empty particles from full particles are needed. Empty particles take up the negative stain (2% uranyl acetate) differently than the full particles making it possible to quantitate the number of full and empty particles in a vector preparation. Figure 4c depicts negative stain TEM images of rAAV1, rAAV2 as well as empty AAV2 particle preparation. The rAAV1 and rAAV2 negative stain TEM images are representative of the other serotypes. The CsCl density gradient purified empty AAV2 capsids were used as a negative staining control and negative stained as empty capsids.31 As shown in Table 1, the overall full to empty particle ratio was no less than 10:1 for all serotypes tested, suggesting that the purification process effectively removes empty particles.
Production of rAAV using WAVE bioreactors
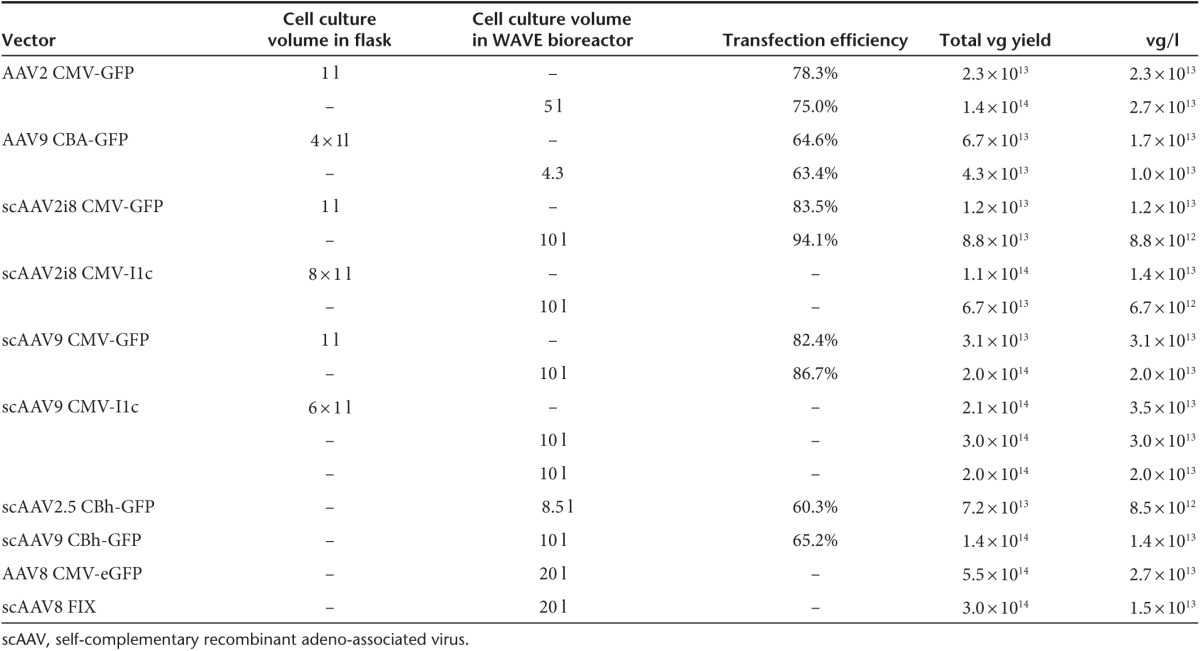
Production parameters for generating rAAV in shaker flasks using the FreeStyle F17 serum-free suspension media have been optimized. The next step was to translate the optimized parameters to the WAVE bioreactor at various cell culture volumes to determine scalability. A number of WAVE bioreactor runs were carried out ranging in size from 4.3 to 20 l of cell culture generating different serotype vectors. When possible, shaker flask cultures were transfected to serve as a control to compare transfection efficiency (if a GFP vector was produced) and vector production per liter of culture postpurification. As shown in Table 2, transfection efficiency was nearly equivalent between flasks and WAVE bioreactor bags. As was shown in flasks, most WAVE bioreactor runs generated at least 1 × 1013 purified rAAV vectors per liter of culture establishing that the parameters optimized for growth, transfection and rAAV production in shaker flasks translates to WAVE bioreactors. In most 10- and all 20-l WAVE bioreactor production runs, greater than 1 × 1014 purified ss and scAAV vectors were recovered.
Table 2. WAVE bioreactor production yields of single-stranded and self-complementary rAAV vectors.
