

Abstract
Duchenne muscular dystrophy (DMD) is a fatal neuromuscular disease caused by mutations in the dystrophin gene, leading to the loss of a critical component of the sarcolemmal dystrophin glycoprotein complex. Galectin-1 is a small 14 kDa protein normally found in skeletal muscle and has been shown to be a modifier of immune response, muscle repair, and apoptosis. Galectin-1 levels are elevated in the muscle of mouse and dog models of DMD. Together, these findings led us to hypothesize that Galectin-1 may serve as a modifier of disease progression in DMD. To test this hypothesis, recombinant mouse Galectin-1 was produced and used to treat myogenic cells and the mdx mouse model of DMD. Here we show that intramuscular and intraperitoneal injections of Galectin-1 into mdx mice prevented pathology and improved muscle function in skeletal muscle. These improvements were a result of enhanced sarcolemmal stability mediated by elevated utrophin and α7β1 integrin protein levels. Together our results demonstrate for the first time that Galectin-1 may serve as an exciting new protein therapeutic for the treatment of DMD.
Introduction
Duchenne muscular dystrophy (DMD) is a common X-linked muscular dystrophy affecting 1 in 3,500 male births. DMD patients suffer from severe, progressive muscle degeneration with clinical symptoms first detected 2–5 years of age. As the disease progresses patients are confined to a wheelchair in their teens and without medical intervention die in their early twenties from cardiopulmonary failure.1 DMD patients and mdx mice (the mouse model for DMD) have mutations in the gene encoding dystrophin. These mutations result in the absence of dystrophin, a 427 kDa cytoskeletal protein located under the sarcolemma of muscle fibers.2 Through the N-terminal region, dystrophin interacts with F-actin of the cell cytoskeleton.3 The C-terminal region of dystrophin interacts with a transmembrane protein complex that includes α- and β-dystroglycans, dystrobrevins, α- and β-syntrophins, sarcospan, sarcoglycans, and nNOS.4 The absence of dystrophin leads to pronounced decrease in dystrophin-associated proteins in skeletal muscle of DMD patients and mdx mice.5 Decreased DGC levels leads to sarcolemmal instability and damage as muscle fibers detach from the laminin rich basal lamina during contraction.2
Galectin-1 is a small 14kDa nonglycosylated protein encoded by the Lectin, Galactoside-binding, Soluble-1 (LGALS1) gene. Galectin-1 protein is expressed by many different tissues with a concentration dependent monomeric or homodimeric structure which determines glycoside binding affinities. Although Galectin-1 binding dynamics are complex, the native dimeric form preferentially binds immobilized extended glycans.6 This binding induces many diverse physiological activities including cell migration, cell growth, angiogenesis, immunomodulation, and immune tolerance.7,8 In skeletal muscle, Galectin-1 aids in the conversion of dermal fibroblasts and stem cells to myogenic cells during muscle repair and regeneration.9,10,11,12,13 It also directly interacts with both laminin and the α7β1 integrin to modulate myoblast fusion during muscle repair.14 In addition Galectin-1 has been shown to modulate inflammation and angiogenesis, both critical modifiers of disease progression in DMD.11,15
In this study, we assessed the therapeutic potential of systemically delivered recombinant mouse Galectin-1 (rMsGal-1) to prevent muscle disease in the dystrophin deficient mdx mouse model of DMD. rMsGal-1-treated mdx mice displayed improved activity and muscle strength with reduced skeletal muscle pathology and kyphosis. Further, we show rMsGal-1 treatments led to elevated levels of two critical sarcolemmal complexes in dystrophin-deficient skeletal muscle which have been shown to modify disease progression; the utrophin glycoprotein complex (UGC) and α7β1 integrin. Together, our preclinical data indicate that Galectin-1 protein has exciting therapeutic potential for the treatment of DMD.
Results
Production of recombinant mouse Galectin-1
Since Galectin-1 is upregulated in skeletal muscle in preclinical models of DMD9 and interacts with the α7β1 integrin, we hypothesized that elevated levels might serve as a modifier of disease progression in DMD. Recombinant mouse Galectin-1 (rMsGal-1) was produced with a C-terminal 6x Histidine tag for purification purposes using the pET23b vector (Figure 1a). Ponceau S (Figure 1b) and western blotting (Figure 1c) confirmed greater than 95% rMsGal-1 purity and that the protein was Galectin-1.
Figure 1.

Purified rMsGal-1 increases disease modifying protein complexes in hDMD myotubes. The cDNA encoding mouse Galectin-1 was cloned into the pET23b vector (a). Purity of rMsGal-1 was determined by Ponceau S (b) and western blot analysis using a LGALS-1 antibody (c). Human DMD (hDMD) myotubes were plated, differentiated for 5–7 days then treated with either phosphate-buffered saline (PBS), 12, or 60 µg/μl of rMsGal-1. Representative immunoblots show expression levels for α7B integrin, α7A integrin, β1D integrin and β-DG (d). Quantification of immunoblots using imageJ software for α7B integrin (e), α7A integrin (f), β1D integrin (g) and β-DG (h) are shown. Statistical analysis used was student t-test between treatment groups (**P < 0.01, *P < 0.05; n = 3 for each concentration and treatment group).
Intramuscular injections of rMsGal-1 reduces muscle pathology in mdx mice
As an initial assessment of the efficacy of rMsGal-1 treatment on skeletal muscle pathology, we performed intramuscular (IM) injections in the tibialis anterior (TA) of mdx mice. Ten-day-old mdx mice were injected with 1.5 µg (n = 4) or 150 µg (n = 3) rMsGal-1 in one TA and a corresponding volume of phosphate-buffered saline (PBS) in the contralateral TA. Muscle tissues were collected between 4–5 weeks of age and analyzed for the presence of centrally located nuclei (CLN) using Hematoxylin and Eosin (H&E) staining. CLN is an indicator of previous muscle damage and repair in mdx muscle. Although no significant difference was observed with rMsGal-1 treatment of 1.5 µg relative to the PBS treated control, a significant ~16% reduction in myofibers containing CLN was observed with 150 µg rMsGal-1 treatment (Supplementary Figure S1). These data suggest that Galectin-1 improves myofiber integrity and prevents muscle damage in the mdx mouse model of DMD.
Galectin-1 increases β-dystroglycan and α7β1 integrin proteins in mouse and DMD myogenic cells
Galectin-1 has been implicated in muscle cell repair through interactions with laminin and α7β1 integrin. We hypothesized that the decreased CLN observed in rMsGal-1-treated mdx mice might be due to enhanced sarcolemmal stability mediated by elevated levels of the UGC and α7β1 integrin protein complexes. To investigate whether rMsGal-1 treatment altered the levels of sarcolemmal protein complexes, we treated C2C12 myoblasts and myotubes using increasing concentrations of rMsGal-1. After a 48-hour treatment with 300 μg/ml rMsGal-1, western blot analysis of C2C12 myoblasts and myotubes displayed a 1.7-fold increase in α7β integrin (Supplementary Figure S2a,b, n = 3). C2C12 myotubes also displayed an increase in β-DG at concentrations above 3 μg/ml of rMsGal-1 (Supplementary Figure S2d, n = 3). Finally, a dramatic increase in β1D integrin levels was observed with rMsGal-1 concentrations above 1.5 μg/ml (9.6-fold; 3 μg/ml) in C2C12 myotubes relative to PBS-treated controls (Supplementary Figure S2c).
