

Abstract
Polyurethane shape memory polymers (SMPs) with tunable thermomechanical properties and advanced processing capabilities have been synthesized, characterized, and implemented in the design of a microactuator medical device prototype. The ability to manipulate glass transition temperature (Tg) and crosslink density in low-molecular weight aliphatic thermoplastic polyurethane SMPs is demonstrated using a synthetic approach that employs UV catalyzed thiol-ene “click” reactions to achieve post-polymerization crosslinking. PUs containing varying C=C functionalization are synthesized, solution blended with polythiol crosslinking agents and photoinitiator and subjected to UV irradiation, and the effects of number of synthetic parameters on crosslink density are reported. Thermomechanical properties are highly tunable, including glass transitions tailorable between 30 and 105°C and rubbery moduli tailorable between 0.4 and 20 MPa. This new SMP system exhibits high toughness for many formulations, especially in the case of low crosslink density materials, for which toughness exceeds 90 MJ/m3 at select straining temperatures. To demonstrate the advanced processing capability and synthetic versatility of this new SMP system, a laser-actuated SMP microgripper device for minimally invasive delivery of endovascular devices is fabricated, shown to exhibit an average gripping force of 1.43 ± 0.37 N and successfully deployed in an in vitro experimental setup under simulated physiological conditions.
Keywords: shape memory polymer, thiol-ene “click” chemistry, structure-property relationships, polyurethane, biomedical device
Graphical abstract
A new platform shape memory polymer system for biomedical device applications is reported that exhibits a unique blend of tunable, high performance mechanical attributes in combination with advanced processing capabilities and good biocompatibility. A post-polymerization crosslinking synthetic approach is employed that combines polyurethane and thiol-ene synthetic processes, and a microactuator medical device prototype is fabricated to demonstrate the processing capability of this new SMP system.
1. Introduction
The materials science of human anatomy constitutes tremendous variations in tissue modulus and architecture. Materials-based avenues to solving biomedical engineering challenges depend both on a material’s ability to be processed into desired geometries and on the extent to which material properties can be tailored to meet the demands of various applications. Shape memory polymers (SMPs) are a class of stimuli-responsive materials that exhibit geometric transformations in response to subjection to external stimuli such as heating or light exposure, and a number of SMP-based biomedical implant devices are currently being proposed.[1] For SMP-based medical devices, the functional utility that arises from a clinician’s ability to trigger geometric transformations after device implantation in the body is both multi-dimensional in nature and complex in conceptualization. Various applications may demand SMPs with tailorable actuation temperature, recoverable strain, recovery stress, modulus at physiological conditions and toughness, in addition to good biocompatibility.[2] Devices also often have complex geometric design requirements, and a material’s ability to be processed using certain fabrication techniques often influences the material selection process in device design.[3] A high performance SMP system with tunable thermomechanical properties, high toughness and good biocompatibility that can be readily synthesized and processed in atmospheric conditions into desired geometries for device fabrication is of importance for a variety of biomedical applications.[4–6] The focus of this work is to introduce an amorphous, thermally actuated shape memory polymer system that exhibits robust, highly tunable material properties and advanced processing capabilities and to demonstrate this SMP system’s viability as a platform system for medical device design through the fabrication of a laser actuated SMP-based microgripper designed to facilitate microcatheter delivery of implantable endovascular devices.
The tunability of material properties, toughness and processability of many SMP systems has been shown to be highly dependent on the nature and extent of crosslinking in the SMPs.[7] Chemical constituents in polymeric backbones or side chains can exhibit physical crosslinking interactions that influence a number of material properties, including glass transition and toughness. Additionally, the environmental conditions required to synthesize polymers comprised of certain chemical functionalities may dictate which fabrication techniques can be employed during polymer processing. From a macromolecular architecture standpoint, covalent crosslinking affords certain advantages in mechanical behavior while simultaneously limiting many aspects of material processing capability. For a thermally actuated, one-way SMP, the polymer constituents that undergo thermal transitions upon heating or cooling across the SMP switching temperature Ttrans are referred to as “switching segments,” and crosslinks, whether covalent, physical, or other are referred to as “netpoints.” Netpoints prevent switching segment chains from permanently sliding past one another during straining to a secondary geometry by effectively acting as anchors that enable shape recovery to occur.[8] Covalently crosslinked SMP systems often exhibit advantages in mechanical behavior over those of physically crosslinked SMP systems, including better cyclic shape memory and greater percent recoverable strains. In contrast, thermoplastic SMPs often possess significant processing advantages over thermosets, which do not flow at elevated temperatures and pressures and do not dissolve in solvents.[9] Thermoplastic processing techniques such as 3D printing, extrusion, injection molding and solution casting are especially useful when high-throughput processing and/or complex prototype fabrication are desired.[10] For medical device applications in particular, a thermoplastic SMP that is capable of being processed into a desired geometry in atmospheric conditions and subsequently crosslinked in a secondary step to tailorable crosslink densities may offer significant advantages over current SMP materials.[11]
Reported herein is an amorphous, aliphatic polyurethane (PU) SMP system designed to exhibit tunable crosslink density and glass transition (Tg), high toughness and good biocompatibility, as well as advanced processing capabilities in atmospheric conditions. One advantage of PUs is the high toughness that results from inter-chain hydrogen bonding between carbamate linkages,[12] and many aliphatic PUs have also been shown to exhibit good biocompatibility.[13] One major disadvantage of PUs is the fact that isocyanate-based PU syntheses must be carried out in moisture-free environments such as glove boxes to prevent isocyanate side reactions, and many commercially available PUs are thermoplastics that have been pre-synthesized by manufacturers to allow for processing into desired geometries in atmospheric conditions. If applications should require the high toughness afforded by urethane chemistry and the mechanical robustness provided by covalent crosslinking while also demanding processability in atmospheric conditions, one strategy that allows for the incorporation of urethane linkages into covalently crosslinked networks in atmospheric synthetic environments is the synthesis of monomers, oligomers, or thermoplastic precursors containing internal carbamate linkages that are functionalized with groups that can be subsequently cured in a secondary step using alternative polymerization methods.[14, 15] Thiol-ene “click” chemistry is a powerful synthetic tool that can proceed to high conversion in atmospheric conditions,[16] and Nair and co-workers from Bowman, Shandas and co-workers have reported an SMP system that is prepared by functionalizing isophorone diisocyanate (IPDI) to afford polyalkene and polythiol monomers with internal (thio)urethane linkages, which are curable using thiol-ene “click” reactions upon UV irradiation.[17] Beigi, et al. have recently investigated the use of alkene-functionalized IPDI as a co-monomer in thiol-ene-methacrylate systems as a resin matrix for dental applications,[18] and a number of studies have also reported the synthesis of thiol-ene crosslinked waterborne PU coatings from alkene end-capped urethane-ester-urethane trimers.[19, 20] Because of the high number of flexible thioether linkages that result from the bulk curing of thiol-ene monomers, even when such monomers contain internal carbamate linkages, a number of poly(thioether-co-urethane) hybrid networks reported in the literature exhibit glass transitions below or near 25°C, and these elastomeric materials may not be suitable for use in certain engineering polymer applications. Although Tg can be increased by increasing crosslink density in thiol-ene polymer systems, this increase in crosslinking is often accompanied by a tradeoff in decreased toughness, and achieving independent control of crosslink density and Tg in thiol-ene systems is often a synthetic challenge.[21] For amorphous, thermally actuated shape memory polymer systems in particular, achieving independent control of Tg and crosslink density (i.e., actuation temperature and recovery stress) is a key material design objective that influences the scope of the SMP system’s potential application range.
We report a synthetic strategy in which thermoplastic PUs are prepared from aliphatic diisocyanates and varying ratios of C=C and non-C=C functionalized diols, as shown in Scheme 1. After thermoplastic synthesis, crosslinking is achieved by solution blending of thermoplastics with polythiol crosslinking agents and photoinitiator and subsequent UV irradiation. This post-polymerization crosslinking approach allows for polyurethane-based SMP devices to be fabricated in atmospheric conditions, and the toughness and thermomechanical properties of these SMPs are primarily influenced by the linkages in the thermoplastic PUs, which statistically significantly outnumber the thioether linkages in the crosslinked materials. Furthermore, the incorporation of C=C linkages into PU side chains by PU step growth polymerization is predicted to result in a fairly uniform distribution of C=C functionalities, which, upon crosslinking with polythiols, is predicted to afford poly(thioether-co-urethane) networks with high network homogeneity and narrow glass transition breadths, which enable precise and rapid shape actuation for SMP-based devices.[22] While previous studies report the syntheses of urethane/thiol-ene hybrid network SMPs from monomeric functionalized precursors that often exhibit mechanical behavior that is more consistent with that of a thiol-ene network than that of a polyurethane, this study employs a synthetic strategy that affords materials that exhibit mechanical behavior consistent with that of polyurethanes. Consequently, the synthetic challenges that have occurred because of the low Tg and high crosslink densities associated with thiol-ene networks can be overcome, and a platform SMP system with widely tunable material properties and advanced processing capability can be developed.