Continuous rAAV harvesting from suspension media
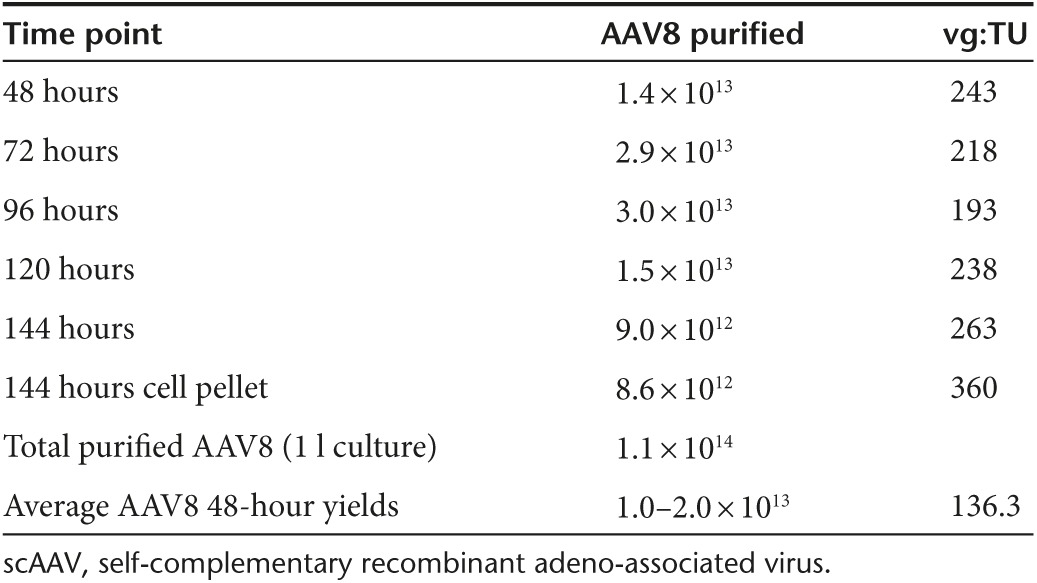
AAV was found to be secreted by an unknown mechanism into cell culture media using adherent HEK293 cells and harvesting rAAV from the media at a late time-point post-transfection.32,33,34 This concept was explored using the suspension HEK293 production system described in this report with the exception that media was harvested and replaced at various time points to reflect a discontinuous harvest of rAAV to maximize the output of the production system from a single transfection. Flasks containing 30 ml cultures of suspension HEK293 cells at 1 × 106 cells/ml were transfected to produce rAAV8 and rAAV9 CMV-eGFP. Forty-eight hours post-transfection, cell cultures were pelleted by low-speed centrifugation and the media removed for titering of DNase-resistant particles by dot blot and qPCR. The cell pellets were then resuspended in fresh media and placed back into the shaker incubator for continued growth and production of rAAV. Media samples were then taken every 24 hours and media was replaced every 48 hours. At the 120-hour time-point, rAAV8 and rAAV9 titers were determined from both the media and cell pellets. As shown in Figure 5, rAAV8 and rAAV9 can be detected and harvested from the media 48 hours post-transfection with increasing amounts of vector found in the media as time progresses and the media is replaced. When comparing the total yield to the 48 hours cell pellet control (Figure 5), the discontinuous harvest method yielded 6.5- and 4.8-fold more rAAV8 and rAAV9 vector respectively. This process was successfully adapted to the WAVE bioreactor using a perfusion set up to remove cell culture media every 24 hours for purification of rAAV followed by supplementing fresh media to keep the cells viable and producing rAAV. A 1 l cell culture in a 2 l perfusion WAVE bioreactor bag was transfected and 48 hours post-transfection 80% of the media was harvested through the perfusion filter and replaced with fresh media every 24 hours. The harvested media containing the rAAV8 CMV-eGFP vector was concentrated by TFF and then purified using the methods described. As shown in Table 3, AAV8 was purified from the cell culture media with yields ranging from 9 × 1012 to 3 × 1013 total vg at each time point. The overall purified yield was 1.1 × 1014, which included the yield from the cell pellet harvested and purified at 144 hours. The yield recovered from the cell pellet comprises only 8% of the overall yield suggesting that processing the cell pellet is not necessary. Producing AAV serotypes (that have low binding affinity to the producer cell line) using the perfusion production method will allow the manufacturer to utilize an animal-component free suspension culture to continuously produce, harvest and purify rAAV from the culture media via a single transfection without the need to lyse cells and introduce additional contaminants (host cell proteins and host cell genomic DNA). This in turn would generate more rAAV from an equal amount of input plasmid than the standard production protocol where the production process is halted when cells are harvested 48 hours post-transfection. This is the first report of continuous rAAV harvested from the cell culture media at numerous time-points post-transfection using a scalable, animal component-free system, suspension HEK293 cell line, and bioreactor.
Figure 5.

Continuous production and harvesting of recombinant adeno-associated virus (rAAV) 8 and rAAV9 from the suspension HEK293 culture medium. Total yield represents all media time-points and 120-hour cell pellet rAAV yields combined. Forty-eight hour cell pellet control represents the yield from a standard harvest at 48 hours post-transfection as described in this report.
Table 3. WAVE bioreactor production of rAAV8 CMV-eGFP using perfusion technology.
Discussion
A number of phase 1 and phase 2 clinical trials utilizing AAV have been performed worldwide for inherited and acquired diseases.1,2 The increase in the number of clinical trials emphasizes the need to establish scalable manufacturing technologies that are universal for all serotypes, GMP compliant as well as user-friendly. This report details a robust scalable transient transfection manufacturing technology (Figure 6) using a HEK293 cell line that can grow in animal component-free and antibiotic-free suspension conditions and generate rAAV yields per cell equivalent to the HSV dual infection technology8,9,10,11,12 and BEVS1 and superior to existing suspension HEK293 systems.20,21,22
Figure 6.