Next, we assessed the effects of rMsGal-1 treatments on sarcolemmal protein levels in human DMD (hDMD) myoblasts and myotubes using western blot analysis. We began by treating hDMD myoblasts with 72 μg/ml rMsGal-1, and found this led to a significant increase in α7B integrin and β-DG (Supplementary Figure S3b,e; 2.3- and 1.6-fold respectively). However, protein levels of β1A integrin and α-DG were not significantly altered in hDMD myoblasts with treatment (Supplementary Figure S3c,d). Treatment of hDMD myotubes with 12 μg/ml rMsGal-1 resulted in an increase in α7B integrin (1.6-fold), α7A integrin (5.6-fold), and β1D integrin (1.8-fold) relative to PBS (Figure 1d, quantitated in Figure 1e–g). Treatment with 60 µg/μl rMsGal-1 was needed to increase the levels of β-DG (1.4-fold) relative to PBS-treated controls (Figure 1d, quantitated in Figure 1e–h). These data suggest that hDMD myotubes are more sensitive to Galectin-1 treatment than myoblasts or C2C12 cells and that Galectin-1 more potently enhances α7β1 integrin levels, but can also act to elevate components of the DGC/UGC complex at higher concentrations. Together, these in vitro experiments show that rMsGal-1 treatment can stabilize and/or increase α7β1 integrin and β-DG, a critical member of the DGC/UGC protein complexes, in skeletal muscle.
rMsGal-1 protein demonstrates a pharmacokinetic and pharmacodynamic profile in mdx mice that supports systemic delivery for the treatment of DMD
Due to the small 14 kDa size of Galectin-1, we predicted intraperitoneal (IP) injections would be an effective delivery method that would ensure bioavailability of the rMsGal-1 to skeletal muscle of mdx mice. Based on the 60 μg/ml dose of rMsGal-1 required to achieve β-DG elevation in human DMD myotubes, we selected a dose in mice that was 5–10 times higher and represented the industry standard in moving from in vitro to in vivo studies. Therefore, we began evaluating mdx mouse serum pharmacokinetics (PK) of the rMsGal-1 after a single 20 mg/kg IP injection (n > 3/time point), a concentration approximately eight times higher than the in vitro concentration used. Enzyme-linked immunosorbent assay (ELISA) was used to assess total mGalectin-1 serum levels prior to injection and at eight time-points between 0.5 and 6 hours post-treatment (Figure 2a). Maximum mGalectin-1 levels were not observed until ~2 hours postinjection, suggesting a slow uptake into serum with IP delivery. Serum clearance was determined from the maximum at two hours onward. The serum half-life (t1/2) for Galectin-1 was determined to be 1.07 hours (Figure 2a). This study shows that rMsGal-1 can be effectively administered systemically into the mdx mouse by IP injections and that elevated serum levels do not persist after ~6 hours postdelivery with a 20 mg/kg treatment.
Figure 2.

Pharmacokinetics and pharmacodynamics of systemically delivered rMsGal-1. Pharmacokinetics of rMsGal-1 using serum taken from mdx mice between 0–6 hours postinjection with 20 mg/kg rMsGal-1 or phosphate-buffered saline (PBS) (n = 7 for 0.5, 2, and 6 hours, n = 4 for 1 hour and n = 3 for 2.5, 3, and 5 hours). For PBS-treated mice, n = 3 for 0, 0.5, 1, 2, and 4 hour (a). Immunoblot quantification of tibialis anterior (TA) lystate from dystrophic muscle treated with PBS or 20 mg/kg/week with rMsGal-1 and probed with LGALS-1 antibody (b). High-resolution confocal microscopic images using TA muscle of rMsGal-1-treated mice versus controls were immunostained with an anti-LGALS-1 antibody (c). Immunofluorescence labeling using an anti-His-Tag verified rMsGal-1 was delivered systemically to the brain, diaphragm, liver, gastrocnemius, and TA of mice receiving a 10-week treatment (d). Scale bar = 100 μm for b and c (*P < 0.05 and **P < 0.01).
Assessment of rMsGal-1 treatment level and schedule in the mdx mouse model
Since the activity of Galectin-1 is known to be concentration dependent, the optimal rMsGal1 dosage and delivery for mdx mice was assessed using IP delivery of rMsGal-1 at 5 mg/kg/week, 20 mg/kg/week, or 20 mg/kg/bi-weekly rMsGal1 (Supplementary Figure S4). While this study used a relatively low number of animals, it should be noted that all treatments regimes showed elevated protein levels of both α7A integrin and β-DG (Supplementary Figure S4). The 20 mg/kg weekly injection provided the greatest difference in α7A integrin and β-DG levels relative to PBS-treatment (Supplementary Figure S4). Therefore, based on this preliminary study, the 20 mg/kg/week delivery schedule was used for further evaluation of the efficacy of Galectin-1 treatment. Total mGalectin-1 in the skeletal muscle of mdx mice treated weekly from 10 to 70 days of age with 20 mg/kg rMsGal-1 or PBS was quantified. Western blot analysis revealed total mGalectin-1 in the TA was increased 1.4-fold increase in rMsGal-1 treated mice compared to PBS-treated animals (Figure 2b). As the final injection had occurred approximately 3–4 days prior to sacrifice, we believe this represents the average level of total mGalectin-1 elevation in skeletal muscle using this treatment regime. Immunofluorescence of total Galectin-1 in the TA confirmed the rMsGal-1 treatments led to elevated mGalectin-1 levels which appeared to be localized to the extracellular matrix (ECM) (Figure 2c). This pattern of localization also observed in PBS-treated mdx mouse TA sections (Figure 2c). Coimmunofluorescence staining of mGalectin-1 and Laminin-α2 verified that the rMsGal-1 localization surrounding the myofiber was primarily in the ECM (Supplementary Figure S5). Together, these data confirm increased total mGalectin-1 levels in the skeletal muscle of rMsGal-1-treated mdx mice relative to PBS-treated controls.
The tissue distribution of the systemically delivered rMsGal-1 protein was assessed using immunofluorescence (IF) to detect the 6x Histidine tag on the exogenously delivered rMsGal-1 protein in mdx mice. The rMsGal-1 protein was observed in all skeletal muscles probed (Diaphragm, TA, and Gastrocnemius) and was localized to the basal lamina (Figure 2d). We also observed rMsGal-1 in the CA1 and CA3 hippocampal regions of the brain (Figure 2d) indicating rMsGal-1 was able to cross the blood-brain barrier. We did not observe exogenously delivered rMsGal-1 in the heart or kidney of treated mdx mice (Figure 2d). Together, these data show that rMsGal-1 can be systemically delivered to the skeletal muscle basal lamina of mdx mice.
rMsGal-1 treatment improves activity and muscle strength in mdx mice
In order to assess the efficacy of rMsGal-1 treatments in mdx mice, treatments of either 20 mg/kg/week (n = 13) or an identical volume of PBS (n = 12) were administered systemically by weekly IP injections beginning at 10 days of age. As functional strength and activity levels are the most critical measurable outcomes for DMD patients,16 individual mouse activity data were collected at 10 weeks of age. Activity levels were measured using an Opto-Varimex-4 computerized system for 30 minutes with wild-type (n = 27) mice included as a controls. The rMsGal-1 treated mdx mice displayed a significant increase in distance traveled (Figure 3a), large ambulatory time (Figure 3c), and vertical sensor breaks (stand-ups) (Figure 3d). Conversely, there was a decrease in the overall resting (nonactive) time of rMsGal-1-treated animals compared to PBS-treated mice (Figure 3b). These results indicate treatment with rMsGal-1 improved the overall activity of mdx mice.
Figure 3.

Improved activity in mdx mice treated with rMsGal-1. Activity levels of rMsGal-1- and phosphate-buffered saline (PBS)-treated mdx mice and wild-type mice were measured for distance traveled (a), resting time (b), large ambulatory time (c), and vertical sensor breaks (d). There were improvements in all activity levels measured in rMsGal-1-treated mice with the greatest difference being a 2.2-fold increase in distance traveled (a). For (a–d), n = 5 for PBS mice, n = 8 for rMsGal-1 mice and n = 27 for wild-type mice (*P < 0.05 and **P < 0.01).
The weights of wild-type and mdx mice treated with rMsGal-1 or PBS was measured weekly (Figure 4a). No significant difference was observed in the average total mouse body mass between any of the groups at any point (Figure 4a). The wet-weights of TA, extensor digitorum longus (EDL), and soleus muscles were also measured. Interestingly, similar to wild-type muscle, we observed a decrease in the average muscle mass of the TA, EDL, and soleus in mdx mice treated with rMsGal-1 relative to PBS alone (Figure 4b,c,f). This suggests that the rMsGal-1 treatment not only restores activity levels of mdx mice but also improves muscle functionality by decreasing hypertrophy in these muscles.
Figure 4.