Scheme 1.
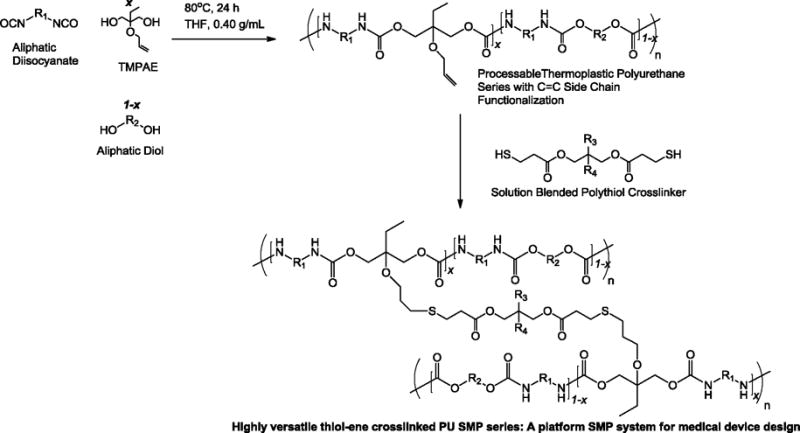
Synthetic strategy utilized in this study. Thermoplastic polyurethanes containing pendant C=C linkages are synthesized, and post-polymerization crosslinking is achieved via UV catalyzed thiol-ene free radical addition upon solution blending of C=C containing thermoplastic polyurethanes with polythiol crosslinking agents, solvent evaporation and subsequent UV irradiation.
To demonstrate the processing capability of this new SMP system, a prototype microactuator device is also synthesized and characterized in this work. One advantage to the solution blending aspect of the synthetic process used to fabricate this device is the solvent’s ability to uniformly disperse non-reactive additives throughout the thermoplastic PU/polythiol solutions. The device fabricated in this study constitutes a new design for a light-actuated SMP microgripper, which is designed for the purpose of delivering endovascular devices via microcatheter. Utilizing the properties of this new SMP system, we seek to improve upon a microactuator device previously reported by our group in 2002.[23] Our new device design, illustrated in Figure 1, offers significantly improved ease of fabrication over our previously reported design. By fabricating this microactuator device, we seek to demonstrate (a) the ease with which non-reactive additives can be blended with this new SMP system; (b) the use of post-polymerization crosslinking to enable the fabrication of a micro-scale medical device; (c) the ability of this new SMP system to be subjected to device fabrication in atmospheric conditions.
Figure 1.
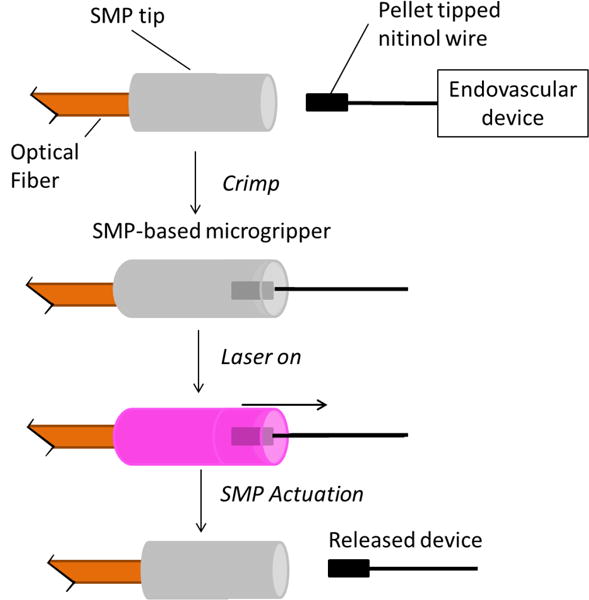
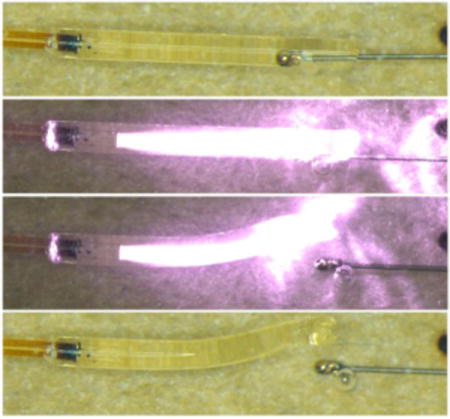
Schematic diagram for laser actuated SMP microgripper. Solutions of thermoplastic PU and polythiol doped with Epolin 4121 were first solution cast over a cleaved optical fiber to form the SMP gripper, after which PU/polythiol solutions were UV cured. Pellet tipped nitinol was then axially crimped into the gripper, and actuation was achieved using laser irradiation, which results in release of embolic device.
2. Results
This study seeks to present a new shape memory polyurethane system as a platform material system for use in medical device applications by (1) reporting materials characterization data that show excellent thermomechanical, shape memory and tensile behavior, as well as indications of biocompatibility and (2) demonstrating the processing advantages of this new SMP system through the fabrication in atmospheric conditions of a laser-activated SMP microgripper device to facilitate the microcatheter delivery of implantable endovascular devices. The device fabricated in this study, although demonstrated to be robust and well-designed, is not reported as a proposed replacement or argued improvement over other devices currently utilized in industry or research studies, but rather as a “proof-of-concept” device that demonstrates the processing capability of the new SMP system reported in this work.
2.1. Achieving Tailorable Crosslink Density
The SMP system reported herein is synthesized through a post-polymerization crosslinking process in which thermoplastic polyurethanes containing pendant C=C functionalities are first synthesized from (alkene)diol and diisocyanate monomers, as shown in Scheme 1. After thermoplastic synthesis, crosslinking is achieved by solution blending of thermoplastic PUs with polythiol crosslinking agents in atmospheric conditions and is shown to be tailorable by varying a number of synthetic parameters, many of which can be employed at the post-polymerization crosslinking step. Control over rubbery modulus was achieved over the range of 0.5 to 10.5 MPa by reacting thermoplastic urethanes comprised of varying TMPAE functionalization with 1:1 equivalents of the trithiol TMPTMP, as shown in the storage modulus data provided in Figure 2(a). Control of rubbery modulus was also demonstrated over the range of 3.0 to 10.5 MPa for a single thermoplastic formulation comprised of 0.9 diol TMPAE fraction by crosslinking this thermoplastic with thiol equivalents ranging from 4.0 C=C :1.0 SH to 1.0 C=C:1.0 SH, as shown in Figure 2(b). Figure 2(c) shows a near-negligible effect of photoinitiator composition on rubbery modulus as DMPA composition is increased over three orders of magnitude, from 0.1 wt% to 10.0 wt%, and Figure 2(d) demonstrates that both glass transition and crosslink density increased slightly over the course of a 24 h post-cure. Figure 2(e) shows that rubbery modulus decreased from 10.5 to 3.0 MPa as THF solvent composition increased from 4% to 300% during curing. The effects of increasing polythiol crosslinker functionality for a 0.1 TMPAE diol component sample are shown in Figure 2(f). As thiol functionality was increased from n=2 to 6, rubbery modulus increased roughly an order of magnitude, from 0.4 to 4.1 MPa.
Figure 2.