Depiction of recombinant adeno-associated virus (rAAV) manufacturing technology. (1) Suspension HEK293 cells in the WAVE bioreactor 20/50EHT are transfected. (2) Approximately 48 hours post-transfection, cells are harvested into centrifuge bottles and pelleted at low-speed centrifugation. (3) Cell pellets are resuspended and then lysed using a sonicator. (4) Lysate is incubated with Benzonase for 45 minutes at 37 °C. (5) Lysate is clarified using low-speed centrifugation. (6) Clarified lysate is loaded onto iodixanol gradient and ultracentrifuged. (7) rAAV collected from the 40%/60% iodixanol interface are loaded onto AKTA FPLC and purified using anion exchange chromatography. (8) rAAV chromatography peak fractions are dialyzed into final formulation buffer. (9) Sterile filtration through 0.2 μm filter.
A strategy was established early on during the development of this technology that influenced the choices made on reagents for deriving an animal component-free and antibiotic-free manufacturing technology. Vigilant screening of chemically defined suspension media to determine derivation of components was the first step followed by adapting the suspension HEK293 cell line to grow in these media. Based on cell growth characteristics, transfection efficiency and rAAV vector production two media were selected for further optimization studies13 SFM4Transfx-293 media and3 Freestyle F17 media. Further optimization of transfection conditions were assessed on these media (Figures 1 and 2) before it was determined that the F17 media was superior to the SFM4Transfx-293 media in regard to transfection efficiency and vector production for our specific suspension HEK293 cell clone. rAAV2 yields per cell for the SFM4Transfx-293 media and F17 media consistently ranged between 3–6 × 104 and 1–2 × 105 respectively (data not shown).
A universal purification strategy was developed using a modified step iodixanol gradient followed by a single ion exchange chromatography step. This purification procedure is considered “universal” due to its ability to purify AAV serotypes 1–6, 8, 9 and all University of North Carolina Gene Therapy Center chimeric capsids following a single protocol with no modifications due to differences in serotype (Figure 4a and Table 1). In addition to generating high-purity vectors, this procedure yields >90% full rAAV particles (Figure 4c and Table 1). The time invested to generate high-purity yields of rAAV using shaker flasks and WAVE bioreactors is nominal, requiring only 1 to 2 weeks. Based on the doses used in the phase 1 Leber's Congenital Amaurosis clinical trial,35 a single 5 l WAVE bioreactor culture would generate several thousand clinical doses (Table 2, AAV2 5 l wave run). Expanding on the utility of this manufacturing technology is its ability to generate and harvest AAV serotypes (with low binding affinity to HEK293 cells) from the suspension culture media. Early studies carried out in 1990s by Xiao and Samulski, demonstrated that AAV particles were secreted in culture media.34 This uncovered anomaly was further characterized by Lock et al. and Vandenberghe et al. to illustrate the partitioning of certain AAV serotypes from the cell during productive infection. As an outcome, this has allowed the manufacturer to utilize an animal component-free/antibiotic-free suspension culture to continuously produce and harvest rAAV from the culture media via a single transfection at all scales without lysing and harvesting AAV from the producer cells. This in turn would generate more AAV from an equal amount of transfected plasmid than the standard transfection protocol where the production process is halted when cells are harvested 48 hours post-transfection without collection of the media. Yields of rAAV using the continuous harvest/perfusion methodology would reach or surpass 1 × 1015 total purified vg for some rAAV vectors from a 10-l WAVE bioreactor culture at low cell density (total vg based on yields in Tables 2 and 3).
While transfection of suspension HEK293 cells requires qualifying plasmids for GMP production of rAAV, these reagents are stable over long durations. When compared to rAAV production systems that require infectious agents like HSV and BEVS, plasmid production is relatively easy, quick, and inexpensive (especially when the continuous harvest of AAV from suspension culture is utilized) to make under GMP compliance with release testing conducted in a good laboratory practices setting. For both the BEVS and rHSV production systems, a significant amount of time, labor, and finances must be invested in generating and characterizing the vectors to generate rAAV at all scales in addition to establishing master and working cell and viral banks. However, once these reagents are established, they are very ameanable to large-scale production and continue to provide the research community a number of viable AAV production approaches going forward. As with all seed stocks that are infectious in nature, potential concern of vector instability is carefully monitored during serial expansion to large volumes. For generation of baculovirus and rHSV master viral banks or working viral banks each expansion requires batch testing prior to use. Similarly to the triple transfection protocol presented here, optimizing these alternative AAV production approaches requires challenges that are being addressed currently. For example current rHSV-based rAAV production requires generating high-titer, infectious rHSV stocks from engineered rHSV vectors that are intentionally designed to be replication deficient that sometimes reduces the overall yield of seed stocks. As these and other identified rate limiting production concerns are systematically addressed, the AAV gene therapy community has access to numerous production alternatives going forward.