Improved ex vivo muscle function in mdx mice treated with rMsGal-1. Mice were weighed each week starting at 10 days of age continuing till 10 weeks with no significant changes in weight (a). Isolated skeletal muscles were excised and wet mass was recorded (b,c,f). Isometric tetanic contractions were performed in vitro in isolated extensor digitorum longus (EDL) muscles from wild type (WT) and mdx PBS and rMsGal-1-treated mice at 10 weeks of age. Isometric tetanic specific force is improved in the EDL (d) and soleus (g) of mdx mice treated with rMsGal-1. Force frequency analysis of EDL (e) and soleus (h) and expressed in specific force. Data represent averages and error bars represent standard deviation of the mean. Statistics were calculated using analysis of variance with a Bonferroni correction (*P < 0.05, **P < 0.01, ***P < 0.001). #P < 0.05 Statistically significant difference from WT to mdx mice treated with rMsGal-1. $P < 0.05 Statistically significant difference from WT to mdx mice treated with PBS.
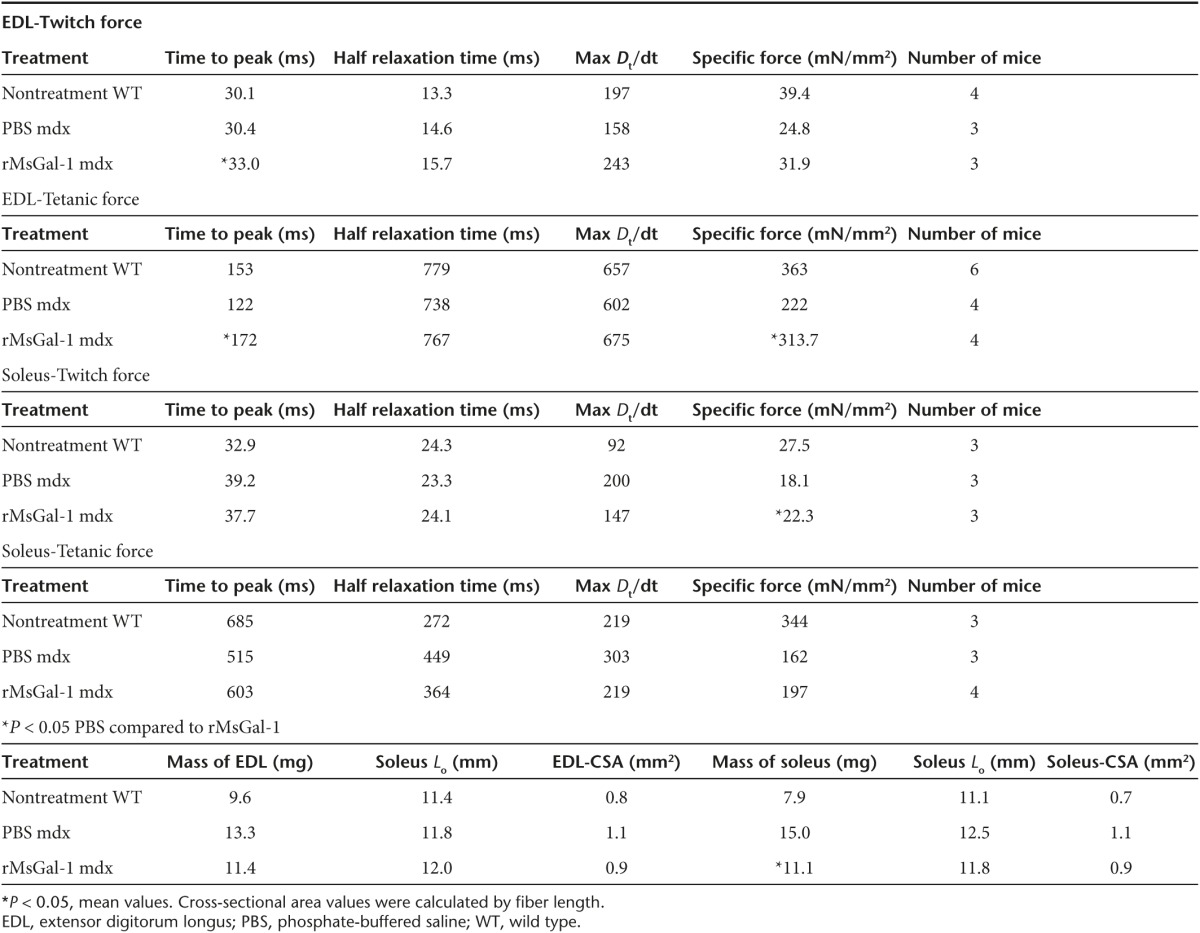
In order to determine whether the activity and changes in muscle mass were due to improved muscle strength, we investigated force production in the EDL and soleus muscles isolated from 10-week-old wild-type and mdx mice treated with PBS or rMsGal-1. The EDL and soleus muscles were subjected to maximum isometric twitch and tetanus followed by force frequency analysis for each treatment group (Table 1 and Figure 4d,e,g,h). The mdx mice treated with rMsGal-1 had EDL and soleus muscles that had improved maximum isometric twitch and tetanus specific force generation relative to PBS-treated mice (Figure 4d,g; Table 1). The EDL force frequency displayed improved force generation beginning at 50 Hz and continuing through 180 Hz (Figure 4e). The rMsGal-1 treatment appeared to be more effective in the EDL muscle than in the soleus where the maximum isometric tetanus specific force did not indicate a significant difference in mdx mouse treatment groups (Figure 4g). While the rMsGal-1 treatment did lead to significantly elevated specific force frequency levels between 50 and 180 Hz relative to PBS, the improvement was not as pronounced as that observed in the EDL (Figure 4h). Time to peak in the EDL for both tetanus and twitch produced significant increases in rMsGal-1-treated mice (Table 1). Together, these data show mdx mice treated with rMsGal-1 protein had improved functional outcome measures including activity, increased muscle strength and decreased muscle hypertrophy.
Table 1. Isometric force measurements for EDL and soleus from wild-type and mdx mice treated with PBS or rMsGal-1.
rMsGal-1 reduces kyphosis in mdx mice
Dystrophic patients and mdx mice have spinal curvature abnormalities which correlate to disease progression.17,18 Previous studies have not examined kyphosis in mdx mice as young as 10 weeks. In order to determine if treatment with rMsGal-1 improved kyphosis relative to controls, left lateral radiographs were taken for wild-type controls (n = 32) along with rMsGal-1 (n = 11) and PBS (n = 7) treated mdx mice at 10 weeks of age (Figure 5a). Spinal curvature was assessed using the kyphotic index (KI) measurement following previously described procedures.17 A significant increase in KI was observed in rMsGal-1 (4.44) treated mdx mice compared to PBS (3.59) controls (Figure 5b), where a higher KI number is indicative of decreased kyphosis. Although previous studies have not examined mice as young as 10 weeks, a KI of ~4.4 was maintained by wild-type control mice from 4 to 18 months of age.17 These data indicate that the rMsGal-1-treated mdx mice maintained paraspinal muscle strength and spinal curvature through 10 weeks of age.
Figure 5.

rMsGal-1 treatment prevents kyphosis in mdx mice. (a) Left lateral radiographs taken at 10 weeks of age indicate an improved kyphotic index for rMsGal-1-treated mice, quantitated and graphed (b). n = 32 for WT, n = 7 for PBS mice, and n = 11 for rMsGal-1 mice (**P < 0.01).