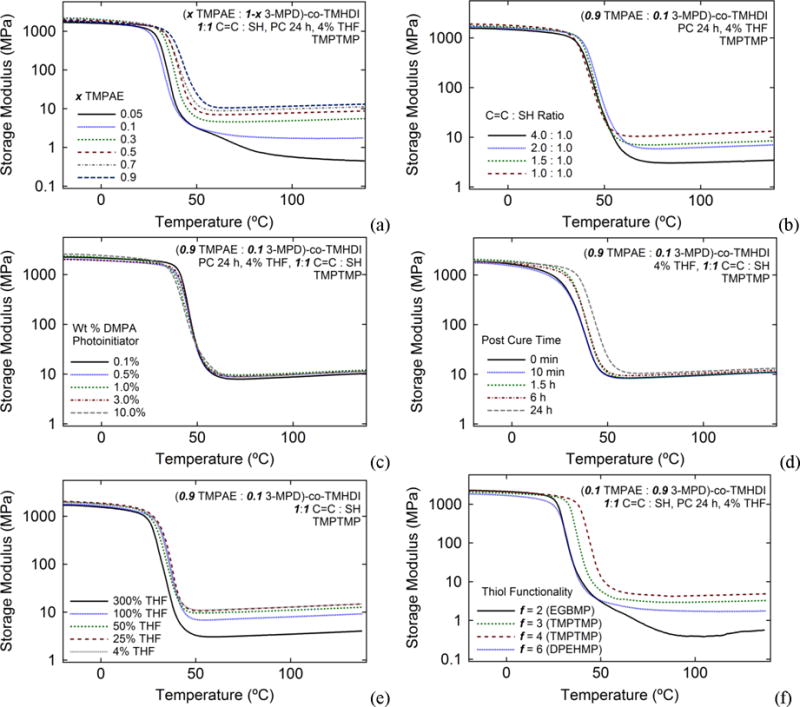
The effects of (a) varying C=C composition, (b) varying C=C : SH ratio, (c) varying DMPA photoinitiator, (d) varying post-cure time at 120°C at 1 torr, (e) varying wt % solvent present during UV curing, and (f) varying thiol crosslinker functionality on crosslink density of PU SMPs prepared from (x TMPAE: 1-x 3-MPD)-co-TMHDI thermoplasticpolyurethanes and polyfunctional thiol crosslinking agents.
2.2. Achieving Tailorable Glass Transition
To achieve synthetic control over glass transition, two approaches are reported. First, control of Tg was attempted by varying diol co-monomer composition for thermoplastics comprised of low C=C diol monomer composition (0.10 TMPAE, 0.90 diol), as shown in Table 1. Instead of selecting a “high-Tg and “Low-Tg” diol and moving glass transition by blending such diols in varying ratios, the 0.10 TMPAE: 0.90 diol varying Tg series was formulated to provide increased network homogeneity and more narrow glass transition breadths.Figures 3(a) and 3(b) show a Tg range of approximately 38 to 70°C and a roughly constant rubbery modulus of approximately 2.1 MPa for all samples, which were prepared from thermoplastics with 0.10 TMPAE and varying 0.90 diol co-monomer constituents. A minimum Tg of 38°C was observed for the diethylene glycol (DEG) co-monomer, and a maximum Tg of 70°C Tg was observed for the cyclohexanedimethanol (CHDM) co-monomer in the 0.10 TMPAE series. Tg breadths were narrow and ranged from 9°C to 12°C (tangent delta full width half maximum, FWHM). This observed sharpness in glass transition breadths indicates that the synthetic strategy of varying Tg by varying the entire saturated diol co-monomer compositions is an effective route to achieving high network homogeneity. To provide a second manner of tailoring glass transition, a series of thermoplastics was prepared from 0.9 TMPAE and varying ratios of HDI and DCHMDI diisocyanate co-monomers. As the DMA data in Figures 3(c) and 3(d) show, as DCHMDI composition increased from 0.0 to 1.0, Tg increased from 56 to 105°C, and rubbery modulus for this SMP series remained roughly constant, in the range of 16.4 to 17.1 MPa. Tg breadth ranged from 16°C to 23°C and was greatest for the 0.5 HDI: 0.5 DCHMDI composition, which was comprised of the most heterogeneous formulation.
Table 1.
Sample compositions and molecular weights as determined by GPC analysis and structures for monomers and polythiol crosslinkers used to prepare thiol-ene crosslinked polyurethane shape memory polymers. Except for the ‘Mn’ and ‘Mw’ columns, all data are unitless.
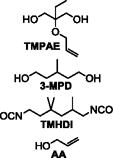
| Varying TMPAE Series (Varying Er) | Equiv. TMHDI | Equiv. TMPAE | Equiv. 3-MPD | Equiv. 2x AA | Mn (kDa) | Mw (kDa) | PDI | |
|---|---|---|---|---|---|---|---|---|
|
TMPAE-0.05 | 1.020 | 0.05 | 0.950 | 0.010 | 15.0 | 26.9 | 1.79 |
| TMPAE-0.1 | 1.020 | 0.100 | 0.900 | 0.010 | 17.2 | 30.7 | 1.79 | |
| TMPAE-0.3 | 1.020 | 0.300 | 0.700 | 0.010 | 1.9 | 5.2 | 2.78 | |
| TMPAE-0.5 | 1.020 | 0.500 | 0.500 | 0.010 | 1.3 | 3.7 | 2.81 | |
| TMPAE-0.7 | 1.020 | 0.700 | 0.300 | 0.010 | 1.1 | 3.5 | 3.01 | |
| TMPAE-0.9 | 1.020 | 0.900 | 0.100 | 0.010 | 2.8 | 9.5 | 2.69 | |
|
| ||||||||
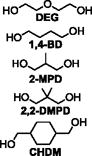
| Varying Diol Series (Varying Tg) | Equiv. TMHDI | Equiv. TMPAE | Equiv Diol | Equiv. 2x AA | Mn | Mw | PDI | |
|
DEG-0.9 | 1.020 | 0.100 | 0.900 | 0.010 | 16.3 | 38.7 | 2.37 |
| 1,4-BD-0.9 | 1.020 | 0.100 | 0.900 | 0.010 | 11.5 | 28.6 | 2.49 | |
| 2-MPD-0.9 | 1.020 | 0.100 | 0.900 | 0.010 | 18.7 | 57.3 | 3.06 | |
| 2,2-DMPD-0.9 | 1.020 | 0.100 | 0.900 | 0.010 | 17.0 | 41.4 | 2.44 | |
| CHDM-0.9 | 1.020 | 0.100 | 0.900 | 0.010 | 11.9 | 31.2 | 2.62 | |
|
| ||||||||
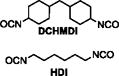
| Varying DCHMDI Series (Varying Tg) | Equiv. DCHMDI | Equiv. HDI | Equiv TMPAE | Equiv. 2x AA | Mn | Mw | PDI | |
|
DCHMDI-0.0 | 0.000 | 1.020 | 1.000 | 0.010 | 21.2 | 65.4 | 3.08 |
| DCHMDI-0.1 | 0.100 | 0.920 | 1.000 | 0.010 | 10.0 | 20.6 | 2.06 | |
| DCHMDI-0.5 | 0.500 | 0.520 | 1.000 | 0.010 | 5.5 | 11.0 | 2.00 | |
| DCHMDI-1.0 | 1.000 | 0.020 | 1.000 | 0.010 | 5.4 | 9.7 | 1.80 | |
Polythiol Crosslinking Agents: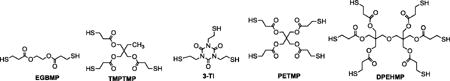
| ||||||||
Figure 3.
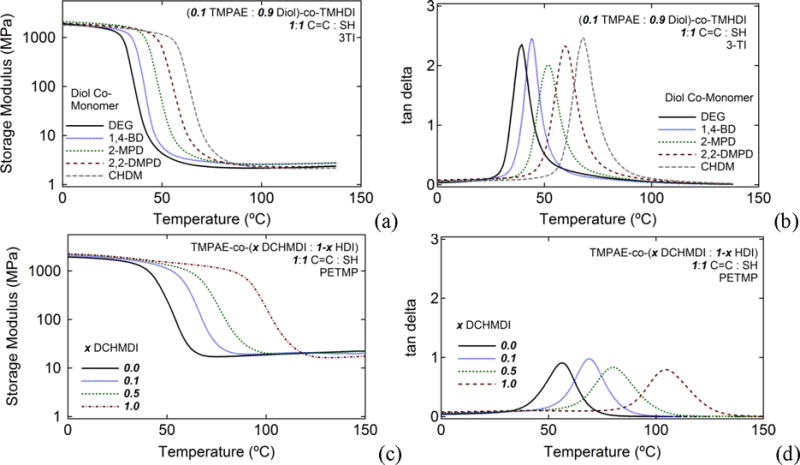
The effects of (a,b) varying diol co-monomer and (c,d) varying diisocyanate co-monomer ratio on glass transition temperature: (a) DMA plots of storage modulus versus temperature for varying diol formulations; (b) DMA plots of tangent delta versus temperature for varying diol formulations; (c) DMA plots of storage modulus versus temperature for varying diisocyanate ratio formulations; (d) DMA plots of tangent delta versus temperature for varying diisocyanate ratio formulations
2.3. Shape Memory Characterization, Tensile Testing, and Biocompatibility Results
One demonstrated advantage of covalently crosslinked SMP systems is good shape memory behavior, which includes high percent recoverable strains during cyclic testing and recovery stresses that are tunable by varying covalent crosslink density. In Figure 4(a), free strain recovery data showing recoverable strain versus temperature over five straining cycles is shown for the (0.3 TMPAE: 0.7 3-MPD)-co-TMHDI SMP crosslinked with TMPTMP, which was subjected to 25% prestrain in each cycle. During Cycle 1, this SMP exhibits a recoverable strain of 94.5%, and during Cycles 2 to 5, its recoverable strain approaches 100%. The shape fixity ratio for all cycles in free strain recovery experiences was 100%. In Figure 4(b), constrained recovery data showing recoverable stress versus temperature for (× TMPAE: 1-x 3-MPD)-co-TMHDI SMPs crosslinked with TMPTMP are shown. These constrained recovery data are for four samples subjected to 25% prestrain with TMPAE compositions of 0.1, 0.3, 0.5, and 0.7, respective rubbery modulus values of 1.7, 4.5, 6.9, and 8.9 MPa and respective max recovery stress values of 0.4, 0.9, 1.2, and 1.4 MPa. As predicted, increasing rubbery modulus/implied increasing crosslink density results in increasing recovery stress, and tailoring crosslink density constitutes an experimentally demonstrated approach to tuning recovery stress.
