

Abstract
The potential host immune response to a nonself protein poses a fundamental challenge for gene therapies targeting recessive diseases. We demonstrate in both dogs and nonhuman primates that liver-directed gene transfer using an adeno-associated virus (AAV) vector in neonates induces a persistent state of immunological tolerance to the transgene product, substantially improving the efficacy of subsequent vector administration targeting the central nervous system (CNS). We applied this approach to a canine model of mucopolysaccharidosis type I (MPS I), a progressive neuropathic lysosomal storage disease caused by deficient activity of the enzyme α-l-iduronidase (IDUA). MPS I dogs treated systemically in the first week of life with a vector expressing canine IDUA did not develop antibodies against the enzyme and exhibited robust expression in the CNS upon intrathecal AAV delivery at 1 month of age, resulting in complete correction of brain storage lesions. Newborn rhesus monkeys treated systemically with AAV vector expressing human IDUA developed tolerance to the transgene, resulting in high cerebrospinal fluid (CSF) IDUA expression and no antibody induction after subsequent CNS gene therapy. These findings suggest that inducing tolerance to the transgene product during a critical period in immunological development can improve the efficacy and safety of gene therapy.
Introduction
The lysosomal storage diseases (LSDs) are a broad class of inherited disorders caused by deficient activity of enzymes involved in the lysosomal catabolism of ubiquitous polysaccharides, glycoproteins, and lipids, leading to intracellular accumulation of these undegraded enzyme substrates and multiorgan pathology. LSDs are excellent targets for gene therapy because many of the associated lysosomal enzymes can be secreted by genetically corrected cells and endocytosed by neighboring cells, allowing for widespread cross-correction even with modest gene transfer efficiency.1,2 Gene therapy may play a particularly important role in treating the central nervous system (CNS) manifestations associated with LSDs, because the CNS cannot be effectively targeted by intravenous (IV) delivery of the deficient enzymes, and chronic direct CNS administration is impractical as a long-term therapy. One LSD in which gene therapy has shown particular promise for treating CNS disease is mucopolysaccharidosis type I (MPS I), which is caused by deficient activity of the lysosomal enzyme α-l-iduronidase (IDUA). Currently, the only treatment capable of curbing the severe cognitive decline experienced by many MPS I patients is hematopoietic stem cell transplantation, which is associated with substantial morbidity and mortality.3,4,5,6,7,8,9 Using a naturally occurring cat model of MPS I, we previously found that a minimally invasive intrathecal injection of an AAV serotype 9 vector into the cerebrospinal fluid (CSF) achieved widespread gene transfer in the brain and sufficient secretion of the therapeutic enzyme into the CSF to correct storage pathology throughout the CNS.10 Intrathecal AAV delivery could, therefore, represent a vast improvement over the current standard of care for CNS disease in MPS I patients.
Despite the promise of intrathecal AAV delivery for MPS I, we found that the efficacy of gene transfer was diminished in some MPS I cats due to the development of antibodies to IDUA, resulting in reduced circulating enzyme in the CSF and less efficient correction of storage lesions. Here, we report that MPS I dogs, another naturally occurring disease model, also develop antibodies to the normal canine enzyme following intrathecal gene therapy leading to less efficient correction of brain lesions. These findings reflect the clinical experience with enzyme replacement therapy in MPS I, as patients treated with recombinant IDUA almost universally develop antibodies to the enzyme, which correlate with a poor response to therapy.11,12
With the goal of developing a safe and effective method for the prevention of antitransgene immune responses, we explored the possibility of exploiting the normal processes by which the immune system learns to distinguish self from nonself. Decades of evidence from transplantation studies suggest that neonatal rodents and humans, unlike adults, are prone to develop tolerance rather than immunity to alloantigens.13,14,15 While this phenomenon has often been ascribed to an immature and poorly functional immune system, it has since become clear that neonates are indeed capable of eliciting functional immune responses, albeit with higher activation thresholds.16 The development of tolerance to neoantigens in newborns is instead an active process involving peripheral anergy and deletion of reactive T- and B-cells, as well as induction of regulatory T-cells.13,14,17 One recent report demonstrated that this phenomenon could be exploited to induce durable tolerance to factor VIII in hemophilic mice by performing gene transfer in neonates.18 While this effect in mice could be attributed to the relative immaturity of the murine immune system at birth, evidence for neonatal tolerance to a foreign transgene has also emerged in studies using retroviral vectors in newborn dogs.19,20,21 These experiments demonstrated sustained expression of relatively immunogenic transgenes after neonatal retroviral vector administration, although the ineffectiveness of these vectors in adults precluded direct comparison of immune responses to the transgene in animals of different ages.