Improved skeletal muscle pathology in mdx mice treated with rMsGal-1 protein
We next wanted to confirm that the physiologic effects observed in the mdx mice treated with 20 mg/kg/week rMsGal-1 were a result of improvements to skeletal muscle integrity. Immunofluorescence for dystrophin and nNOS (Figure 6a) was used to verify animal genotyping and showed that Galectin-1 treatment did not increase the number of revertant dystrophin expressing myofibers. The lack of restored nNOS localization suggests that the functional and histological improvements in rMsGal1 treated mdx are in the absence of a normally localized nNOS protein (Figure 6a). The TA muscles from wild-type and mdx mice treated with rMsGal-1 or PBS were stained with Hematoxylin and Eosin or Alexafluor-488 labeled Wheat Germ Agglutinin and 4',6-diamidino-2-phenylindole (DAPI) (Figure 6a) and analyzed for the presence of CLN (Figure 6b) and Feret's diameter (Figure 6c). Previously damaged muscle fibers were determined by quantifying the percentage of myofibers with CLN (Figure 6a). A twofold decrease in CLN percentage was observed in the rMsGal-1-treated mdx TA muscle (29.9%) compared to those treated with PBS (58.0%) and wild-type (3.2%) (Figure 5b). Minimum Feret's diameter of the muscle fibers was used to assess hypertrophy in the mdx treatment groups. We found the average myofiber diameter for the rMsGal-1 (34.6 µm) treated mdx mice was only slightly lower compared to PBS-treated animals (35.2 µm) (Supplementary Figure S6). However, mice treated with rMsGal-1 had a slight change in the percentage of myofibers between 21–30 μm (Figure 6c). This change in fiber size distribution causes a minor left shift in the Feret's diameter fiber distribution curve away from PBS-treated mdx mice toward wild-type indicative of more myofibers with a smaller diameter and less hypertrophy. Evans blue dye (EBD) positive fibers were counted in order to assess the sarcolemmal integrity. We observed approximately twofold decrease in the percentage of myofibers positive for EBD with rMsGal-1 (15.0%) treatment compared to PBS (28.4%) treated animals (Figure 6d; wild-type 3.0%). Together, these data indicate that rMsGal-1 treatment improves sarcolemmal stability in dystrophin deficient muscle and prevents muscle fiber damage and normalizes myofiber size.
Figure 6.

Reduced muscle pathology in mdx mice treated with rMsGal-1. Tibialis anterior (TA) muscle cryosections from mdx mice treated with PBS or rMsGal-1 and untreated WT mice were stained with hematoxylin and eosin, immunostained with Alexafluor-488 labeled wheat germ agglutinin (WGA) or WGA and Evans blue dye (a, rows 3–5). Subjects from each treatment group were analyzed for the presence of dystrophin to ensure disease genotype and lack of revertant fibers. Immunofluorescence for nNOS localization displayed a lack of sarcolemma restoration in either mdx treatment group (a, rows 1 & 2). Centrally located nuclei (CLN), minimum Feret's diameter, and EBD percentage analysis were performed using the same composite WGA stained TA images. rMsGal-1 treatment resulted in a decrease in CLN (PBS n = 12, rMsGal-1 n = 13, WT n = 12 mice) (b). rMsGal-1-treated mice had a minimum Feret's diameter percent myofiber curve which shifted left toward less hypertrophic myofibers (n = 4 mice, n = 6 mice, and n = 3 mice) (c). A poststain with EBD indicated a decrease in EBD-positive fibers (d). Average hydroxyproline content of quarter costal diaphragm muscles in WT (n = 13), PBS (n = 5), or rMsGal-1-treated mdx mice (n = 10) was analyzed and graphed wild-type (n = 10) (e). Statistical analysis used was unpaired Student's t-test. P = 0.10. Scale bar = 100 μm (*P < 0.05, ***P < 0.001, ***P < 0.0001).
Previous studies have shown Galectin-1 can suppress fibrosis,19 but can also induce TGF-β1 signaling which enhances fibrosis.20 Therefore, a hydroxyproline assay was used to determine the collagen content of costal diaphragm muscle from the mdx treatment groups and wild-type mice. The results showed no difference in collagen content in the costal diaphragms from either mdx treatment group (Figure 6e). However, as expected, all mdx diaphragms contained approximately twofold more collagen than wild-type (Figure 6e). While this result suggests Galectin-1 treatment did not reduce diaphragm fibrosis, it also did not accelerate the process.
Mdx mice treated with rMsGal-1 have increased sarcolemmal levels of the UGC and α7β1 integrin complexes
To investigate the mechanism underlying the improvements in mdx mice, we examined if rMsGal-1 increased sarcolemmal stabilizing complexes in skeletal muscle. Western analysis was used to quantify α- and β-dystroglycan (DG), α7A and α7B integrin, utrophin (Utr), α- β-, δ- and ɛ-sarcoglycans (SG), sarcospan (SSPN), and β1D integrin (Figure 7a). The rMsGal-1 treatment led to an average protein increase in α- and β-dystroglycans (DG) and α7A and α7B integrin of 1.4 or greater (Figure 7c,g,i,j). rMsGal-1 treatment led to elevated utrophin, SGs (α-, β-, δ-, and ɛ-SGs), SSPN, and β1D integrin protein with a fold increase between 3–5.4-fold (Figure 7b,e,h,k,f,l,d). While it is unclear whether these elevated proteins are a result of protein/complex stabilization or altered signaling leading to transcriptional activation, the observation that UCG and α7β1 integrin receptor complexes are increased with rMsGal-1 treatments suggests that sarcolemmal stabilization is a factor in preventing damage in dystrophin deficient muscle.
Figure 7.

Increased sarcolemmal localization of UGC and α7β1 integrin protein complexes in mdx mice treated with rMsGal-1. Immunoblot analysis of α- and β-dystroglycans (DG), α7A and α7B integrin, utrophin (Utr), α- β-, δ- and ɛ-sarcoglycans (SG), sarcospan (SSPN), and β1D integrin (a). Representative example immunoblots are shown for each protein tested (b–l). All immunoblots were quantified using ImageJ analysis software and were normalized as indicated on graphs (*P < 0.05, **P < 0.01). High-resolution confocal images were taken of transverse sections of tibialis anterior from mdx mice treated with either phosphate-buffered saline or rMsGal-1. Immunofluorescence images show increased correct localization of UGC and α7β1 Integrin proteins (m–z).
To determine whether the increased levels of the UGC and α7β1 integrin complexes were localized to the sarcolemma, immunofluorescence (IF) of key members of these protein complexes was performed. Results show increased sarcolemmal localization of utrophin (Figure 7m,n), α-DG (Figure 7o,p), β-DG (Figure 7q,r), α7 integrin (Figure 7s,t), α-SG (Figure 7u,v), β-SG (Figure 7w,x), β1D integrin (Figure 7y,z) in rMsGal-1-treated mdx mice compared to PBS-treated animals. This study confirms that rMsGal-1 treatment of mdx mice leads to increased sarcolemma localization of the UGC and α7β1 integrin protein complexes in mdx mice.
Discussion
DMD is a fatal neuromuscular disease for which there is no cure and limited treatment options.21 Current treatments for DMD involve the daily use of glucocorticoids (prednisone or deflazacort), which provide only transient benefits by temporarily improving muscle strength. However, high-dose, long-term steroid treatment is associated with numerous negative side effects especially in children.22 Several novel approaches are being developed for the treatment of DMD including gene replacement, exon skipping, gene repair, and use of embryonic and adult muscle stem cells.23,24,25 These approaches face several challenges including the large size of the dystrophin gene, immune response, efficient stem cell engraftment, and feasibility in treating all muscles affected in DMD. Targeting disease modifiers including Utrophin, GalNac, Biglycan, and α7β1 integrin have shown promise as potential therapeutics for DMD.26,27,28
Biologics are an exciting new therapeutic area for DMD. Recently, protein therapy using TAT-utrophin, biglycan, MG53, and laminin-111 have shown efficacy in DMD preclinical models.29,30,31,32,33 These protein therapeutics all have roles in muscle related to sarcolemma stability, membrane repair, or enhanced regeneration. In this study, we have identified Galectin-1 as a novel protein therapeutic for DMD. Galectin-1 is a nonglycosylated 14 kDa protein which can be produced and purified in large quantities using a bacterial expression system. We show rMsGal-1-treated mdx mice exhibit reduced muscle damage as measured by percentage of myofibers containing CLN and Evans blue dye uptake of rMsGal-1-treated mice. Given its known role in muscle repair, it is possible the presence of exogenous Galectin-1 may promote more efficient myogenic fusion of satellite cell populations. Although this mechanism may provide benefits in DMD muscle repair and modify disease progression, these cells would still harbor the dystrophin mutation and would not prevent the underlying DMD pathogenesis. These results are more likely due to a role for Galectin-1 protein therapy in protecting the sarcolemma of dystrophin-deficient muscle from progressive damage. Utrophin and α7β1 integrin have been identified as major DMD disease modifiers and these complexes act in part through sarcolemmal stabilization.28 Studies have shown sarcospan, a component of the UGC and DGC, can act to link utrophin and α7β1 integrin complexes in skeletal muscle.34 We show that Galectin-1 acts to increase levels of both utrophin and α7β1 integrin complexes in mdx mice. In addition, we observed increased sarcospan levels in Galectin-1 treated mdx mice suggest treatment promotes a sarcospan-mediated formation of α7β1 and utrophin macromolecular complexes that might act to protect myofibers from contraction induced muscle injury and prevent progressive muscle disease.