Figure 4.
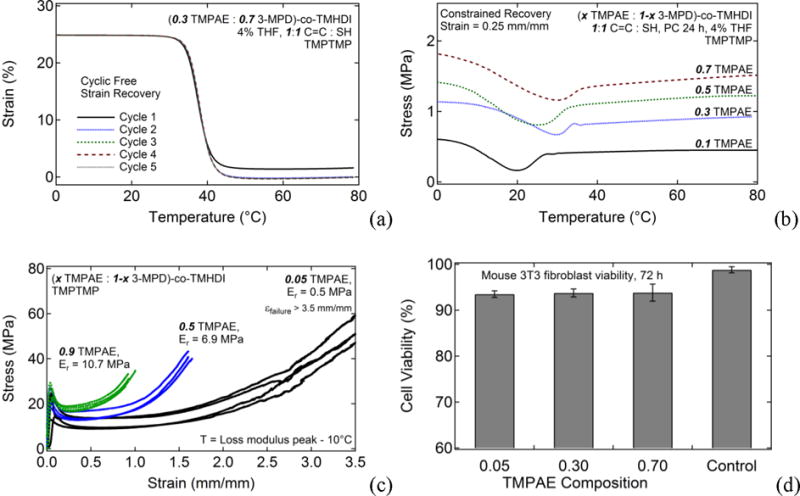
Materials characterization data demonstrating the material advantages of this polymer system. (a) 5-cycle free strain recovery shape memory characterization results for (0.3 TMPAE: 0.7 3-MPD)-co-TMHDI SMP; (b) constrained recovery for samples with increasing TMPAE composition; (c) Strain-to-Failure results for thiol-ene crosslinked PU SMP samples made from TMHDI and varying ratios of TMPAE and 3-MPD (i.e., varying C=C composition); (d) Cytotoxicity results showing mouse 3T3 fibroblast viabilities greater than 93% were observed for TMPAE-0.05, TMPAE-0.3, and TMPAE-0.7 thiol-ene crosslinked polyurethanes for 72 h direct contact studies.
Strain-to-failure data were obtained by conducting experiments at T = Tloss modulus -10°C for (x TMPAE: 1-x 3-MPD)-co-TMHDI samples crosslinked with TMPTMP and are provided in Figure 4(c). TMPAE compositions of 0.05, 0.5, and 0.9, which exhibited rubbery moduli of 0.5, 6.9, and 10.5 MPa, respectively, were selected for strain to failure testing at equivalent temperatures relative to each material’s loss modulus peak temperature as determined by DMA. Average toughness was calculated to be > 90.0 MJ/m3 for the 0.05 TMPAE sample, which did not break in the extension range of the tensile tester, 36.3 ± 3.9 MJ/m3 for the 0.5 TMPAE sample and 20.9 ± 2.1 MJ/m3 for the 0.9 TMPAE sample. As predicted, toughness was shown to decrease with increasing crosslink density.
Cell viability data for mouse 3T3 fibroblasts after direct contact exposure for 72 h to (x TMPAE: 1-x 3-MPD)-co-TMHDI samples crosslinked with TMPTMP are provided in Figure 4(d). Samples with TMPAE compositions of 0.05, 0.3, and 0.7 exhibited average 72 h percent cell viabilities of 93.5 ± 0.7%, 93.8 ± 0.8%, and 93.8 ± 1.9%, while the control exhibited a cell viability of 98.8 ± 0.7%.
2.4. SMP Microgripper Device Characterization Results
In order to approximate the maximum forces to which the crimped microgripper/ball-tipped assemblies could be subjected during microcatheter delivery of endovascular devices, tensile testing experiments were run to measure device gripping forces in an MTS tensile testing system as pictured in Figure 5(c). Using the immersion chamber of the MTS system, strain-to-failure experiments were carried out on seven devices in water at 37°C. Figure 5(d) shows a table of gripping force at failure values for each of seven devices tested, and the average gripping force was 1.43 ± 0.37 N.
Figure 5.

(a) Device fabrication showing liquid solution of thermoplastic PU, photoinitiator, and polythiol after injection into glass mold before UV vuring; (b) crimping process for ball-tipped simulated embolic device and UV cured microgripper device; (c) experimental setup for tensile testing experiments for crimped device assemblies; (d) maximum stresses measured in tensile testing experiments for seven crimped devices.
Figures 6(a) and 6(b) show the experimental setup for the PDMS vascular model that was used for in vitro microcatheter delivery experiments. Figure 6(c) shows images of a successful in vitro device deployment triggered by laser actuation through a fiber optic cable run through a microcatheter. This experiment was repeated five times and resulted in successful release each time. Figure 6(d) shows higher-resolution images of the laser-induced actuation of the SMP microgripper in 25°C aqueous conditions without flow, which were taken using an optical microscope.
Figure 6.

Illustration of setup for in vitro microcatheter delivered microgripper deliver experimental setup, (a) zoomed out and (b) zoomed in; (c) photographs of in vitro actuation of microcathether delivered device deployed in the experimental setup shown in (a) and (b); (d) high-resolution microscope images of microgripper device at 25°C in water
3. Discussion
The work reported in this study is first a materials engineering endeavor, second a biomedical materials development and third a medical device development project. From a materials engineering standpoint, the objectives of developing a new SMP system exhibiting high toughness, tunable glass transition and crosslink density and good shape memory behavior that also possesses post-polymerization crosslinking capability in atmospheric conditions were successful. Thermomechanical characterization experiments demonstrated actuation temperatures (glass transitions) that were tunable over the range of 38 to 70°C and 56 to 105°C by varying diol and diisocyanate monomer constitutents, respectively. For the varying diol series, diol monomer compositions were selected that were predicted to afford a variation in polymer backbone rigidity in order to create a polymer system with tailorable glass transition. Flexible C-O and C-C linkages generally afforded lower glass transitions, and the addition of methyl groups to polymer backbone methylene and methyne linkages were observed to increase glass transition. The incorporation of sterically constrained cyclohexane linkages into polyurethane backbones was also observed to increase glass transition. For the varying diisocyanate series, the sterically constrained isophorone diisocyanate monomer increased glass transition when it replaced hexamethylene diisocyanate as a comonomer.
For the varying diol co-monomer series, rubbery modulus was shown to be tunable over the order-of-magnitude range of approximately 0.4 to 10.4 MPa by varying a number of synthetic factors, and the ability to manipulate crosslink density in this newly reported SMP system was shown to be a successful approach to tailoring recovery stress using constrained recovery shape memory characterization experiments. The DMA data in Figures 2(b), 2(d) and 2(f) demonstrate that, for a single thermoplastic formulation, rubbery modulus can be tailored over a roughly order-of-magnitude range by varying the synthetic conditions of C=C to SH ratio, solvent composition at curing time and polythiol additive functionality, respectively. The ability to achieve a variety of material properties upon crosslinking of a single thermoplastic formulation is a synthetic advantage of this versatile polyurethane SMP system, as it provides synthetic flexibility at the materials engineering level to the process of medical device fabrication. It is important to note, however, that in the case of the materials whose thermomechanical data are shown in Figure 2(e), the ability to control crosslink density by varying solvent composition at curing time most likely occurs because solvent dilution of reactants decreases conversion of reactive species and effectively causes unreacted thiols to remain in the final crosslinked materials. If solvent dilution is to be seriously considered as a means of tailoring mechanical properties, stress-strain and shape memory behavior should be evaluated for these materials, as well as biocompatibility, which may be influenced by the presence of increased residual thiol functionality. Also, the evaporation of THF after solution blending of thermoplastic PUs with polythiol crosslinking agents could potentially alter the miscibility of polythiols with thermoplastic polyurethanes. The narrow glass transitions observed for the thiol-ene crosslinked materials prepared in this work that contained 4% THF or less during polymerization serve as evidence that polythiol/PU miscibility was sufficient enough to enable the formation of homogeneous covalently crosslinked networks upon polymerization, and the high gel fractions of these crosslinked materials, which approached 1.0 for most formulations, indicate that minimal polythiol crosslinking agents remained unincorporated in the final crosslinked networks, therein providing further evidence of sufficient polythiol/PU miscibility after THF evaporation to afford robust and homogeneous PU networks exhibiting good shape memory behavior.