In this study, we evaluated the potential for neonatal AAV-mediated systemic expression of a therapeutic protein to induce immunological tolerance that could subsequently allow for safe and effective CNS-directed gene therapy. We found that MPS I dogs treated intravenously in the first week of life with an AAV vector expressing canine IDUA from a liver-specific promoter did not develop antibodies to the transgene, and following subsequent intrathecal gene transfer at 1 month of age exhibited 3- to 100-fold higher IDUA levels in CSF than naive dogs, with complete resolution of brain storage lesions. Likewise, rhesus monkeys administered liver-directed human IDUA gene transfer at birth exhibited tolerance to the protein, allowing for robust CSF IDUA expression without antibody induction after intrathecal AAV injection 1 month later. These findings suggest a potential approach to prevent immune responses to a nonself transgene through neonatal gene transfer, which could significantly improve the efficacy of gene therapy for many recessive diseases.
Results
Antibody induction to canine IDUA after intrathecal AAV9-mediated gene transfer in MPS I dogs
The canine model of MPS I faithfully recapitulates many of the manifestations of the human disease.22,23 These animals have no detectable IDUA activity due to a splice site mutation that results in retention of the first intron of IDUA.24 Given the absence of detectable IDUA expression in these animals, we anticipate that they will model the immune response to intrathecal gene therapy that would occur in patients with the severe form of MPS I, as these individuals generally carry alleles that produce no full-length IDUA, leaving them immunologically naive to the protein.25 The brains of MPS I dogs show the characteristic pathology associated with MPS I, including widespread storage of gangliosides such as GM3 in neurons, as well as abnormal accumulation of cholesterol and lysosomal membrane proteins including LIMP2.23 MPS I dogs also exhibit prominent storage of glycosaminoglycans (GAGs) in the meninges, resulting in significant meningeal thickening, a process which contributes to spinal cord compression in some MPS I patients.26,27,28