The improvements in muscle pathology in dystrophin deficient mdx mice treated with rMsGal-1 may also be attributed to the role Galectin-1 plays in the immunomodulation of leukocytes.9 Studies suggest that the binding of dimeric Galectin-1 to activated T helper (Th) cells will promote Regulatory Type 1 (Tr1) cell differentiation. Tr1-mediated secretion of interleukin-27 and interleukin-10 efficiently dampens inflammation in mouse models of autoimmunity, and immune evasion or tolerance in murine cancer models.8 Furthermore, Galectin-1 can suppress T-cell immune response in Mesenchymal stem cells.35 Galectin-1 facilitated interactions reducing immune response, apoptosis and/or cell turnover.8 Chronic skeletal muscle damage in DMD patients and mdx mice leads to increased immune response and inflammation. Preventing the immune response to muscle damage in mdx mice has been shown to limit muscle damage and lead to elevated strength,36 similar to what we have observed with Galectin-1 treatment. Thus the immune-modulatory actions of Galectin-1 could be responsible for many of the observed improvements, although this remains unclear from the current study.
Studies suggest that potential DMD therapeutics should use health-related quality of life (HrQOL) outcomes to assess efficacy of the therapeutic potential.37,38 For patients in the ambulatory stage of DMD, the primary goal reported to healthcare providers is to maintain ambulation for as long as possible.39,40,41 The 6 Minute Walking Distance (6MWD) is considered one the most relevant endpoint measures for ambulatory DMD patients and has a strong correlation to self-reported HrQOL.37 In this study, we show that Galectin-1 protein therapy improved the activity of mdx mice compared to PBS treatment. This included increased distance traveled and ambulatory time. Interestingly, this improvement took place without the restoration of nNOS localization which has been previously shown to critically impact muscle fatigue.42,43 Galectin-1 treatment was also shown to improve overall strength and specific muscle force relative to PBS. It is interesting that the specific force improvement was greater in the EDL which contains a greater number of fast-twitch fibers than in the Soleus which contains a greater number of slow-twitch. Together, these data indicate Galectin-1 treatment may have a positive impact on the HrQOL measures that include maintaining muscle strength and ambulation in DMD patients.
Spinal deformity or kyphosis is a major clinical problem for DMD patients which is caused by degeneration of the musculature supporting the vertebral column.17,18 Severe kyphosis in DMD patients can cause chest compression limiting respiratory function as well as reducing patient mobility often resulting in wheelchair confinement.18 Loss in respiratory function due to severe kyphosis in DMD patients is thought to be caused by reduced lung and chest wall compliance that restricts diaphragm and ribcage activity.44 Progressive and severe kyphosis is therefore a major contributing factor to the increased morbidity due to respiratory dysfunction observed in DMD patients.44 The mdx mouse also exhibits progressive spinal deformity which can be measured using x-ray imaging.17 In our study, we show that Galectin-1-treated mdx mice exhibit an improved kyphotic index. These results indicate Galectin-1 treatment slows progressive spinal deformity in the mdx mouse model, which may improve respiratory outcome measures for DMD patients.
DMD patients exhibit changes in the CNS as a result of loss of dystrophin.21 These include neuronal loss, neurofibrillary tangles and changes in the blood–brain barrier. Recent studies show prednisone treatments are able to alleviate blood–brain barrier fragility in mdx mice.45 Our results show rMsGal-1 could be detected in the brain of mdx mice after treatment indicating the exogenous protein was able to cross from the blood compartment into the brain parenchyma. Although the current study did not examine if the presence of rMsGal-1 in the brain improved CNS preclinical outcomes, the presence of rMsGal-1 may re-establish components of the dystrophin-associated protein complex and restore pathways involved in CNS function.
DMD patients often have severe progressive cardiomyopathy which is a critical factor in life span. Galectin-1 has been shown to be elevated shortly after myocardial infarction.46 Therefore, it is somewhat surprising that little to no rMsGal-1 or endogenous Galectin-1 was observed in the cardiac tissue of the mdx mice. However, the mdx mouse model does not show any observable signs of cardiac dysfunction until around 9 months of age. Perhaps rMsGal-1 is targeted to damaged tissue and would therefore be present in the cardiac tissue with treatment of older mdx mice.
In this study we have identified Galectin-1 as a novel small protein therapeutic for the treatment of DMD. There are still numerous questions that remain to be answered concerning the therapeutic potential of Galectin-1: (i) Can recombinant Galectin-1 protein therapy prevent cardiomyopathy associated with DMD disease? (ii) Will long-term Galectin-1 protein therapy prevent DMD muscle disease? (iii) Will Galectin-1 protein treatment be an effective therapeutic after disease onset? (iv) Can recombinant Galectin-1 manufacturing be scaled up to treat DMD patients? (v) Can Galectin-1 improve muscle repair and engraftment of myogenic cells? (vi) Can Galectin-1 improve CNS function in DMD patients? (vii) Is Galectin-1 unique or do other Galectins (e.g., Galectin-3) also have therapeutic potential for the treatment of DMD? Although future research is needed to answer these questions, this study indicates Galectin-1 may represent a novel class of protein therapeutic for DMD which may have applications for the treatment of other muscle diseases.
Materials and Methods
Recombinant mouse Galectin-1 production. The mouse Galectin-1 cDNA was produced using standard reverse transcriptase (Superscript III, Invitrogen, Grand Island, NY) from mouse muscle total RNA (Trizol, Invitrogen) followed by PCR using Platinum Taq Supermix (Invitrogen). This PCR product was then subcloned into the pGEM T-Easy vector, sequenced and compared to NCBI database sequence, and finally cloned into the pET23b vector (EMD Millipore, Billerica, MA) in frame with the 6x Histidinetag. This vector was then transfected into Rosetta E. coli (EMD Millipore), grown and induced with 0.4 mmol/l IPTG (Invitrogen) to express mGalectin-1. Galectin-1 was then purified as described in the pET vector handbook using the cobalt Talon Metal Affinity Resin (Clontech 635502, Mountain View, CA) in a disposable column (Thermo Scientific, Grand Island, NY) and imidazole (Sigma-Aldrich, Saint Louis, MO) buffer for elution. Purified rMsGal-1 was filtered through a 0.4 µm filter, dialyzed three times for 1 hour in PBS at 4 °C and used in various experiments.
Tissue culture. C2C12 myoblasts and myotubes were grown as previously described33 Human DMD myoblast and myotubes from the same patient were originally isolated and maintained as described.47 Briefly, C2C12 myoblasts were grown and maintained in DMEM without phenol red (GIBCO, Grand Island, NY), 20% FBS (Atlanta Biologicals, Lawrenceville, GA), 0.5% chick-embryo extract (CEE, Seralab, West Sussex, UK), 1% L-glutamine (GIBCO, Grand Island, NY) and 1% penicillin/streptomycin (PS) (GIBCO). All myoblasts were maintained below 70% confluence until use in assay. C2C12 myoblasts were differentiated into myotubes in DMEM without phenol red (GIBCO), 1% horse-serum, and 1% Penicillin/Streptomycin (P/S) + L-Glutamine. All cells were maintained in Heracell 150i tissue culture (Thermo Scientific) incubators at 37 °C with 5% CO2.
Recombinant mouse Galectin-1 in vitro treatment. C2C12 and human DMD myoblasts were treated with various amounts of recombinant mouse Galectin-1 (rMsGal-1) for 48 hours, washed in PBS, and lysed in RIPA buffer with protease inhibitor cocktail set III (Calbiochem, Billerica, MA) for western blotting. C2C12 and human myotubes were treated with different amounts of rMsGal-1 subsequent to 3–5 days in differentiation media after myotubes were formed.