As described in this report, this user-friendly manufacturing process can easily be adapted and used in all academic AAV laboratories and serve as the workhorse technology to bring additional investigators' work into preclinical and clinical phases of research. This approach is ideally suited when efforts to optimize vector of choice (e.g., protmoter tweaking, optimization modifications) are an active process since the time from plasmid to vector is extremely quick. More importantly, the process of producing large quantities rAAV can be completed within 1–2 weeks, provides yields and purities acceptable for clinical applications and is universal with respect to all serotypes and chimeric capsids tested to date. With respect to use by numerous investigators, the manufacturing technology does not require the use of infectious agents or the capacity to learn another biological system (e.g., BEVS), but however does require an understanding of AAV biology and transient transfection. Currently, the Vector Core facility at the University of North Carolina is generating research, preclinical and clinical grade lots of rAAV using the manufacturing technology detailed in this report. Greater than 100 large-scale research and preclinical lots of different rAAV serotypes have been administered into mice, rats, cats, dogs, pigs, horses, and nonhuman primates. Several cGMP phase 1 clinical manufacturing campaigns have been successfully carried out using the manufacturing technology including ongoing scAAV8 FIX clinical trial carried out by Baxalta. As the described transfection-based scalable manufacturing technology is further developed, manufacturing capacities and efficiencies will increase in addition to critical starting reagents (input plasmids, transfection reagent, and media) and overall cost of manufacturing will decrease. Our recent transfection experiments utilizing a modestly higher cell density (4 × 106 cells/ml) conducted in small scale shaker flasks and 3 l stir tank biroeactors demonstrated a linear increase in vector production (~1 × 1015 vg/l of lysate) using less than-linear plasmid amount (data not shown). Optimization of feed strategies for high cell density (~1 × 107 cells/ml) transfection and AAV vector manufacturing are currently underway in addition to an optimized centrifugation-free, scalable purification process to accommodate larger batch sizes of 250 l or greater. This coupled with the AAV perfusion approach described above allowing one to generate essentially 5–10× amount of vector from same amount of starting plasmid suggest that current improvement of AAV vector production is on stable footing and transition to clinical demand should become routine due to user friendly protocols such as the one described here.
Materials and Methods
Derivation of suspension HEK293 cells from an adherent HEK293 qualified master cell bank. The derivation of the suspension cell line from the parental HEK293 master cell bank (MCB) was performed in a Class 10,000 clean room facility. The derivation of the suspension cell line was carried out in a two-phase process that involved first weaning the cells off of media containing bovine serum and then adapting the cells to SFM4Transfx-293 serum-free suspension media (Hyclone, Logan, UT). The suspension cell line was created as follows. First, a vial of qualified master cell bank was thawed and placed into culture in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and cultured for several days to allow the cells to recover from the freeze/thaw cycle. The MCB cells were cultured and passaged over a 4-week period while the amount of fetal bovine serum in the DMEM was gradually reduced from 10 to 2.5%. The cells were then transferred from DMEM 2.5% fetal bovine serum into SFM4Transfx-293 serum-free suspension media and grown in shaker flasks. The cells were then cultured in the SFM4Transfx-293 media for another 3 weeks while their growth rate and viability were monitored. The adapted cells were then expanded and frozen down. A number of vials from this cell bank were subsequently thawed and used during process development studies to create a scalable manufacturing process using shaker flasks and wave bioreactor systems to generate rAAV vectors. Suspension HEK293 cells were maintained in serum-free suspension F17 media (Thermo Fisher Scientific, Waltham, MA) that supports both growth and high transfection efficiency in shaker flasks and WAVE bioreactor bags. Multitron Shaker Incubators (Infors USA Inc., Laurel, MD) were used for maintenance of the cells and generation of rAAV vectors at specific rpm shaking speeds (based on cell culture volumes), 80% humidity, and 5% CO2. Cells were passaged and maintained between densities of 2 × 105 viable cells/ml and 3.5 × 106 viable cells per ml. Cell viabilities of > 95% were obtained consistently during routine cell passaging.
Transfection of suspension HEK293 cells (optimized protocol). On the day of transfection, the cells were counted using a ViCell XR Viability Analyzer (Beckman Coulter) and diluted to 1 × 106 viable cells/ml. To mix the transfection cocktail, the following reagents were added in this order: plasmid DNA (CsCl purified), Optimem (Thermo Fisher Scientific), Optipro or F17 media, Polyethyleneimine Max (MW 25,000). The plasmid DNA was used in a ratio of 2:1.5:1 for pXX680:pXR2:TR plasmid and 1.5 µg total plasmid was used per 1 × 106 total cells. The pXR series of helper plasmids were used in these studies to generate multiple rAAV serotype vectors.36 PEI Max was used at a PEI:DNA ratio of 2:1. Optimem I, Optipro, or F17 media was added such that the entire transfection mix cocktail made up 5% of the total flask final volume. The cocktail was vortexed for 5–10 seconds prior to being incubated at RT for 10–15 minutes. The transfection cocktail was then pipetted into the flasks and placed back in the shaker/incubator. Three hours post-transfection, the flasks were supplemented with CDM4HEK293 media (HyClone, Logan, UT) at 10% of final total cell culture volume to halt the transfection and prevent further PEI-related cell aggregation. All optimization studies were carried out at 30 ml culture volumes followed by validation at larger culture volumes. Cells were harvested 48 hours post-transfection with viabilities ranging between 75 and 90% depending on input plasmids.