We initially treated three dogs at 1 month of age with an intrathecal injection of an AAV9 vector carrying the canine IDUA sequence under the control of a ubiquitous promoter (Table 1). The injection was well tolerated in all animals; no clinical signs were observed throughout the study. CSF analyses were generally unremarkable, with only a mild transient elevation of CSF lymphocytes occurring in two animals (Supplementary Table S1). A single CSF sample in one animal showed a marked pleocytosis consisting primarily of monocytoid cells. A subsequent tap showed no evidence of pleocytosis, and at the time of euthanasia, there was no histological evidence of inflammation in the brain or spinal cord of any treated animal.
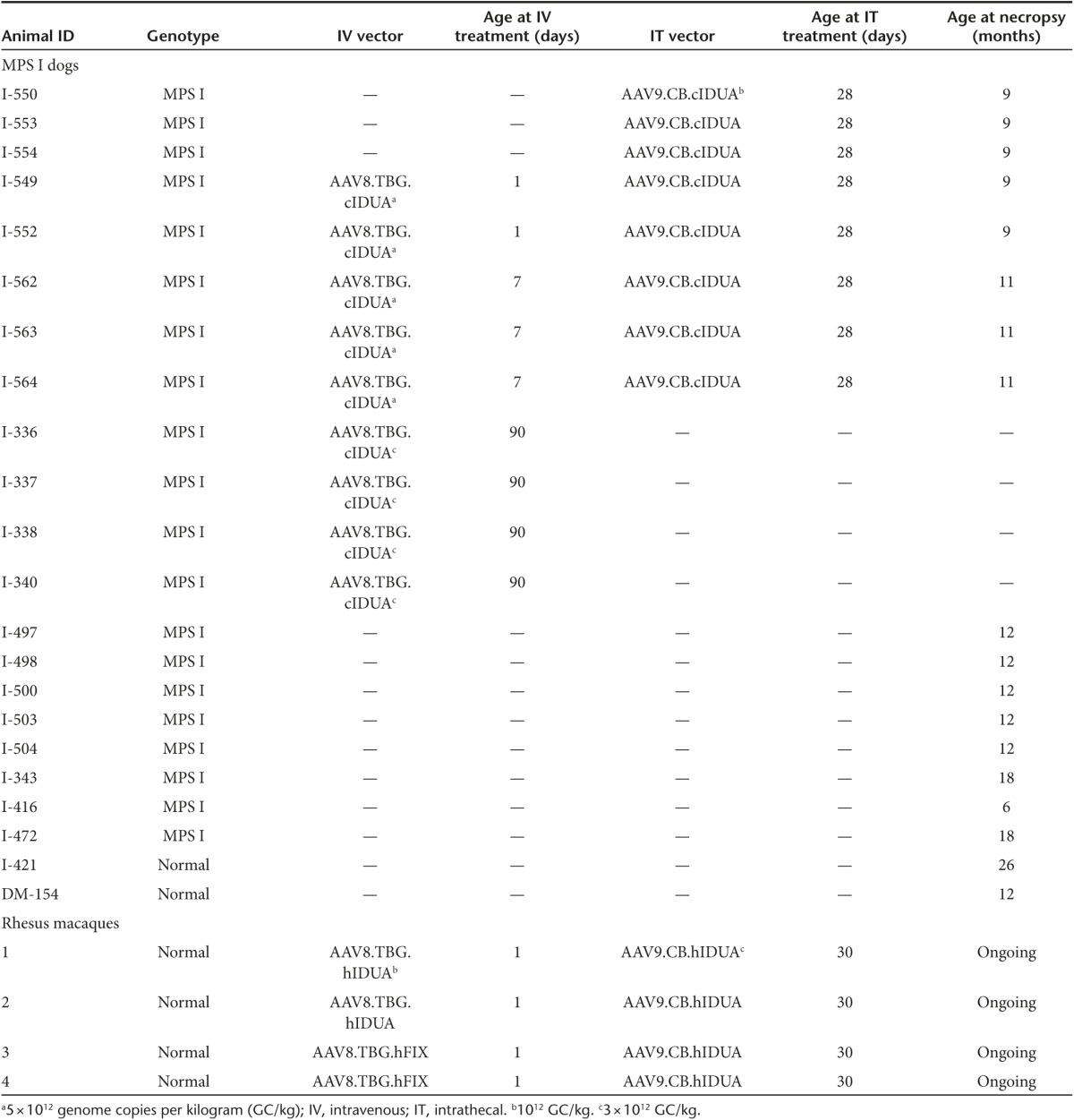
Table 1. Summary of study subjects.
The vector was distributed throughout the CNS, transducing cells in all analyzed regions of the brain and spinal cord (Supplementary Table S2). All animals exhibited supraphysiologic expression of IDUA in CSF, which declined to the normal range in one animal (I-550) and to below normal levels in two animals (I-553 and I-554), after which CSF enzyme levels were essentially stable for 5 months until the animals were euthanized (Figure 1a). The absence of clinical signs, vector genome loss, or histological evidence of encephalitis indicated that the decline in CSF IDUA activity was not due to killing of transduced cells by cytotoxic T lymphocytes, which was also supported by persistent residual CSF IDUA activity. Instead, the decline in CSF IDUA activity was associated with the induction of high titer antibodies against canine IDUA in CSF (Figure 1b).
Figure 1.