Mouse maintenance and treatments. In this study mdx mice (strain C57BL/10ScSn-DMDmdx/J) and wild-type mice (strain C57BL/10ScSn/J) were housed and experiments performed under an approved protocol from the University of Nevada, Reno Institutional Animal Care and Use Committee under guidelines set forth by NIH. Genotype of mdx mice were verified as described48 or by dystrophin immunofluorescence on all mdx mice. 100 µl volume rMsGal-1 (1.5 or 150 µg) was delivered into the left mouse TA muscle by IM injection with an equal volume of PBS delivered to the right at 10 days of age. Mice were then sacrificed at 4–5 weeks of age and the TA muscles were removed for use in other experiments. IP treatments were started at 10 days of age and given weekly with either 5 mg/kg/weekly rMsGal-1, 20 mg/kg/weekly rMsGal-1, 20 mg/kg/bi-weekly rMsGal-1 or with corresponding volume of PBS as controls. Mice received weekly treatments from 10–70 days of age, were sacrificed and tissue harvested at the end of study. All treatments have been well tolerated with no observed side-effects.
ELISA. The mouse Galectin-1 DuoSet ELISA development kit (R & D Systems, Minneapolis, MN) was used to determine the levels of mGalectin-1 in serum of mdx and wild-type mice following manufacturer's directions. Plates were read on the Victor V plate reader made by Perkin Elmer (Waltham, MA). Half-life was determined using GraphPad Prism 5 software nonlinear curve fit one-phase decay with the plateau constrained at 394 (pg/ml average noninjected). As mGalectin-1 concentration peaked at 2 hours; only points at or beyond 2 hours were used to determine curve (Figure 2a).
Physiological assays. Mouse body mass was measured weekly prior to treatment. Distance traveled, resting time, large ambulatory time, and vertical sensor breaks were assessed using the Opto-Varimex-4 System with Auto-track v4.96 software for a 30-minute time period (Columbus Instruments, Columbus, OH).
Muscle contractile properties: muscle preparation and force measurements. Mice were deeply anesthetized using isoflurane. The EDL and soleus muscles were prepared following as described.49 Briefly, muscles were mounted in an oxygenated tissue bath in a physiologic salt solution49 and hung from a computer-controlled Aurora Scientific servomotor. After 10 minutes of acclimation in bath without electrical stimulation each muscle received three isometric twitch and three isometric tetanus contractions at 150 Hz followed by force frequency protocol at 10, 30, 50, 65, 80, 100, 120, and 150 Hz (Treat-NMD SOP, DMD_M.1.2.002). There was a 10-minute rest between each set of experiments. L0 was determined as the muscle length at which the maximal twitch force was elicited and was usually 1 g. For all experiments, an electrical stimulation of 7 V with a 200 μs pulse duration (701A Stimulator; Aurora Scientific, Aurora, Ontario, Canada) were delivered by a pair of platinum electrodes. Data were analyzed as described in Treat-NMD SOP, DMD_M.1.2.002.
Mouse digital radiography. Digital radiography was performed blinded by a member of the University of Nevada Reno's Lab Animal Medicine staff on 10-week-old mdx mice using a Sound-eklin tru/Digital radiography machine. Spinal curvature was assessed using the kyphotic index (KI) measurement, which is defined as the measure of the caudal margin of the last cervical vertebra to the caudal margin of the sixth lumbar vertebra divided by the perpendicular distance to the dorsal edge of the vertebra at the point of greatest curvature.17 Measurements from the radiographs were taken using both the Sound-eklin eSeries software (to draw 90 °angles, lines and scale bars) and ImageJ software to convert measurements to millimeters.
Histology. Using a Leica CM1850 cryostat 10-μm sections of Tissue-TEK Optimal cutting Temperature compound (Sakura Finetek USA, Torrance, CA) embedded tissues from mice were placed on Surgipath microscope slides (Surgipath Medical Industries, Richmond, IL). Hemotoxylin and eosin (H&E) staining and CLN was performed on the TA of IM-treated mice as previously described50 and images were taken using an Olympus Fluoview FV1000 Laser Confocal Microscope. TA muscle sections from IP-injected mice were poststained with a 0.003% EBD (Sigma) solution for 20 minutes at room temperature. Tissue was then fixed using 3.7% paraformaldehyde, washed and myofibers were outlined using Oregon Green-488 labeled wheat germ agglutinin conjugates (WGA, W6748, 1:250, Molecular Probes, Invitrogen detection technologies). Pictures of an entire TA section were taken at 100× using a Zeiss Axioskop 2 Plus fluorescent microscope, Zeiss AxioCam HRc digital camera (San Diego, CA), and Axiovision 4.8 software or an Olympus FluoviewFV1000 Laser scanning biological confocal microscope using the Olympus micro FV10-ASW 3.1 software. Compiled images were used to reconstruct a view of the entire TA muscle. This compilation was used for calculating CLN, number of fibers and minimum Feret's diameter. There was a minimum of n = 3 mice for each treatment group and n > 6,200 fibers counted. IP-treated H&E slides were used for qualitative assessment of treated tissue.
Western blotting. Protein concentrations of extracts from myoblast, myotubes and mouse TA tissue were isolated and extracted as previously described.51 Protein was quantified using the Pierce BCA Assay kit (Thermo Scientific), loaded at identical concentrations into 4–12% SDS-PAGE gels and separated under standard conditions. Proteins were then transferred to nitrocellulose (GE Healthcare Life Sciences, Whateman, Pittsburgh, PA) and probed using the following rabbit or goat polyclonal or mouse monoclonal antibodies: α7A Integrin,52 α7B Integrin,52 Galectin-1 (LGALS, Aviva System Biology 1:500, San Diego, CA), β1D integrin,53 α-dystroglycan (IIH6 C4, 1:50, Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA), glyceraldehyde 3-phosphatae dehydrogenase (GAPDH V-18, sc-20357, 1:200), β-dystroglycan H-242 (sc-28535, 1:200), α-sarcoglycan, (IVD3(1) A9 c, DSHB,1:100), β-sarcoglycan H-98 (sc-28279, 1:200), γ-sarcoglycan Z-24 (sc-133984, 1:100), δ-sarcoglycan H-55 (sc-28281, 1:100), ɛ-sarcoglycan H-67 (sc-28282, 1:100, all sc antibodies are from SantaCruz Biotechnology, Dallas, TX), sarcospan (gift from Dr. Rachelle H. Crosbie-Waston, UCLA, 1:5), dystrophin (MANDRA1,1:50, DSHB), utrophin (MANCHO3, 1:50, DSHB), and α-tubulin (DM1A, ab7291, Abnova, Walnut, CA 1:500). Primary antibodies were detected using Alexa Fluor 680 goat anti-rabbit IgG, Alexa Fluor 800 donkey anti-rabbit IgG, Alexa Fluor 800 goat anti-mouse-IgG, Alexa Fluor 800 or 680 donkey anti-goat-IgG (1:5,000, Li-Cor Biosciences or Molecular Probes, Invitrogen detection technologies) in 2.5% milk, 0.02% Sodium Azide solution for 1 hour. Prior to blocking a representative number of immunoblots were treated with Swift Membrane Stain (G. Biosciences, Saint Louis, MO) or Ponceau S stain to normalize for sample loading. Band intensities for all antibodies were determined using ImageJ software and normalized to bands visualized using either α-tubulin or GAPDH as indicated.