The decrease in stress with increasing temperature prior to the glass transition onset of the materials for which constrained recovery data are reported in Figure 4(b) is hypothesized to be a combined artifact of the experimental technique used and the thermal expansion of the polymers being subjected to constrained recovery characterization.
From a biomedical materials development standpoint, the cell viability data shown in Figure 4(d) provide promising initial evidence of the biocompatibility of multiple formulations in the reported SMP system. The three formulations subjected to mouse 3T3 fibroblast cell viability studies are comprised of polymer formulations whose compositions vary over the range of 0.05, 0.30 and 0.70 C=C units per polymer repeat unit that are crosslinked using 1:1 SH to C=C ratios. As the DMA data in Figure 2(a) show, these three formulations constitute a rubbery modulus variation of 0.4 to 8.9 MPa, and each chemical formulation utilized to achieve this variation in crosslink density is shown to exhibit a cell viability greater than 90% in direct contact studies with mouse 3T3 fibroblasts. These initial findings suggest that the three SMP formulations are non-cytotoxic to this line of mammalian cells over the course of 72 hour exposure and consequently provide preliminary evidence that this class of SMPs could be a promising candidate material system for use in the fabrication of medical devices designed for implantation in the body. However, further cytotoxicity, sensitization, and hemotoxicity studies on human cell lines are required to support these initial findings.
Concerning the significance of the laser actuated SMP-based microgripper device reported herein within the broader scope of laser-actuated SMP devices reported in the literature, a number of laser activated SMP-based medical device prototypes have been reported, including SMP stents, mechanical thrombectomy devices, diffusers and embolic foams. The microgripper device prototype reported in this study is designed to facilitate minimally invasive delivery and release of embolic coils for treating aneurysms and is modeled after the SMP microgripper reported by Maitland and co-workers in 2002.23 One motivation for developing a new microgripper is to provide improved and more efficient routes to delivery of aneurysm occlusion devices. Guglielmi detachable coils (GDCs), the clinical standard for embolic treatment of aneurysms, utilize electrolytically cleavable polymer bridges to detach metal coils from delivery cables. The detachment process for a single GDC coil may take 1 to 5 minutes or longer, and up to 20 coils may be necessary to occlude an aneurysm. Prolonged treatment times may increase a number of risk factors for patients, including increased exposure to ionizing radiation. Both the Maitland 2002 SMP microgripper and the SMP microgripper reported in this study exhibit release times of 10 seconds or less and consequently offer advantages in delivery times in comparison with those of GDC coils.
In comparison with the Maitland 2002 microgripper, the device in this study possesses a number of advantages, including ease of fabrication. The 2002 device required etching the cladding off an optical fiber using hydrofluoric acid, dip-coating the etched fiber in an epoxy resin, bead blasting the epoxy to make a diffuse surface and attaching an SMP tube over the epoxy. These steps were required in order to increase the coupling efficiency of the light into the SMP tube. In contrast, the microgripper in this study requires fewer and more facile steps to fabricate. By casting the polymer directly over the SMP tip, laser radiation is coupled directly into the gripper, and additional techniques are not required to guide light into the SMP. In a similar manner to that of the 2002 device, because the index of refraction of the polymer is higher than that of water, light is guided through the SMP to the distal tip where the nitinol ball is crimped. In order to enable laser-induced heating and actuation of the SMPs in each microgripper under flow conditions, the SMPs were doped with a laser absorbing dye. For the polymer system reported herein, incorporation of light absorbing dye is achieved by solution blending with thermoplastic precursors and crosslinking additives prior to crosslinking. Solution blending in the manner reported herein affords homogeneous distribution of functional additives such as light absorbing dye particles, and homogeneous distribution enables a homogenous heating throughout the volume of the device upon laser irradiation. Other functional additive blending techniques such as solvent swelling into a network matrix or thermoplastic melt blending afford less uniform additive dispersion. While both the Maitland 2002 and present study microgripper devices utilize solution blending to disperse light absorbing dye particles throughout thermoplastic polymer chains, the device in this study is subjected to an additional post-polymerization crosslinking step after dye blending. Covalent crosslinking fixes dispersed additive positions in polymer matrices and reduces dispersed particle rearrangements associated with thermoplastic polymer mobility. From a material performance standpoint, covalent crosslinking often enables more high integrity shape memory behavior than physical crosslinking because covalent crosslinking sites constitute more permanent netpoints than physical crosslinks. The device in this study is predicted to exhibit better shape fixity and shape recovery over a prolonged storage time in a programmed geometry than the 2002 device. For SMP materials used to fabricate “off-the-shelf” medical devices that may be stored for months or years before use, maintaining integrity of shape memory behavior over an extended time frame is a key material design criterion.
By determining the minimum optical power required for actuation, and by comparison to similar devices reported in literature,23 the internal temperature of the microgripper at the distal tip when irradiated with 3.1W of optical power was approximately 73°C. While the high laser powers required for actuation may appear to pose thermal hazards to tissues, the convective cooling that occurs in the body is very efficient, minimizing this risk. We have previously described a method to estimate the local temperature rise due to similar microgrippers,23 and using this previously described procedure, the estimated temperature rise due to this study’s microgripper in vivo would be 0.30°C.
4. Conclusions
This study constitutes notable developments in the fields of shape memory polymers and thiol-ene polymers research that together portray a new polymer system as a promising candidate material system for biomedical engineering and other applications. In comparison with other shape memory polymer systems that have been reported, the thiol-ene crosslinked polyurethane SMP system reported in this study offers a unique blend of material attributes including high toughness, tunable and narrow glass transitions, tunable shape memory behavior and suitability for processing in atmospheric conditions. In comparison with a number of previously reported thiol-ene polymers, this polyurethane/poly(thioether) hybrid exhibits higher glass transitions and lower crosslink densities, which are achieved using a post-polymerization crosslinking synthetic approach that also enables thermoplastic urethane/polythiol blends to be processed into desired geometries in atmospheric conditions. Good biocompatibility was also observed in preliminary cell viability studies, and together these mechanical attributes, material processing capabilities and biocompatibility results support this polymer system’s presentation as a platform SMP system for medical device applications. The feasibility of implementing this polymer system in a medical device was demonstrated via the fabrication of an embolic coil microgripper release system used in the treatment of cerebral aneurysms, and the facile device fabrication process for this microgripper device indicates that this material system may be suitable for fabricating other medical devices.