Neonatal systemic α-l-iduronidase (IDUA) gene transfer induces tolerance to subsequent CNS gene therapy in MPS I dogs. MPS I dogs were treated with an intrathecal injection of an AAV9 vector encoding canine IDUA at 1 month of age (I-550, I-553, I-554) or were first treated with an intravenous injection of an AAV8 vector encoding IDUA under control of a liver specific promoter on postnatal day 1 (I-549, I-552) or postnatal day 7 (I-562, I-563, I-564) followed by intrathecal vector injection at 1 month of age. (a) Cerebrospinal fluid (CSF) was serially collected from treated animals and assayed for IDUA enzyme activity. The dotted line represents the mean CSF IDUA activity in normal control animals. (b) Antibodies against canine IDUA were detected in CSF samples at baseline and on day 80 after intrathecal vector injection by indirect enzyme-linked immunosorbent assay. Error bars represent standard error of replicate wells. The dashed line is the upper limit of pretreatment samples. The median CSF antibody titers in the animals treated with IT AAV9 alone and those pretreated systemically as neonates were 348 and 2.61, respectively; the distributions in the two groups differed significantly (Mann–Whitney U = 0, n1–n2 = 345.5, P < 0.05 two-tailed). (c) Serum antibodies against canine IDUA were evaluated in the IT-treated animals as well as 4 MPS I dogs treated at 3 months of age with an IV injection of the AAV8 vector alone. The dashed line is the upper limit of naive samples. There was a statistically significant difference in serum anti-IDUA titers between the groups (Kruskal-Wallis H test, χ2(2) = 8.26, P = 0.0027), with a mean rank titer of 8.3 for dogs treated with IT AAV9 at 1 month of age, 3.0 for dogs treated with IV AAV8 as neonates followed by IT AAV9 at 1 month of age, and 9.5 for dogs treated with IV AAV8 at 3 months of age. Serum titers were significantly greater in the animals treated with IV AAV8 at 3 months of age than those treated as neonates (Dunn's test, P < 0.05).
Induction of tolerance to IDUA by neonatal gene transfer
To determine whether neonatal expression of canine IDUA could induce immune tolerance to the enzyme in MPS I dogs, we treated six animals with an IV injection of an AAV serotype 8 vector expressing canine IDUA from a liver selective promoter on either the first (N = 3) or the seventh (N = 3) day after birth (Table 1). One of the dogs treated on postnatal day 1 died 2 days after treatment. Overall survival of neonates was similar to historical data for untreated MPS I dogs, which have approximately 20% mortality in the first 2 weeks of life.19 The cause of this early mortality in MPS I dogs has not been determined; in this treated animal postmortem examination showed systemic lesions typical of MPS I as well as possible evidence of a systemic bacterial infection. Treated animals demonstrated an elevation in serum IDUA followed by a rapid decline (Supplementary Figure S1). This is consistent with observations of transient expression due to vector genome loss during hepatocyte division in previous studies utilizing nonintegrating vectors for hepatic gene transfer in newborns.29
At 1 month of age, the five surviving dogs that received IV AAV8 in the first week of life were given an injection of an AAV9 vector using an intrathecal approach. All five animals exhibited greater than 30-fold normal peak levels of IDUA in CSF following intrathecal vector injection, with long-term CSF enzyme levels 3- to 100-fold higher than those achieved in naive animals (Figure 1a). Antibodies to canine IDUA in the CSF were reduced nearly 100-fold compared to the animals treated with IT AAV9 alone, with undetectable CSF antibodies in the dogs treated on postnatal day 1, and low but detectable responses in the animals treated on postnatal day 7 (Figure 1b). All animals treated as neonates exhibited no detectable serum antibodies against canine IDUA, whereas the animals treated with IT AAV9 alone at 1 month of age had elevated serum anti-cIDUA titers (Figure 1c).
Induction of tolerance to canine IDUA in neonates is not due to hepatic expression
Previous studies in both mice and dogs have demonstrated tolerance induction to foreign proteins through AAV-mediated hepatic expression.30,31,32,33 To determine whether immune tolerance in the animals treated with intravenous AAV8 as neonates was due to liver targeted expression rather than the age of the animal at the time of treatment, we treated four MPS I dogs at 3 months of age with an intravenous injection of the AAV8 vector expressing canine IDUA from a liver-specific promoter. All four animals developed serum antibodies against the transgene product (Figure 1c). This indicates that the lack of antibody response to IDUA in the neonatal gene transfer cohort is related to the age of the animal at the time of exposure to the transgene product, rather than the presence of hepatic expression.
Correction of biochemical and histological abnormalities in the CNS of MPS I dogs
The lysosomal enzyme hexosaminidase (Hex) is upregulated in tissues of MPS I animals, and the elevated Hex activity in both brain tissue and CSF serves as a useful marker for the aberrant cellular processes occurring downstream of IDUA deficiency.10 Measurement of CSF Hex activity at the time of intrathecal vector delivery (~1 month postnatal) revealed abnormally elevated Hex activity in all MPS I dogs (Figure 2a). The animals treated with intrathecal AAV9 alone exhibited modest reductions in CSF Hex activity, with only the animal with the highest residual IDUA expression (I-550) reaching the normal range. All five animals treated with neonatal systemic gene transfer followed by intrathecal vector administration demonstrated complete normalization of CSF Hex. Hex activity in brain tissue samples showed a greater response to therapy than CSF Hex, with substantial reductions in brain Hex activity in all treated animals, although the effect was slightly diminished in the two intrathecal-only treated animals with the lowest CSF IDUA levels (Figure 2b).