Production of rAAV using WAVE bioreactor systems. WAVE bioreactor bags were seeded at a volume that was 2.5-fold less than the maximum cell culture volume of the WAVE bag. For example, 2 and 4 l for 10 and 20 l WAVE bioreactor bags respectively. Seeding cell density was between 7 × 105 and 9 × 105 viable cells/ml. The F17 media was supplemented with 0.2% Pluronic-F68 to allow for growth in WAVE bioreactor bags. Two days postseeding, the WAVE bioreactor bag, cell culture counts were taken and the cell culture was then expanded to 85% of the final cell culture volume. The WAVE bioreactor cell culture was then transfected. The transfection cocktail mix contains the following with a final volume of 5% of the final cell culture volume in the WAVE bioreactor bag: 1.5 μg of DNA/ml of cell culture, 2:1.5:1 plasmid ratios of XX680:pXR:TR plasmid, Polyethyleneimine Max (Polysciences) to DNA ratio of 2:1 and Opti-Mem I media (Thermo Fisher Scientific), Optipro (Thermo Fisher Scientific) or F17 media. After 10–15-minute incubation at room temperature, the transfection cocktail is pumped into the WAVE bioreactor bag. Three hours post-transfection, CDM4HEK293 media (HyClone) was pumped in at a volume of 10% of the final volume of cell culture. Cell culture was harvested from the WAVE bioreactor bag at least 48 hours post-transfection.
Analyzing transfection efficiency/GFP expression using flow cytometry. Approximately 24 hours post-transfection, 1 ml of cell culture was removed from each flask or WAVE bioreactor bag as well as an untransfected control. Samples were analyzed using a Dako Cyan (Carpinteria, CA) flow cytometer.
Harvesting suspension HEK293 cells from shaker flasks and WAVE bioreactor bags. Fourty-eight hours post-transfection, cell cultures were collected into 500 ml polypropylene conical tubes (Corning, Corning, NY) either by pouring from shaker flasks or pumping from WAVE bioreactor bags. The cell culture was then centrifuged at 655 × g for 10 minutes using a Sorvall RC3C plus centrifuge and H6000A rotor. The supernatant was discarded, the cells were resuspended in 1× phosphate-buffered saline, transferred to a 50 ml conical tube and centrifuged at 655 × g for 10 minutes. At this point, the pellet could either be stored in a −80 °C freezer or continued through purification.
Titering rAAV from cell lysate using qPCR. Ten milliliters of cell culture were removed and centrifuged at 655 × g for 10 minutes using a Sorvall RC3C plus centrifuge and H6000A rotor. The supernatant was decanted from the cell pellet. The cell pellet was then resuspended in 5 ml of DNase buffer (5 mmol/l CaCl2, 5 mmol/l MgCl2, 50 mmol/l Tris-HCl pH 8.0) followed by sonication to lyse the cells efficiently. Three hundred microliters were then removed and placed into a 1.5 ml microfuge tube. One hundred and forty units of DNaseI were then added to each sample and incubated at 37 °C for 1 hour. To determine the effectiveness of the DNase digestion, 4–5 µg of TReGFP plasmid was spiked into a nontransfected cell lysate with and without the addition of DNase. Fifty microliters of ethylenediaminetetraacetic acid (EDTA)/Sarkosyl solution (6.3% sarkosyl, 62.5 mmol/l EDTA pH 8.0) were then added to each tube and incubated at 70 °C for 20 minutes. Fifty microliters of Proteinase K (10 mg/ml) were then added and incubated at 55 °C for at least 2 hours. Samples were then boiled for 15 minutes to inactivate the Proteinase K. An aliquot was removed from each sample to be analyzed by qPCR. Two qPCR reactions were carried out in order to effectively determine how much rAAV vector was generated per cell. One qPCR reaction was set up using a set of primers designed to bind to a homologous sequence on the backbones of plasmids XX680, pXR2, and TReGFP. The second qPCR reaction was set up using a set of primers to bind and amplify a region within the eGFP gene. qPCR is conducted using Sybr green reagents and Light cycler 480 from Roche. Samples are denatured at 95 °C for 10 minutes followed by 45 cycles (90 °C for 10 seconds, 58 °C for 10 seconds, and 72 °C for 10 seconds) and melting curve (1 cycle 99 °C for 30 seconds, 65°C for 1 minute continuous).
Purification of rAAV from crude lysate. Each cell pellet was adjusted to a final volume of 10 ml using ddH2O. The pellets were vortexed briefly and sonicated for 4 minutes at 30% amplitude in 1 second on, 1 second off intervals. After sonication, 550 U of DNase was added and incubated at 37 °C for 45 minutes. Three hundred microliters of 5M NaCl were added to the pellets to free any virus bound/interacting to the cell debris and then briefly vortexed. The pellets were then centrifuged at 9,400 × g using the Sorvall RC5B centrifuge and HS-4 rotor to pellet the cell debris and the clarified lysate was transferred to a Type70Ti centrifuge tube (Beckman 361625).