5. Experimental Section
5.1. Materials
To provide a monomer-based synthetic approach to tailoring crosslink density in the thiol-ene crosslinked PU SMP system in this study, the alkene diol trimethylolpropane allyl ether (TMPAE, > 98%) and the end-capping agent allyl alcohol (AA, > 99%) were purchased from Sigma Aldrich Corporation. To tailor glass transition by varying aliphatic diol co-monomer, diethylene glycol (DEG, > 99%), 3-methyl-pentanediol (3-MPD, > 99%), 1,4-butanediol (1,4-BD, > 99%), 2-methylpropanediol (2-MPD, > 99%), 2,2′-dimethylpropanediol (2,2-DMPD, > 99%), and 1,4-cyclohexanedimethanol, mixture of cis and trans (CHDM, > 99%) were purchased from Sigma Aldrich Corporation. To tailor glass transition by varying diisocyanate monomer composition, hexamethylene diisocyanate (HDI, > 98%), trimethylhexamethylene diisocyanate (TMHDI, > 97%), and dicyclohexylmethane 4,4′-diisocyanate (DCHMDI, > 90%) were purchased from TCI America. To absorb water during polyurethane polymerizations, 4 Å molecular sieves were purchased from Sigma Aldrich and added to polymerization mixtures. The catalyst zirconium (IV) acetylacetonate (Zr Cat, > 99%), which has been shown to favor urethane formation over urea formation when water is present during urethane polymerizations, was purchased from Alfa Aesar and used in thermoplastic polyurethane synthesis. Anhydrous tetrahydrofuran (THF, > 99.98%) was purchased from EMD Chemicals and used as a polymerization and blending solvent. The polythiol crosslinking agents ethylene glycol bis(3-mercaptopropionate) (EGBMP, > 97%) and dipentaerythritol hexakis(3-mercaptopropionate) (DPHMP, > 97%) were purchased from Wako Chemicals, and trimethylolpropane tris(3-mercaptopropionate) (TMPTMP, > 95%), tris[2-(3-mercaptopropionyloxy)ethyl] isocyanurate (3TI, > 95%), and pentaerithritol tetrakis(3-mercaptopropionate) (PETMP, > 95%), as well as the photoinitiator 2,2′-dimethoxy-2-phenylacetophenone (DMPA) were purchased from Sigma Aldrich. All monomers, solvents, additives, catalysts, and crosslinking agents were purchased from the specified distributors and used as received without further purification. For microgripper device fabrication, Epolight™ 4121 platinum dye was purchased from Epolin, Inc., and 200/220/240 μm core/cladding/buffer optical fiber was purchased from Polymicro Technologies (FVP200220240).
5.2. Thermoplastic Polyurethane Synthesis, Purification and Solvent Removal
Formulations for all thermoplastic polyurethanes synthesized in this study are provided in Table 1. Each thermoplastic polyurethane formulation was synthesized in a 100 g scale polymerization batch in a 0.40 g/mL solution in anhydrous THF in a previously flame dried 240 mL glass jar in the presence of approximately 80 mL of 4 Å molecular sieves, which were also previously flame dried. All monomers, solvents and catalysts were mixed under dry air in a LabConco glove box. Using a 1.02:1.00 NCO:OH ratio, all diisocyanate and hydroxyl starting materials were added to the molecular sieve-containing glass jars in the glove box, after which anhydrous THF and the Zr catalyst (0.010 overall wt % Zr Cat) were added. The polymerizations were carried out in sealed jars using a LabConco RapidVap instrument at 80 °C for 24 h at a vortex setting of 150 RPM. The RapidVap was used to heat and mix the monomer solutions. After 24 h, the viscous polymer solutions were diluted with additional THF to afford 0.10 g/mL concentrations and filtered to remove molecular sieve particulates, residual monomers and catalyst using a 15 cm tall, 10 cm diameter flash chromatography silica column. After filtration, the purified, diluted polymer solutions were decanted into 30 cm × 22 cm rectangular polypropylene (PP) dishes for solvent removal. The solution-containing PP dishes were heated in a vacuum oven to 50°C at ambient pressure using a house air purge for 24 h, after which the oven temperature was increased to 80°C for an additional 24 h. The vacuum oven was then evacuated to a pressure of 1 torr at 80°C for 72 h, after which neat, amorphous, thermoplastic polyurethane films approximately 0.5 mm in thickness were removed from the PP dishes and stored under desiccation until future use.
5.3. Characterization of Thermoplastic Molecular Weight by Gel Permeation Chromatography
To determine the molecular weights of all thermoplastic polyurethane formulations, gel permeation chromatography (GPC) experiments were performed on a Waters Chromatography, Inc., 1515 isocratic HPLC pump equipped with an inline degasser, a model PD2020 dual angle (15° and 90°) light scattering detector, a model 2414 differential refractometer (Waters, Inc.), and four PLgel polystyrene-co-divinylbenzene gel columns (Polymer Laboratories, Inc.) connected in series: 5 μm Guard (50×7.5 mm), 5 μm MixedC (300×7.5 mm), 5 μm 104 (300×7.5mm), and 5 μm 500Å (300×7.5 mm) using the Breeze (version 3.30, Waters, Inc.) software. The instrument was operated at 35°C with THF as eluent (flow rate set to 1.0 mL/min). Data collection was performed with Precision Acquire 32 Acquisition program (Precision Detectors, Inc.) and analyses were carried out using Discovery32 software (Precision Detectors, Inc.). A system calibration curve generated from plotting molecular weight as a function of retention time for a series of broad polydispersity poly(styrene) standards was used to determine the molecular weight values.
5.4. Polythiol and Photoinitiator Solution Blending, Film Casting and Sample Curing
To prepare blended mixtures of thermoplastic polyurethane, polythiol and photoinitiatior to be subjected to UV curing, approximately 5 g of each thermoplastic PU formulation were dissolved in THF solutions in 40 mL amber glass vials to afford solutions of approximately 0.13 g/mL concentrations. Polythiol crosslinking agents and photoinitiator were then added to each solution in specified quantities. Unless otherwise stated, a 1:1 C=C to SH stoichiometric ratio, a 3.0 wt% DMPA photoinitiator additive composition, a post cure time of 24 h, less than 4 wt% THF solvent at cure, and TMPTMP polythiol selection were used. After pouring the mixtures into 10 cm × 5 cm × 5 cm PP containers, the THF was evaporated by placing the PP containers in a vacuum oven at 50°C at ambient pressure using a house air purge for 24 h for all series except for the varying THF series, for which THF evaporation times were varied to afford the specified THF compositions.
“Percent THF” as reported in this study refers to the mass ratio of THF to (PU + polythiol + DMPA) at curing time. Percent THF was determined by taking the masses of the PP containers before and after pouring the blended solutions into them and again after THF evaporation for the varying THF series only. For the varying THF series, the amount of solvent evaporation time required to afford THF concentrations of 100%, 50%, 25% and 4% was 2.17 h, 2.42 h, 4.42 h, and 19.17 h, respectively. The 300% THF sample was prepared separately without THF evaporation by preparing a solution of THF, PU, polythiol and DMPA quantities to afford a 3:1 THF: PU/polythiol/DMPA ratio. The 300% THF solution was UV cured immediately without solvent evaporation after injecting the solution between glass slides separated by 1 mm glass spacers. For all other polymers synthesized in this study that were not part of the varying THF series, percent THF at cure time is reported as <4% because these polymers were subjected to solvent evaporation times of 24 h, approximately 5 h longer than that required to afford THF evaporation such that 4% THF remained in the samples in the varying THF series.
All thiol-ene blends were irradiated using 365 nm UV light in a UVP CL-1000L crosslinking chamber for 45 min, and after UV curing, all samples were post-cured at 120°C at 1 torr for 24 h, except for samples in the varying post cure time series, for which post cure time was varied as specified. For the varying C=C to SH stoichiometric ratio series, C=C to SH ratios of 4.0:1.0, 2.0:1.0, 1.5:1.0, and 1.0:1.0 were used. For the varying photoinitiator composition series, DMPA compositions of 0.1, 0.5, 1.0, 3.0, and 10.0 wt% were used. For the varying post-cure time series, post cure times of 0 min, 10 min, 1.5 h, 6 h, and 24 h were used. For the varying THF at cure series, THF compositions of 300, 100, 50, 25, and 4 wt% at cure were used. For the varying polythiol functionality series, EGBMP, TMPTMP, PETMP, and DPHMP were used (f = 2, 3, 4, and 6, respectively). For the varying diol co-monomer and varying diisocyanate co-monomer ratio Tg variation series, 3TI and PETMP polythiol crosslinkers were used, respectively. After post-curing, all crosslinked films were stored under desiccation until future use.
5.5. Materials Characterization Methods
To determine the percent of thermoplastic PU chains and polythiol crosslinking additives incorporated into each network after UV curing and post curing, sol/gel analysis experiments were run on select formulations. Dry, ~50 mg samples were massed in triplicate for each formulation and placed in 20 mL glass vials, after which THF was added in approximately a 150:1 solvent to polymer mass ratio. The vials were capped and vortexed at 50 RPM at 50°C for 48 h using a LabConco RapidVap instrument. After 48 h, the solvent swollen polymer samples were removed from the THF/sol fraction solutions and placed in new 20 mL glass vials, dried at 80°C for an additional 48 h at 1 torr and then re-massed to provide sufficient mass data to determine the gel fractions of each irradiated sample.