Figure 2.

Biochemical markers are normalized following intrathecal gene therapy in MPS I dogs. (a) Cerebrospinal fluid (CSF) samples from treated dogs and untreated controls were analyzed for Hexosaminidase (Hex) activity at the time of intrathecal vector injection (pre) and at the time of tissue harvest (post). The dotted line indicates the upper limit of CSF Hex activity in normal control samples. Changes from baseline were not statistically significant for either group (Wilcoxon signed-rank test). (b) Hex activity was also measured in brain lysates. Values are the mean ± SEM of samples collected from six brain regions (frontal cortex, temporal cortex, occipital cortex, hippocampus, medulla, and cerebellum). There was a statistically significant difference in brain Hex activity between the groups (Kruskal-Wallis H test, χ2(2) = 7.47, P = 0.0076), with a mean rank of 6.3 for dogs treated with IT AAV9 at 1 month of age, 3.4 for dogs treated with IV AAV8 as neonates followed by IT AAV9 at 1 month of age, and 10 for untreated MPS I dogs. Hex activity was significantly reduced in animals treated with IV AAV8 as neonates followed by IT AAV9 at 1 month of age compared to untreated controls (Dunn's test, P < 0.05). (c) Pathogenic GAGs were measured in CSF at the time of intrathecal vector injection and on day 21 and 112 post injection. A two-way analysis of variance showed significant effects of both time relative to IT vector administration (F(1,6) = 15.4, P < 0.05) and neonatal treatment status (F(1,6) = 11.0, P < 0.05) on CSF pGAG concentration, with the animals treated as neonates having significantly lower CSF pGAG concentrations than the IT-only group on day 112 postinjection (Holm-Sidak, P < 0.05). Insufficient data were available from normal dogs to establish a reference range for CSF pGAG concentration.
GAG concentrations in CSF were measured using an assay specific for the nonreducing end of the pathologic GAGs (pGAG) that accumulate due to IDUA deficiency (Figure 2c).34 All animals exhibited a marked reduction in CSF pGAG concentration 3 weeks after intrathecal AAV injection. This reduction was sustained at day 112, although the dogs that were not immune tolerant to IDUA maintained higher residual CSF pGAG than immune tolerant dogs.
Histological analysis revealed severe storage lesions throughout the brains of untreated MPS I dogs, with widespread neuronal accumulation of GM3, cholesterol, and LIMP2 (Figure 3). The animals treated with intrathecal AAV9 alone demonstrated substantial improvements in storage lesions, although only the animal with the highest CSF IDUA (I-550) experienced complete resolution of neuronal storage. The other two intrathecal-treated dogs had residual storage lesions. CNS storage lesions were completely reversed in all five dogs treated with neonatal AAV8 systemic gene transfer followed by intrathecal AAV9 administration.
Figure 3.

CNS storage lesions are more effectively cleared by intrathecal gene therapy in MPS I dogs tolerized to α-l-iduronidase (IDUA) as neonates. (a) Representative brain sections are shown for normal, untreated MPS I, and tolerized intrathecal treated MPS I dogs. For the three animals treated with AAV9 alone, images are shown for each animal. Immunostaining was performed on cortical brain sections for the ganglioside GM3 and the lysosomal membrane protein LIMP2. Unesterified cholesterol was detected by filipin stain. GAG storage in meninges is stained with Alcian blue. Scale bar = 200 µm. (b) Automated quantification was performed on GM3, and (c) LIMP2, and filipin stained sections. Values are the mean ± SEM of 10 sections. There was a statistically significant difference in GM3 storage between the groups (Kruskal-Wallis H test, χ2(2) = 9.0, P = 0.0019), with a mean rank of 5.7 for dogs treated with IT AAV9 at 1 month of age, 3.8 for dogs treated with IV AAV8 as neonates followed by IT AAV9 at 1 month of age, and 11 for untreated MPS I dogs. GM3 was significantly reduced relative to MPS I controls in animals treated with IV AAV8 as neonates followed by IT AAV9 at 1 month of age (Dunn's test, P < 0.05). Insufficient control sections were available for statistical comparisons of LIMP2 or filipin staining.