Immunofluorescence. Tissues were embedded in Tissue-Tek OCT and 10 µm transvers sections were cut using a Leica CM 1850 cryostat (Leica, Wetzal, Germany). Sections were placed on precleaned Surgipath slides (Surgipath Medical Industries, Richmond, IL) and fixed using methanol, acetone, 4% paraformaldehyde (PFA) and/or 4% formaldehyde. The Mouse on Mouse (M.O.M.) kit was used with all mouse antibodies according to the package instructions (FMK-2201, Vector Laboratories, Burlingame, CA). Mouse primary antibodies were applied overnight followed by a fluorescein isothiocyanate (FITC)-conjugated rabbit-anti-mouse-IgG secondary antibody (1:5,000; Li-Cor Biosciences). All other sections were blocked in 5% bovine serum albumin (BSA, Fisher Scientific, Grand Island, NY) and allowed to incubate overnight at 4 °C. Antibodies were diluted in 1% BSA, except α7 integrin (CA5.5) which was applied for 1 hour at room temperature. The following primary antibodies were used: FITC-α7 (CA5.5, 1:1,000, Sierra Biosource), 6x His-Tag (Thermo Scientific, 1:25), nNOS (Abcam, Cambridge, MA 1376, 1:100), laminin-α2 (Sigma L0663, 1:200), and β1D integrin (1900 Millipore, 1:40) in addition to antibodies previously listed. Secondary antibodies were applied for 1 hour followed by a FITC-conjugated anti-rabbit-IgG (1:5,000; Li-Cor Biosciences, Lincoln, NE). All immunofluorescence experiments were performed with secondary only antibody controls in order to test auto-fluorescence. Slides were mounted using Vectashield Hard Set with DAPI (Vector Laboratories, Burlingame, CA). Images were captured using a Zeiss Axioskop 2 Plus fluorescent microscope, Zeiss AxioCam HRc digital camera, and Axiovision 4.8 software or an Olympus FluoviewFV1000 Laser scanning biological confocal microscope using the Olympus micro FV10-ASW 3.1 software.
Hydroxyproline assay. Hydroxyproline assay was performed following the procedure outlined in Giordano et al.54 All chemicals were purchased from Sigma-Aldrich. Briefly, between 5–10 mg of trimmed costal diaphragm muscle was homogenized in 0.5 mL 0.5M acetic acid and dried in a speed vac. The tissue was then transferred into a glass vial and incubated in 1 ml 6N HCl O/N at 110 °C. The next day the vial caps were removed and HCl evaporated at 110 °C, then brought up in citrate-acetate buffer at 50 μl/1 mg and hydroxyproline analyzed with Chloramine T/Ehrlich's buffers as previously described.54 Results were compared to standards ranging from 1 to 10 μg hydroxyproline. Results were graphed using Graphpad Prism as previously described.
Statistical analysis. All statistical analysis was performed using GraphPad Prism 5 software. Averaged data are reported as the mean ± the standard error of the mean. Individually reported data points were reported at mean ± standard deviation. Comparison for two groups was performed using a Student's t-test and between multiple groups using one-way analysis of variance with Bonferroni post-test with P < 0.05 considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. rMsGal-1 delivered intramuscularly to dystrophin deficient muscle decreased Centrally Located Nuclei (CLN). Figure S2. rMsGal-1 treatment enhanced levels of α7B and β1D integrin in mouse skeletal. Figure S3. hDMD myoblast have increased levels of α7B integrin and β-DG with rMsGal-1 treatment. Figure S4. Skeletal muscle protein outcome measures to determine optimal dosage concentration and regiment in rMsGal-1 treated mdx mice. Figure S5. Immunofluorescent staining for Galectin-1 staining (red) shows it co-localizes with Laminin-α2 staining (green). Figure S6. Myofibers in mdx mice treated with rMsGal-1 were less hypertrophic than those treated with PBS.
Acknowledgments
The authors thank Robert Grange, Virginia Tech University for training and advice with the muscle contraction studies, Priscilla Henson, Annemarie Vogedes, Suzanne Duan, Jordan Tice, Vivian Cruz, and Rebecca Evans for technical assistance. This study was supported by NIH/NIAMS R01AR053697, R01AR064338, R41AR067014, CureCMD, and SAM to D.J.B. P.M.V. is supported by a post-doctoral fellowship from Orphan Disease Center at University of Pennsylvania. R.D.W. was supported by a MDA Development Grant #240684. P.M.V.R., R.D.W., and D.J.B. developed the concept for the studies and designed the experiments. P.M.V.R., R.D.W., and M.K. participated in the production of the recombinant mouse Galectin-1 protein. P.M.V.R. and R.D.W. performed in vitro assays. P.V.R. performed in vivo assays. M.K. and P.V.R. performed functional assays. P.M.V.R., R.D.W., and D.J.B. wrote the manuscript (all authors are affiliated with the University of Nevada School of Medicine). The University of Nevada, Reno, has a patent pending on the therapeutic use of Galectin-1 which has been has been licensed to StrykaGen Corp. The patent inventors are D.J.B., R.D.W., and P.M.V.R. D.J.B. and R.D.W. are the founder and cofounders respectively of StrykaGen Corp.
Supplementary Material
References
- Davies, KE, Smith, TJ, Bundey, S, Read, AP, Flint, T, Bell, M et al. (1988). Mild and severe muscular dystrophy associated with deletions in Xp21 of the human X chromosome. J Med Genet 25: 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, KP (1995). Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80: 675–679. [DOI] [PubMed] [Google Scholar]
- Senter, L, Luise, M, Presotto, C, Betto, R, Teresi, A, Ceoldo, S et al. (1993). Interaction of dystrophin with cytoskeletal proteins: binding to talin and actin. Biochem Biophys Res Commun 192: 899–904. [DOI] [PubMed] [Google Scholar]
- Suzuki, A, Yoshida, M, Hayashi, K, Mizuno, Y, Hagiwara, Y and Ozawa, E (1994). Molecular organization at the glycoprotein-complex-binding site of dystrophin. Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur J Biochem 220: 283–292. [DOI] [PubMed] [Google Scholar]
- Ohlendieck, K, Ervasti, JM, Snook, JB and Campbell, KP (1991). Dystrophin-glycoprotein complex is highly enriched in isolated skeletal muscle sarcolemma. J Cell Biol 112: 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppänen, A, Stowell, S, Blixt, O and Cummings, RD (2005). Dimeric galectin-1 binds with high affinity to alpha2,3-sialylated and non-sialylated terminal N-acetyllactosamine units on surface-bound extended glycans. J Biol Chem 280: 5549–5562. [DOI] [PubMed] [Google Scholar]
- Camby, I, Le Mercier, M, Lefranc, F and Kiss, R (2006). Galectin-1: a small protein with major functions. Glycobiology 16: 137R–157R. [DOI] [PubMed] [Google Scholar]
- Cedeno-Laurent, F and Dimitroff, CJ (2012). Galectin-1 research in T cell immunity: past, present and future. Clin Immunol 142: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerri, DG, Rodrigues, LC, Stowell, SR, Araujo, DD, Coelho, MC, Oliveira, SR et al. (2008). Degeneration of dystrophic or injured skeletal muscles induces high expression of Galectin-1. Glycobiology 18: 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J, O'Donoghue, K, Gavina, M, Torrente, Y, Kennea, N, Mehmet, H et al. (2006). Galectin-1 induces skeletal muscle differentiation in human fetal mesenchymal stem cells and increases muscle regeneration. Stem Cells 24: 1879–1891. [DOI] [PubMed] [Google Scholar]
- Goldring, K, Jones, GE, Thiagarajah, R and Watt, DJ (2002). The effect of galectin-1 on the differentiation of fibroblasts and myoblasts in vitro. J Cell Sci 115(Pt 2): 355–366. [DOI] [PubMed] [Google Scholar]
- Goldring, K, Jones, GE, Sewry, CA and Watt, DJ (2002). The muscle-specific marker desmin is expressed in a proportion of human dermal fibroblasts after their exposure to galectin-1. Neuromuscul Disord 12: 183–186. [DOI] [PubMed] [Google Scholar]
- Georgiadis, V, Stewart, HJ, Pollard, HJ, Tavsanoglu, Y, Prasad, R, Horwood, J et al. (2007). Lack of galectin-1 results in defects in myoblast fusion and muscle regeneration. Dev Dyn 236: 1014–1024. [DOI] [PubMed] [Google Scholar]
- Gu, M, Wang, W, Song, WK, Cooper, DN and Kaufman, SJ (1994). Selective modulation of the interaction of alpha 7 beta 1 integrin with fibronectin and laminin by L-14 lectin during skeletal muscle differentiation. J Cell Sci 107 (Pt 1): 175–181. [DOI] [PubMed] [Google Scholar]