To determine key thermomechanical data for each sample, including rubbery moduli and glass transitions, dynamic mechanical analysis (DMA) experiments were run on every crosslinked sample synthesized in this study. Rectangular 25.0 × 4.0 × 0.4 mm specimens were machined using a Gravograph LS100 40 W CO2 laser machining system using a power setting of 15, a speed setting of 12, and a pass multiplicity of n = 2. Using a TA Instruments Q800 dynamic mechanical analyzer in the DMA Multifrequency/Strain mode, DMA experiments were run in tension at 1 Hz from −20 to 140°C using a heating rate of 2°C/min, a Preload Force of 0.01 N, a Strain of 0.1%, and a Force Track of 150%. DMA results were recorded using TA Instruments QSeries software and analyzed using TA Instruments Universal Analysis software. DMA experiments were run on select formulations in triplicate and reported a glass transition (tangent delta peak) average standard deviation of 0.85°C and a minimum rubbery modulus average standard deviation of 0.26 MPa.
To determine percent recoverable strain and recovery stress for select samples, shape memory characterization experiments were performed using a TA Instruments Q800 DMA on laser machined 25.0 × 4.0 × 0.4 mm rectangular specimens prepared using the same laser machining parameters as those used to prepare the DMA samples in the above paragraph. In the DMA Strain Rate Mode in tension, rectangular specimens were heated to Tg + 25°C (glass transitions were determined by the peak of the tangent deltas from the previous DMA results), allowed to equilibrate for 30 min, and then strained to deformations of 25, 50, or 100% at a strain rate of 20%/min. The strained samples were then cooled to 0°C and allowed to equilibrate for an additional 30 min. For constrained recovery experiments, which were used to measure the recovery stress of the materials, the drive force of the DMA instrument was maintained, and the samples were heated to 120°C at 2°C/min. For free strain recovery experiments, which were used to measure the percent recoverable strains of the SMPs, the drive force was set to zero after equilibration at 0°C, the samples were re-heated to 120°C at 2°C/min, and the amount of recoverable deformation was recorded using TA Instruments QSeries software and analyzed using TA Instruments Universal Analysis software.
Multi-temperature strain-to-failure experiments were performed to determine ultimate tensile properties and toughness for select crosslinked samples. ASTM Type V dog bone samples were machined using a Gravograph LS100 40 W CO2 laser machining device using a power setting of 15, a speed setting of 12, and a pass multiplicity of n = 2. All laser machined samples were sanded around the edges using 400, then 800, then 1200 grit sandpaper. Strain-to-failure experiments were conducted on select samples (n ≥ 5) using an Instron Model 5965 electromechanical, screw driven test frame, which was equipped with a 500 N load cell, 1 kN high temperature pneumatic grips, and a temperature chamber that utilizes forced convection heating. An Instron Advanced Video Extensometer with a 60 mm field-of-view lens was used to optically measure the deformation of the samples by tracking parallel lines applied at the ends of the gauge length. The samples were heated to T = Tloss modulus peak − 10°C, which corresponds to a thermal state of high toughness for this polyurethane system as observed in previous experiments (loss modulus peak temperatures were determined from previously obtained DMA data). The samples were heated under zero loading, which was achieved by keeping the bottom grip unclamped during thermal equilibration. The temperature was held at the target temperature for 30 min to allow for thermal equilibrium, after which the bottom grip was clamped, and then experiments were started thereafter using a deformation rate of 10 mm/min. Data were recorded using Instron Bluehill 3 software.
To determine percent cell viability for select crosslinked samples, four 9.0 × 4.0 × 0.4 mm specimens of each sample that would be subject to cytotoxicity testing were laser cut using a Gravograph LS100 40 W CO2 laser machining system using the laser cutting parameters specified in the previous paragraph. After laser machining, all specimens were washed with soap and water, rinsed with isopropanol, and then dried in a vacuum oven at 80°C for 24 hours and subsequently subjected to EtO sterilization. For the cell culturing protocol, mouse 3T3 fibroblasts were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2. Polymer samples were placed in 12 well plates with sterile forceps and covered with 500μL of culture media. The cells were trypsinized, centrifuged, and re-suspended in 500 μL of culture media, after which they were placed into each well of the 12 well plate at a density of approximately 66,000 cells/cm2. At 24 and 72 hour time points, culture media was aspirated from the wells, and approximately 500 μL of Calcein AM stain solution was added to each well before incubation at 37°C and 5% CO2 for 60 min. Calcein AM is a cell-permeable dye, which fluoresces green upon hydrolysis catalyzed by esterases found in the cytoplasm of living cells. Dead cells do not contain the viable esterases to catalyze this reaction and do not fluoresce green. During incubation, a 5 μM bisbenzimide (BBI, Hoechst 33258) stain solution was prepared by dilution with PBS. Before imaging, the cells were washed once with PBS, and approximately 500 μL of BBI solution was added to each well, after which the wells were covered with aluminum foil when not being imaged to limit light exposure. BBI was used as a nuclear stain, which allowed visualization of cells that did not stain with Calcein AM. In this way, live cells were counted as those fluorescing green, while dead cells were counted as those displaying nuclear BBI staining (blue) with no Calcein AM (green). Three to four replicates of each polymer formulation were imaged and analyzed as guided by ISO 10993 Part 5. Wide field fluorescence images were taken of each well at 10× magnification, and the images were analyzed using ImageJ computer software and its “Cell Counter” plug-in.
5.6. Microgripper Device Fabrication
To prepare Microgrippers, 1 g of CHDM-0.9 PU was dissolved in 2 g of THF. A portion of the THF added was doped with Epolin. The amount of doped THF added was tailored to achieve the desired wt% of 0.15% of Epolin in the microgrippers. DMPA and 3TI were then added to the dissolved PU solution. Molds consisting of Rain-X coated silica capillary tubing (ID 700 μm, Polymicro Technologies, Phoenix, AZ) and Teflon spacers used to center the optical fiber in the gripper were fabricated to achieve our desired geometry, as pictured in Figure 5(a). Cleaved optical fiber tips were prepared (FVP200220240, Polymicro Technologies, Phoenix, AZ), inserted into the mold, and dissolved SMP was injected into the distal end of the mold, filling the tube and expelling any bubbles. The microgrippers were then UV crosslinked for 20 minutes and post-cured at 120°C under vacuum overnight. In this step the solvent was removed from the polymer through evaporation and the SMP shrank from the initial OD of 700 μm to our desired OD of approximately 450 μm. After post-cure, the mold was removed and the microgripper was inspected for defects, such as bubbles, improper fiber placement, or particulates within the gripper.
The crimping process for the microgripper/ball-tipped assemblies is shown in Figure 5(b). To load the coil into the microgripper, the gripper was placed into a capillary tube with an ID slightly larger than the OD of the gripper. The gripper was then heated above its Tg and the ball tip of the wire coil was axially forced into the distal end of the SMP. All of the components were then cooled to room temperature, fixing the SMP in the deformed configuration, locking in the ball tipped coil.
5.7. Microgripper Device Characterization
In order to provide indicative data about the gripping strength capacity of the crimped microgripper/ball-tipped assembles during microcatheter delivery of endovascular devices, the maximum gripping forces of the crimped assembles were determined using tensile testing experiments. As pictured in Figure 5(c), these experiments were set up and run using an MTS tensile testing system. Using the immersion chamber of the MTS system, strain-to-failure experiments were carried out on seven crimped seven devices in water at 37°C. Additionally, to demonstrate proof-of-concept of successful device deployment, in vitro measurements were carried out within a flow loop held at 37°C and using a flow rate of 190 mL/min, which is similar to flow rates seen in vivo, using the experimental setup pictured in Figures 6(a) and 6(b). To actuate the grippers, approximately 3.1 W of laser irradiation was delivered through the optical fiber to the gripper using an 808 nm diode laser (Jenoptik AG, Jena, Germany) for approximately five seconds. 3.1 W was the lowest optical power necessary to actuate the microgripper, as determined by incrementally increasing the power from approximately 0.25W to 3.1W. Five crimped devices were subjected to these in vitro laser-actuated deployments to test for consistent release.
Table 2.