In addition to the storage lesions in the brain parenchyma, untreated MPS I dogs showed accumulation of GAGs in meninges visible by Alcian blue stain (Figure 3). This meningeal GAG accumulation and the resulting thickening of the meninges is implicated in many cases of spinal cord compression requiring surgical intervention, and also likely contributes to the development of communicating hydrocephalus in some MPS I patients by interfering with normal routes of CSF resorption. All treated animals showed evidence of improvement in meningeal GAG storage. While the meninges appeared almost completely normal in all tolerant dogs and one nontolerant dog, the two nontolerant animals with the lowest CSF IDUA activity retained some meningeal GAG storage.
Induction of tolerance to human IDUA in newborn Rhesus Macaques
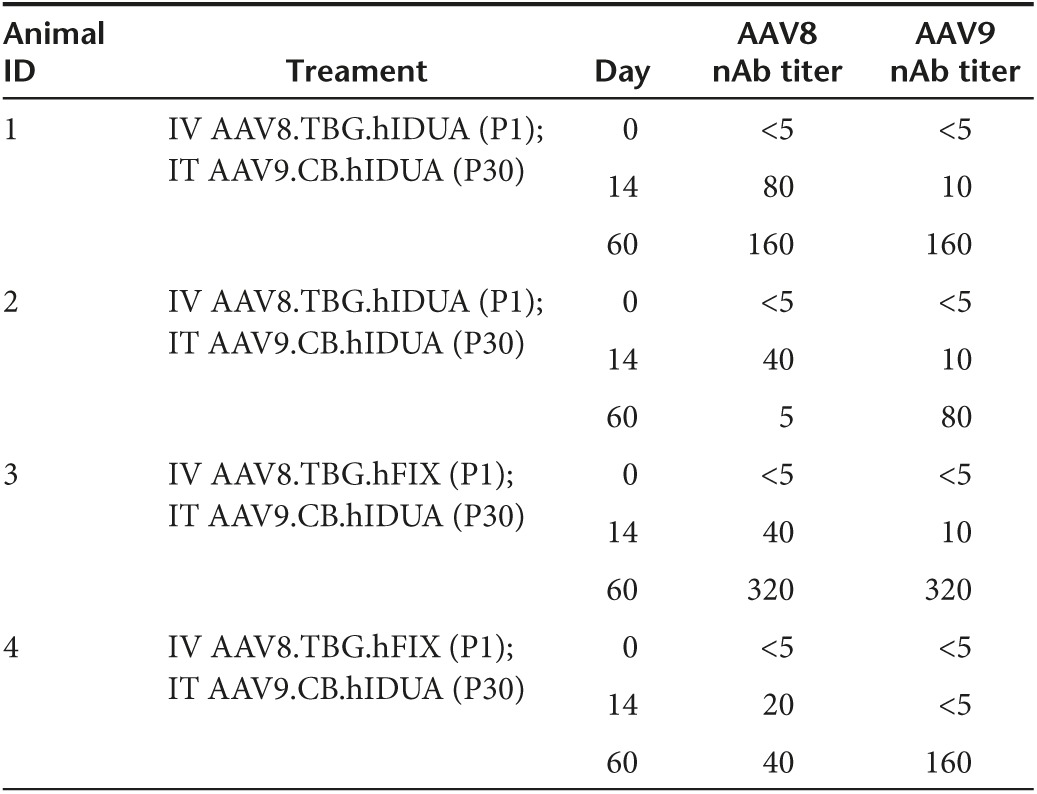
To assess whether the neonatal window for immune tolerance induction that was observed in MPS I dogs could also be found in primates, a similar study was performed in newborn rhesus monkeys (N = 4). Because these animals are not IDUA deficient, the human IDUA transgene was used to model the immune response that might be expected against a species-specific transgene in a patient lacking the endogenous protein. Two newborn rhesus monkeys were administered AAV8 vector expressing human IDUA from a liver-specific promoter IV at birth. Both demonstrated a brief increase in serum IDUA activity (Supplementary Figure S2). Two additional newborns were administered an AAV8 vector expressing an irrelevant transgene (human factor IX) IV at birth. All four animals were administered AAV9 vector expressing human IDUA at 1 month postnatal age by intrathecal injection. Similar to the MPS I dogs, the IDUA naive animals exhibited declining CSF IDUA activity 3 weeks after injection, with a return to near baseline levels by 2 months postadministration (Figure 4a). These animals also developed transgene-specific antibodies in the CSF (Figure 4b). The two animals administered IDUA gene transfer IV at birth did not develop antibodies to human IDUA in CSF (Figure 4b), and maintained CSF enzyme activity greater than 10-fold normal (Figure 4a) 2 months after intrathecal AAV9 administration. Unlike the kinetics of CSF IDUA expression observed in dogs, CSF IDUA activity continued to increase 60 days after IT vector administration, although overall expression was lower in nonhuman primates (NHPs) as previously described.35 The animals developed neutralizing antibodies to the vector capsids following both the IV administration at birth and the subsequent IT injection (Table 2). All animals remained robust and healthy during the study period with no evidence of adverse effects, normal growth trajectories, and complete blood counts and chemistry panels within normal limits based on age and when compared to historical controls.
Figure 4.