- Grossi, A, Lametsch, R, Karlsson, AH and Lawson, MA (2011). Mechanical stimuli on C2C12 myoblasts affect myoblast differentiation, focal adhesion kinase phosphorylation and galectin-1 expression: a proteomic approach. Cell Biol Int 35: 579–586. [DOI] [PubMed] [Google Scholar]
- McDonald, CM, Henricson, EK, Abresch, RT, Florence, JM, Eagle, M, Gappmaier, E et al.; PTC124-GD-007-DMD Study Group. (2013). The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve 48: 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws, N and Hoey, A (2004). Progression of kyphosis in mdx mice. J Appl Physiol (1985) 97: 1970–1977. [DOI] [PubMed] [Google Scholar]
- Karol, LA (2007). Scoliosis in patients with Duchenne muscular dystrophy. J Bone Joint Surg Am 89 Suppl 1: 155–162. [DOI] [PubMed] [Google Scholar]
- Okano, K, Tsuruta, Y, Yamashita, T, Takano, M, Echida, Y and Nitta, K (2010). Suppression of renal fibrosis by galectin-1 in high glucose-treated renal epithelial cells. Exp Cell Res 316: 3282–3291. [DOI] [PubMed] [Google Scholar]
- Lim, MJ, Ahn, J, Yi, JY, Kim, MH, Son, AR, Lee, SL et al. (2014). Induction of galectin-1 by TGF-β1 accelerates fibrosis through enhancing nuclear retention of Smad2. Exp Cell Res 326: 125–135. [DOI] [PubMed] [Google Scholar]
- Pichavant, C, Aartsma-Rus, A, Clemens, PR, Davies, KE, Dickson, G, Takeda, S et al. (2011). Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther 19: 830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxley, RT 3rd, Pandya, S, Ciafaloni, E, Fox, DJ and Campbell, K (2010). Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol 25: 1116–1129. [DOI] [PubMed] [Google Scholar]
- Berardi, E, Annibali, D, Cassano, M, Crippa, S and Sampaolesi, M (2014). Molecular and cell-based therapies for muscle degenerations: a road under construction. Front Physiol 5: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg, UT, Nicolas-Métral, V, Challet, C, Bernard-Hélary, K, Dorchies, OM, Wagner, S et al. (2002). Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul Disord 12 Suppl 1: S155–S161. [DOI] [PubMed] [Google Scholar]
- Shieh, PB (2013). Muscular dystrophies and other genetic myopathies. Neurol Clin 31: 1009–1029. [DOI] [PubMed] [Google Scholar]
- Tinsley, JM, Potter, AC, Phelps, SR, Fisher, R, Trickett, JI and Davies, KE (1996). Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384: 349–353. [DOI] [PubMed] [Google Scholar]
- Amenta, AR, Yilmaz, A, Bogdanovich, S, McKechnie, BA, Abedi, M, Khurana, TS et al. (2011). Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci USA 108: 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin, DJ, Wallace, GQ, Milner, DJ, Chaney, EJ, Mulligan, JA and Kaufman, SJ (2005). Transgenic expression of {alpha}7{beta}1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am J Pathol 166: 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisleder, N, Takizawa, N, Lin, P, Wang, X, Cao, C, Zhang, Y et al. (2012). Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med 4: 139ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnemann, KJ, Heun-Johnson, H, Turner, AJ, Baltgalvis, KA, Lowe, DA and Ervasti, JM (2009). Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med 6: e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado, ML, Amenta, AR, Hagiwara, H, Rafii, MS, Lechner, BE, Owens, RT et al. (2006). Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J 20: 1724–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta, AR, Creely, HE, Mercado, ML, Hagiwara, H, McKechnie, BA, Lechner, BE et al. (2012). Biglycan is an extracellular MuSK binding protein important for synapse stability. J Neurosci 32: 2324–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney, JE, Gurpur, PB and Burkin, DJ (2009). Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc Natl Acad Sci USA 106: 7991–7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, JL, Chou, E, Oh, J, Kwok, A, Burkin, DJ and Crosbie-Watson, RH (2012). Dystrophin and utrophin expression require sarcospan: loss of α7 integrin exacerbates a newly discovered muscle phenotype in sarcospan-null mice. Hum Mol Genet 21: 4378–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sioud, M, Mobergslien, A, Boudabous, A and Fløisand, Y (2011). Mesenchymal stem cell-mediated T cell suppression occurs through secreted galectins. Int J Oncol 38: 385–390. [DOI] [PubMed] [Google Scholar]
- Ruegg, UT (2013). Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr Opin Neurol 26: 577–584. [DOI] [PubMed] [Google Scholar]
- McDonald, CM, Henricson, EK, Abresch, RT, Florence, J, Eagle, M, Gappmaier, E et al.; PTC124-GD-007-DMD Study Group. (2013). The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 48: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricson, E, Abresch, R, Han, JJ, Nicorici, A, Goude Keller, E, de Bie, E et al. (2013). The 6-minute walk test and person-reported outcomes in boys with duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr 5:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke, MH, Fenichel, GM, Griggs, RC, Mendell, JR, Moxley, R, Florence, J et al. (1989). Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology 39: 475–481. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9: 177–189. [DOI] [PubMed] [Google Scholar]
- Bushby, K, Finkel, R, Birnkrant, DJ, Case, LE, Clemens, PR, Cripe, L et al.; DMD Care Considerations Working Group. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9: 77–93. [DOI] [PubMed] [Google Scholar]
- Heydemann, A and McNally, E (2009). NO more muscle fatigue. J Clin Invest 119: 448–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini, C and Tasca, E (2012). Fatigue in muscular dystrophies. Neuromuscul Disord 22 Suppl 3: S214–S220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman, M (2002). Duchenne muscular dystrophy. J Am Acad Orthop Surg 10: 138–151. [DOI] [PubMed] [Google Scholar]
- Tamma, R, Annese, T, Capogrosso, RF, Cozzoli, A, Benagiano, V, Sblendorio, V et al. (2013). Effects of prednisolone on the dystrophin-associated proteins in the blood-brain barrier and skeletal muscle of dystrophic mdx mice. Lab Invest 93: 592–610. [DOI] [PubMed] [Google Scholar]
- Al-Salam, S and Hashmi, S (2014). Galectin-1 in early acute myocardial infarction. PLoS One 9: e86994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebbles, RD, Sarathy, A, Kornegay, JN and Burkin, DJ (2013). Levels of α7 integrin and laminin-α2 are increased following prednisone treatment in the mdx mouse and GRMD dog models of Duchenne muscular dystrophy. Dis Model Mech 6: 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalfitano, A and Chamberlain, JS (1996). The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the mdx allele. Muscle Nerve 19: 1549–1553. [DOI] [PubMed] [Google Scholar]
- Wolff, AV, Niday, AK, Voelker, KA, Call, JA, Evans, NP, Granata, KP et al. (2006). Passive mechanical properties of maturing extensor digitorum longus are not affected by lack of dystrophin. Muscle Nerve 34: 304–312. [DOI] [PubMed] [Google Scholar]
- Rooney, JE, Welser, JV, Dechert, MA, Flintoff-Dye, NL, Kaufman, SJ and Burkin, DJ (2006). Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci 119(Pt 11): 2185–2195. [DOI] [PubMed] [Google Scholar]
- Rooney, JE, Gurpur, PB, Yablonka-Reuveni, Z and Burkin, DJ (2009). Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am J Pathol 174: 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, WK, Wang, W, Sato, H, Bielser, DA and Kaufman, SJ (1993). Expression of alpha 7 integrin cytoplasmic domains during skeletal muscle development: alternate forms, conformational change, and homologies with serine/threonine kinases and tyrosine phosphatases. J Cell Sci 106 (Pt 4): 1139–1152. [DOI] [PubMed] [Google Scholar]
- Rooney, JE, Knapp, JR, Hodges, BL, Wuebbles, RD and Burkin, DJ (2012) Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol 180: 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano, C, Mojumdar, K, Liang, F, Lemaire, C, Li, T, Richardson, J et al. (2015). Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum Mol Genet 24: 2147–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.