Sol/Gel analysis data for thiol-ene crosslinked polyurethane shape memory polymers prepared using various synthetic parameters
| Series | Sample | Polythiol Crosslinker | % DMPA | % THF | Post Cure @ 120°C | Gel Fract-ion | Error |
|---|---|---|---|---|---|---|---|
| Varying TMPAE | TMPAE-0.1 | TMPTMP | 3.00% | < 4.00% | 24 h | 0.998 | ± 0.004 |
| TMPAE-0.5 | TMPTMP | 3.00% | < 4.00% | 24 h | 0.967 | ± 0.005 | |
| TMPAE-0.9 | TMPTMP | 3.00% | < 4.00% | 24 h | 0.957 | ± 0.005 | |
|
| |||||||
| Varying Diol | 1,4-BD-0.9 | 3TI | 3.00% | < 4.00% | 24 h | 0.964 | ± 0.012 |
| CHDM-0.9 | 3TI | 3.00% | < 4.00% | 24 h | 0.893 | ± 0.015 | |
| DEG-0.9 | 3TI | 3.00% | < 4.00% | 24 h | 0.932 | ± 0.010 | |
| 2,2-DMPD-0.9 | 3TI | 3.00% | < 4.00% | 24 h | 0.831 | ± 0.057 | |
|
| |||||||
| Varying DCHMDI Co-Monomer | DCHMDI-0.0 | PETMP | 3.00% | < 4.00% | 24 h | 0.985 | ± 0.012 |
| DCHMDI-0.1 | PETMP | 3.00% | < 4.00% | 24 h | 0.998 | ± 0.016 | |
| DCHMDI-0.5 | PETMP | 3.00% | < 4.00% | 24 h | 0.981 | ± 0.011 | |
| DCHMDI-1.0 | PETMP | 3.00% | < 4.00% | 24 h | 0.986 | ± 0.015 | |
|
| |||||||
| Varying % Solvent During Cure | TMPAE-0.9 | TMPTMP | 3.00% | 300% | 24 h | 0.987 | ± 0.014 |
| TMPAE-0.9 | TMPTMP | 3.00% | 100% | 24 h | 0.992 | ± 0.007 | |
| TMPAE-0.9 | TMPTMP | 3.00% | 50% | 24 h | 0.985 | ± 0.017 | |
| TMPAE-0.9 | TMPTMP | 3.00% | 25% | 24 h | 0.992 | ± 0.009 | |
| TMPAE-0.9 | TMPTMP | 3.00% | 10% | 24 h | 0.997 | ± 0.008 | |
| TMPAE-0.9 | TMPTMP | 3.00% | 4% | 24 h | 0.998 | ± 0.004 | |
|
| |||||||
| Varying Polythiol | TMPAE-0.1 | EGBMP | 3.00% | < 4.00% | 24 h | 0.818 | ± 0.031 |
| TMPAE-0.1 | TMPTMP | 3.00% | < 4.00% | 24 h | 0.946 | ± 0.006 | |
| TMPAE-0.1 | PETMP | 3.00% | < 4.00% | 24 h | 0.997 | ± 0.007 | |
| TMPAE-0.1 | DPEHMP | 3.00% | < 4.00% | 24 h | 0.917 | ± 0.049 | |
|
| |||||||
| Varying Photoinitiator | TMPAE-0.9 | TMPTMP | 0.10% | < 4.00% | 24 h | 0.981 | ± 0.021 |
| TMPAE-0.9 | TMPTMP | 1.00% | < 4.00% | 24 h | 0.979 | ± 0.015 | |
| TMPAE-0.9 | TMPTMP | 10.00% | < 4.00% | 24 h | 0.974 | ± 0.009 | |
Acknowledgments
This work was partially performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. This material is also based upon work supported by the National Science Foundation Graduate Research Fellowship #2011113646 and by the National Institutes of Health/National Institute of Biomedical Imaging and Bioengineering Grant R01EB000462. The authors also acknowledge financial support from the National Science Foundation (CHE-1057441 and CHE-1410272), and the Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry (A-0001).
Contributor Information
Keith Hearon, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA.
Mark A. Wierzbicki, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA
Landon D. Nash, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA
Todd L. Landsman, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA
Christine Laramy, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA.
Alexander T. Lonnecker, P.O. Box 30012, Department of Chemistry, Department of Chemical Engineering, Texas A&M University, College Station, Texas 77842-3012 USA
Michael C. Gibbons, Building 13, Room 263, Biomedical & General Engineering Department, California Polytechnic State University, San Luis Obispo, CA 93407 USA
Sarah Ur, Building 13, Room 263, Biomedical & General Engineering Department, California Polytechnic State University, San Luis Obispo, CA 93407 USA.
Kristen O. Cardinal, Building 13, Room 263, Biomedical & General Engineering Department, California Polytechnic State University, San Luis Obispo, CA 93407 USA
Thomas S. Wilson, 7000 East Avenue, Lawrence Livermore National Laboratory, Livermore, CA 94550 USA
Karen L. Wooley, P.O. Box 30012, Department of Chemistry, Department of Chemical Engineering, Texas A&M University, College Station, Texas 77842-3012 USA
Duncan J. Maitland, Email: djmaitland@tamu.edu, 5045 Emerging Technologies Building, Department of Biomedical Engineering, 3120 Texas A&M University, College Station, TX 77843-3120 USA.
References
- 1.Small W, IV, Singhal P, Wilson TS, Maitland DJ. J Mater Chem. 2010;20:3356. doi: 10.1039/B923717H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Safranski DL, Smith KE, Gall K. Polym Rev. 2013;53:76. [Google Scholar]
- 3.Ware T, Simon D, Hearon K, Liu C, Shah S, Reeder J, Khodaparast N, Kilgard MP, Maitland DJ, Rennaker RL, Voit WE. Macromol Mater Eng. 2012;297:1193. doi: 10.1002/mame.201200241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Julich-Gruner KK, Löwenberg C, Neffe AT, Behl M, Lendlein A. Macromol Chem Phys. 2013;214:527. [Google Scholar]
- 5.Hearon K, Singhal P, Horn J, Small W, Olsovsky C, Maitland KC, Wilson TS, Maitland DJ. Polym Rev. 2013;53:41. doi: 10.1080/15583724.2012.751399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yakacki C, Gall K. Adv Polym Sci. Vol. 226. Springer-Verlag; Berlin/Heidelberg, Germany: 2010. (Shape-Memory Polymers for Biomedical Applications). (A. Lendlein) [Google Scholar]
- 7.Safranski DL, Gall K. Polymer. 2008;49:4446. [Google Scholar]
- 8.Lendlein A, Kelch S. Angew Chem Int Ed. 2002;41:2035. [PubMed] [Google Scholar]
- 9.Liu C, Qin H, Mather PT. J Mater Chem. 2007;17:1543. [Google Scholar]
- 10.Hearon K, Smith SE, Maher CA, Wilson TS, Maitland DJ. Radiat Phys Chem. 2013;83:111. doi: 10.1016/j.radphyschem.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hearon K, Gall K, Ware T, Maitland DJ, Bearinger JP, Wilson TS. J Appl Polym Sci. 2011;121:144. doi: 10.1002/app.33428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q, Zhou H, Wicks DA, Hoyle CE, Magers DH, McAlexander HR. Macromolecules. 2009;42:1824. [Google Scholar]
- 13.Rodriguez JN, Clubb FJ, Wilson TS, Miller MW, Fossum TW, Hartman J, Tuzun E, Singhal P, Maitland DJ. J Biomed Mater Res Part A. 2013 doi: 10.1002/jbm.a.34782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harkal UD, Muehlberg AJ, Webster DC. Prog Org Coat. 2012;73:19. [Google Scholar]
- 15.Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 16.Hoyle CE, Lowe AB, Bowman CN. Chem Soc Rev. 2010;39:1355. doi: 10.1039/b901979k. [DOI] [PubMed] [Google Scholar]
- 17.Nair DP, Cramer NB, Scott TF, Bowman CN, Shandas R. Polymer. 2010;51:4383. doi: 10.1016/j.polymer.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beigi S, Yeganeh H, Atai M. Dent Mater. 2013;29:777. doi: 10.1016/j.dental.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Wicks DA, Hoyle CE, Pu H, Yuan J, Wan D, Liu Y. Polymer. 2009;50:1717. [Google Scholar]
- 20.Otts DB, Heidenreich E, Urban MW. Polymer. 2005;46:8162. [Google Scholar]
- 21.Kasprzak SE, Martin B, Raj T, Gall K. Polymer. 2009;50:5549. doi: 10.1016/j.polymer.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson TS, Bearinger JP, Herberg JL, J.E.M III. Wright WJ, Evans CL, Maitland DJ. J Appl Polym Sci. 2007;106:540. [Google Scholar]
- 23.Maitland DJ, Metzger MF, Schumann D, Lee A, Wilson TS. Laser Surg Med. 2002;30:1. doi: 10.1002/lsm.10007. [DOI] [PubMed] [Google Scholar]
- 24.Hearon K, Besset CJ, Lonnecker AT, Ware T, Voit WE, Wilson TS, Wooley KL, Maitland DJ. Macromolecules. 2013;22:8905–8916. doi: 10.1021/ma4018372. [DOI] [PMC free article] [PubMed] [Google Scholar]