Neonatal gene transfer induces tolerance in nonhuman primates. Four newborn rhesus monkeys were administered an AAV8 vector expressing either human α-l-iduronidase (IDUA) (Animal #s 1 and 2) or human factor IX (3 and 4) intravenously. All animals were administered AAV9 expressing human IDUA using the intrathecal approach at 1 month postnatal age. (a) IDUA activity was measured in cerebrospinal fluid (CSF) after intrathecal vector injection. The dotted line represents the mean IDUA activity in pretreatment CSF samples. (b) Antibodies were detected in CSF by indirect enzyme-linked immunosorbent assay (ELISA) 2 months after intrathecal vector administration. The dashed line indicates the upper limit of baseline samples. Error bars represent SEM. ELISA values were significantly lower for animals treated as neonates with an IDUA vector compared to those treated with the control vector (unpaired t-test with Welch's correction, t = 38.72, P < 0.05 two-tailed).
Table 2. AAV neutralizing antibody titers in NHP serum.
Discussion
Immune activation to a wild-type therapeutic protein is a potential obstacle to the successful treatment of any recessive disease. Antibody responses to protein replacement therapy have been particularly challenging for some LSDs, as antibodies can interfere with the distribution and uptake of the intravenously delivered enzyme.12 Antibodies may be equally problematic for gene therapies targeting these disorders, as they can interfere with cross-correction mediated by enzyme secreted from transduced cells.
In this study, we demonstrated that intrathecal AAV9 delivery can effectively target cells throughout the CNS in dogs and achieve sufficient expression to correct the biochemical and histological abnormalities associated with MPS I in the brain of a large animal. Previous work has demonstrated that even doses of intrathecal AAV9 much greater than those employed in this study result in transduction of a small fraction of cells in the canine brain, suggesting that the widespread reduction in storage pathology observed was due to cross-correction by secreted enzyme.36 However, of the three animals treated with intrathecal vector alone, two developed sufficiently robust antitransgene antibody responses to prevent complete resolution of CNS storage lesions. Only the animal that maintained near-normal CSF IDUA activity after antibody induction to the transgene demonstrated a complete response to CNS gene therapy. From this outcome, we conclude that IDUA activity in CSF is a reasonable predictor of efficacy following intrathecal gene transfer, with approximately normal levels required for full therapeutic benefit. This is consistent with our findings with intrathecal gene therapy in MPS I cats.10 MPS I cats generally exhibited weaker antibody responses to intrathecal gene transfer and more stable CSF IDUA activity than MPS I dogs. This may relate to the underlying mutation in the two models, as MPS I cats express an inactive mutant IDUA, potentially rendering them partially immunologically tolerant to the enzyme. Importantly, the present data in MPS I dogs indicate that even for MPS I patients with severe disease who, like the dogs, have no residual IDUA expression, the antitransgene antibody response that may occur after intrathecal gene transfer does not result in adverse clinical events, and substantial efficacy is retained despite the antibody response. However, these data also suggest that preventing antibody responses against IDUA in the CNS could improve the efficacy of intrathecal gene therapy for MPS I.