Discontinuous iodixanol gradient. Iodixanol concentrations were made according to the protocol established by Zolothukin et al.28 substituting Tris pH 9.0 to a final concentration of 25 mmol/l instead of 1× phosphate-buffered saline. 17, 25, 40, and 60% at 6, 6, 7, and 5 ml, respectively under-layered the clarified lysate using a Watson Marlow peristaltic pump at 35 rpm to form the discontinuous gradient. The gradients were then centrifuged at 70K for 1 hour in a Type70Ti rotor (Beckman). After centrifugation, the tubes were removed and the 40% layer was extracted using a 10 ml syringe and 18-gauge needle.
Ion exchange chromatography. Using an AKTA FPLC (GE Healthcare, Little Chalfont, Buckinghamshire, UK), 1 ml Q Hyper-D columns (Pall) were equilibrated with 10 ml of 0.5M NaOH followed by 10 ml 2M NaCl in 25 mmol/l Tris pH 9.0 (Buffer B), and finally 20 ml of 25 mmol/l Tris pH9.0 (Buffer A). The extracted iodixanol fraction was adjusted to a specific volume with Buffer A, then loaded onto AKTA FPLC at 0.5 ml/minute and unbound sample was washed with 7 ml of Buffer A. The elution consisted of a 4 part step gradient using Buffer B at 12.5, 25, 50, and 100% intervals in 10 ml steps. When larger rAAV preparations were generated, the 5 ml HiTrap Q HP columns (GE Healthcare) were utilized for purification.
Dialysis of purified rAAV. Anion exchange chromatography peak fractions were dialyzed at 4 °C overnight into a buffer composed of 1× phosphate-buffered saline, 5% Sorbitol, and 350 mmol/l NaCl.
Titering rAAV using dot blot. Hundred microliters of DNase buffer (140 units DNase, 5 mmol/l CaCl2, 5 mmol/l MgCl2, 50 mmol/l Tris-HCl pH 8.0) were added to each well of a 96-well microtiter plate. One to three microliters or serial dilutions of virus were added to each well and incubated at 37 °C for 30 minutes. The samples were then supplemented with 15 µl Sarkosyl/EDTA solution (6.3% sarkosyl, 62.5 mmol/l EDTA pH 8.0) and placed at 70 °C for 20 minutes. Next, 15 µl of Proteinase K (10 mg/ml) was added and incubated at 50 °C for at least 2 hours. One hundred and twenty-five microliters of NaOH buffer (80 mmol/l NaOH, 4 mmol/l EDTA pH 8.0) were added to each well. A series of transgene specific standards were created through a dilution series. NaOH buffer was then added and incubated. Nylon membrane was incubated at RT in 0.4M Tris-HCl, pH 7.5 and then set up on the dot blot apparatus. After a 10–15-minute incubation in NaOH buffer, the samples and standards were loaded into the dot blot apparatus onto the GeneScreen PlusR hybridization transfer membrane (PerkinElmer). The sample was then applied to the membrane using a vacuum. The nylon membrane was soaked in 0.4M Tris-HCl, pH 7.5 and then crosslinked using UV strata linker 1800 (Stratagene, La Jolla, CA) at 60,000 ujoules. The membrane was prehybridized in CHURCH buffer (1% BSA, 7% sodium dodecyl sulfate, 1 mmol/l EDTA, 0.5M Na3PO4, pH 7.5). After prehybridization, the membrane was hybridized overnight with a 32P CTP-labeled transgene probe (Roche Random Prime DNA labeling kit). The following day, the membrane was washed with low stringency saline sodium citrate buffer (1× saline sodium citrate, 0.1% sodium dodecyl sulfate) and high stringency (0.1× saline sodium citrate, 0.1% sodium dodecyl sulfate). It was then exposed on a phosphorimager screen and analyzed for densitometry using a STORM840 scanner (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Analyzing rAAV vector purity using silver stain method. Samples from purified vector were loaded onto NuPage 10% Bis-Tris gels (Invitrogen) and run using 1× NuPage running buffer. Typically, 1 × 1010 genome containing particles were loaded per well. The gels were treated with SilverXpress Silver staining kit #LC6100 (Invitrogen).
Analysis of self-complementary genomes using alkaline gel electrophoresis and Southern blot. Briefly, purified self-complementary rAAV was added to 200 µl of DNase I buffer (140 units DNase, 5 mmol/l CaCl2, 5 mmol/l MgCl2, 50 mmol/l Tris-HCl pH 8.0) incubated at 37 °C for 60 minutes, followed by inactivation of the DNase by adding 30 µl of EDTA Sarkosyl/EDTA solution (6.3% sarkosyl, 62.5 mmol/l EDTA pH 8.0) and placed at 70 °C for 20 minutes. Twenty microliters of Proteinase K (10 mg/ml) were then added to the sample and incubated for a minimum of 2 hours at 50 °C. Phenol/chloroform was added in a 1:1 ratio, followed by ethanol precipitation of the viral vector DNA. The pelleted DNA was then resuspended in alkaline buffer (50 mmol/l NaOH, 1 mmol/l EDTA) for denaturation, loaded onto a 1% alkaline agarose gel, and run at 25V overnight at 4 °C. The gel was then equilibrated in alkaline transfer buffer (0.4 M NaOH, 1M NaCl) and a Southern blot was performed for 8 hours to overnight transfer of the vector DNA to a GeneScreen PlusR hybridization transfer membrane (PerkinElmer, Waltham, MA). The membrane was then neutralized using 0.5 M Tris pH 7.5 with 1M NaCl, and was hybridized overnight with a 32P CTP-labeled transgene probe. After washing the membrane as previously described, the membrane was exposed to a phosphorimager screen and analyzed using a STORM840 scanner.
Preparing samples for mass spectrometry. Twenty microliters of the elution fraction containing the identified contaminant were loaded onto a 10% Bis/Tris gel (Invitrogen) using a 1× 3-(N-morpholino)propanesulfonic acid buffer/sodium dodecyl sulfate running buffer (Invitrogen). The gel was then stained with Coomassie Brilliant Blue R250 (Bio-Rad) to visualize the contaminant around 25 kDa. The contaminant was then removed from the gel using a razor blade and stored in 250 μl dH2O. The sample was shipped to Applied Biomics for tryptic digestion and sequencing.
Transduction assays. HeLaRC-32 cells30 were plated at 2 × 105 cells/well of a 24-well plate and incubated at 37 °C overnight. The cells were observed for 90–100% confluence. Fifty milliliters of DMEM with 2% fetal bovine serum, 1% Pen/Strep were prewarmed, and Adenovirus (dl309) was added at an MOI of 10. The dl309 containing media was aliquoted in 900 µl fractions and used to dilute the rAAV in a series of 10-fold dilutions. The rAAV was then plated at 400 µl and allowed to incubate for 48 hours at 37 °C. For rAAV containing the eGFP transgene, the GFP-positive cells were counted using fluorescence microscopy. Transducing units (TU) are then calculated based upon the dilution and multiplying the average number of counts/field by the total number of fields per well at the specific magnification.
TEM of negatively stained rAAV particles. Electron microscopy allows a direct visualization of the viral particles. Purified dialyzed rAAV vectors were placed on a 400-mesh glow-discharged carbon grid by inversion of the grid on a 20 µl drop of virus. The grid was then washed two times by inversion on a 20 µl drop of ddH2O followed by inversion of the grid onto a 20 µl drop of 2% uranyl acetate for 30 seconds. The grids were blotted dry by gently touching the edges of the grids to whattman paper. Each vector was visualized using a Zeiss EM 910 electron microscope.
Acknowledgments
This project was funded by North Carolina Biotechnology Center 2007-CFG-8012. J.C.G. holds patents that have been licensed by University of North Carolina to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Asklepios Biopharmaceutical and has received payment for consultation. R.J.S. is the founder and a shareholder at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking.
References
- Aucoin, MG, Perrier, M and Kamen, AA (2008). Critical assessment of current adeno-associated viral vector production and quantification methods. Biotechnol Adv 26: 73–88. [DOI] [PubMed] [Google Scholar]
- Mueller, C and Flotte, TR (2008). Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther 15: 858–863. [DOI] [PubMed] [Google Scholar]
- Atchison, RW, Casto, BC and Hammon, WM (1965). Adenovirus-associated defective virus particles. Science 149: 754–756. [DOI] [PubMed] [Google Scholar]
- Buller, RM, Janik, JE, Sebring, ED and Rose, JA (1981). Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J Virol 40: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, X, Li, J and Samulski, RJ (1998). Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72: 2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm, D, Kern, A, Rittner, K and Kleinschmidt, JA (1998). Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 9: 2745–2760. [DOI] [PubMed] [Google Scholar]
- Matsushita, T, Elliger, S, Elliger, C, Podsakoff, G, Villarreal, L, Kurtzman, GJ et al. (1998). Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5: 938–945. [DOI] [PubMed] [Google Scholar]
- Booth, MJ, Mistry, A, Li, X, Thrasher, A and Coffin, RS (2004). Transfection-free and scalable recombinant AAV vector production using HSV/AAV hybrids. Gene Ther 11: 829–837. [DOI] [PubMed] [Google Scholar]
- Conway, JE, Rhys, CM, Zolotukhin, I, Zolotukhin, S, Muzyczka, N, Hayward, GS et al. (1999). High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther 6: 986–993. [DOI] [PubMed] [Google Scholar]
- Hwang, KK, Mandell, T, Kintner, H, Zolotukhin, S, Snyder, R and Byrne, BJ (2003). High titer recombinant adeno-associated virus production using replication deficient herpes simplex viruses type 1. Mol Ther 7: S14–S15. [Google Scholar]
- Kang, W, Wang, L, Harrell, H, Liu, J, Thomas, DL, Mayfield, TL et al. (2009). An efficient rHSV-based complementation system for the production of multiple rAAV vector serotypes. Gene Ther 16: 229–239. [DOI] [PubMed] [Google Scholar]
- Thomas, DL, Wang, L, Niamke, J, Liu, J, Kang, W, Scotti, MM et al. (2009). Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum Gene Ther 20: 861–870. [DOI] [PubMed] [Google Scholar]
- Aslanidi, G, Lamb, K and Zolotukhin, S (2009). An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc Natl Acad Sci USA 106: 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini, S, Negrete, A and Kotin, RM (2008). Toward exascale production of recombinant adeno-associated virus for gene transfer applications. Gene Ther 15: 823–830. [DOI] [PubMed] [Google Scholar]
- Kohlbrenner, E, Aslanidi, G, Nash, K, Shklyaev, S, Campbell-Thompson, M, Byrne, BJ et al. (2005). Successful production of pseudotyped rAAV vectors using a modified baculovirus expression system. Mol Ther 12: 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrete, A and Kotin, RM (2008). Large-scale production of recombinant adeno-associated viral vectors. Methods Mol Biol 433: 79–96. [DOI] [PubMed] [Google Scholar]
- Negrete, A, Yang, LC, Mendez, AF, Levy, JR and Kotin, RM (2007). Economized large-scale production of high yield of rAAV for gene therapy applications exploiting baculovirus expression system. J Gene Med 9: 938–948. [DOI] [PubMed] [Google Scholar]
- Urabe, M, Ding, C and Kotin, RM (2002). Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther 13: 1935–1943. [DOI] [PubMed] [Google Scholar]
- Urabe, M, Nakakura, T, Xin, KQ, Obara, Y, Mizukami, H, Kume, A et al. (2006). Scalable generation of high-titer recombinant adeno-associated virus type 5 in insect cells. J Virol 80: 1874–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher, Y, Pham, PL, St-Laurent, G, Jacob, D, Cass, B, Chahal, P et al. (2007). Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J Virol Methods 144: 32–40. [DOI] [PubMed] [Google Scholar]
- Hildinger, M, Baldi, L, Stettler, M and Wurm, FM (2007). High-titer, serum-free production of adeno-associated virus vectors by polyethyleneimine-mediated plasmid transfection in mammalian suspension cells. Biotechnol Lett 29: 1713–1721. [DOI] [PubMed] [Google Scholar]
- Park, JY, Lim, BP, Lee, K, Kim, YG and Jo, EC (2006). Scalable production of adeno-associated virus type 2 vectors via suspension transfection. Biotechnol Bioeng 94: 416–430. [DOI] [PubMed] [Google Scholar]
- Grieger, JC, Choi, VW and Samulski, RJ (2006). Production and characterization of adeno-associated viral vectors. Nat Protoc 1: 1412–1428. [DOI] [PubMed] [Google Scholar]
- Thomas, M, Lu, JJ, Ge, Q, Zhang, C, Chen, J and Klibanov, AM (2005). Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci USA 102: 5679–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff, AM, Ng, CY, Sleep, S, Gray, J, Azam, S, Zhao, Y et al. (2004). Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J Virol Methods 121: 209–215. [DOI] [PubMed] [Google Scholar]
- Kaludov, N, Handelman, B and Chiorini, JA (2002). Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Hum Gene Ther 13: 1235–1243. [DOI] [PubMed] [Google Scholar]
- Zolotukhin, S, Potter, M, Zolotukhin, I, Sakai, Y, Loiler, S, Fraites, TJ Jr et al. (2002). Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28: 158–167. [DOI] [PubMed] [Google Scholar]
- Zolotukhin, S, Byrne, BJ, Mason, E, Zolotukhin, I, Potter, M, Chesnut, K et al. (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973–985. [DOI] [PubMed] [Google Scholar]
- McCarty, DM, Monahan, PE and Samulski, RJ (2001). Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther 8: 1248–1254. [DOI] [PubMed] [Google Scholar]
- Chadeuf, G, Favre, D, Tessier, J, Provost, N, Nony, P, Kleinschmidt, J et al. (2000). Efficient recombinant adeno-associated virus production by a stable rep-cap HeLa cell line correlates with adenovirus-induced amplification of the integrated rep-cap genome. J Gene Med 2: 260–268. [DOI] [PubMed] [Google Scholar]
- Sonntag, F, Schmidt, K and Kleinschmidt, JA (2010). A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci USA 107: 10220–10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock, M, Alvira, M, Vandenberghe, LH, Samanta, A, Toelen, J, Debyser, Z et al. (2010). Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21: 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe, LH, Xiao, R, Lock, M, Lin, J, Korn, M and Wilson, JM (2010). Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum Gene Ther 21: 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, X (1992). Characterization of adeno-associated virus replication and integration. Ph.D. Thesis. Univ. Pittsburgh. [Google Scholar]
- Maguire, AM, Simonelli, F, Pierce, EA, Pugh, EN Jr, Mingozzi, F, Bennicelli, J et al. (2008). Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 358: 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz, JE, Rolling, F, Li, C, Conrath, H, Xiao, W, Xiao, X et al. (2002). Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol 76: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]