Using liver-directed gene transfer, we tested the effect of early exposure to IDUA on subsequent immune responses following intrathecal gene therapy. Neonatal IDUA expression induced tolerance to the enzyme in MPS I dogs, which markedly increased CSF enzyme levels achieved with intrathecal gene therapy at 1 month of age. The high CSF IDUA levels in the immune tolerant group consistently resulted in complete reversal of neuropathology, providing a strong example of the efficacy that is possible with intrathecal gene therapy for LSDs when interfering antibody responses are overcome. The finding that this neonatal window for induction of immune tolerance to a transgene also exists in nonhuman primates appears promising for translation to the clinic. Interestingly, neutralizing antibodies were elicited against the vector capsid in newborn NHPs, demonstrating that while neonates may be relatively tolerant to foreign proteins, they are fully capable of eliciting functional immune responses to pathogens. This finding has important clinical implications; while neonatal gene transfer may provide a method to circumvent immunity to the transgene, it does not provide a means of avoiding the antibody response to the AAV capsid. Thus vector readministration—a likely necessity in the setting of a life-saving gene therapy administered to a newborn—will depend on the development of alternative serotypes or other methods to evade capsid antibodies. In the case of intrathecal AAV delivery, readministration may be possible due to the remarkable insensitivity of this approach to pre-existing capsid antibodies.36
Previous studies in mice, cats, and nonhuman primates have demonstrated vector escape to the peripheral circulation following intrathecal AAV delivery, often resulting in significant hepatic gene transfer and therapeutic levels of systemic transgene expression.10,36,37 We did not directly evaluate somatic disease correction in treated MPS I dogs due to the confounding effects of anti-IDUA antibodies in the naive cohort or previous systemic gene transfer in the immune-tolerant cohort. Notably we observed minimal hepatic gene transfer following intrathecal AAV9 injection, consistent with the relatively low permissivity of canine liver to AAV-mediated transduction.38 Given the low level of hepatic gene transfer and serum IDUA activity in treated dogs, improvement in somatic disease is unlikely. However, it should be emphasized that this inefficient peripheral transduction is inconsistent with that observed following intrathecal AAV delivery in other species, and may be specific to the canine model.
There are several limitations of the present study. Due to the increased risks associated with performing intrathecal vector injections in newborn MPS I pups, we chose to use systemic gene transfer as a means of inducing tolerance rather than performing CNS directed gene therapy in neonates. We therefore cannot conclude whether route of administration plays a role in the induction of tolerance. Hepatic gene transfer has often been associated with transgene-specific tolerance, although we observed antibody responses against IDUA in 3-month-old MPS I dogs treated with AAV8-mediated hepatic gene therapy, indicating that liver-directed gene transfer alone is insufficient to induce tolerance in this model.30,31,32,33 In this study, we also did not rule out the possibility that prior liver directed gene therapy contributed to the improved correction of brain pathology in immune tolerant animals, although this appears unlikely given that IDUA was undetectable in CSF in these animals at the time of intrathecal vector injection, and CSF Hex activity and pGAG concentration showed no evidence of correction before intrathecal gene transfer. This is consistent with our studies in MPS I cats, in which even extremely high serum IDUA activity had no impact on brain lesions.39 Additionally, while we focused this study entirely on the observation of neonatal tolerance and its potential utility for therapeutic applications, we did not evaluate the mechanisms responsible for this phenomenon. This model could serve as a powerful tool for better understanding the development of immunological recognition of self and nonself, and future studies should explore possible mechanisms. Finally, the present study did not define the temporal window in which tolerance induction is possible. Based on the observation that detectable antibody responses began to appear in the MPS I dogs treated on postnatal day 7, we estimate that this period lasts no more than 1 to 2 weeks, which could serve as a useful starting point for human studies.
If human neonates are found to exhibit the same potential for transgene-specific immunological tolerance that we have demonstrated in dogs and nonhuman primates, neonatal gene transfer could have enormous potential to treat many genetic disorders for which immune responses limit the safety or efficacy of therapy. In order for clinical trials to be feasible, newborn screening will be essential for identifying patients sufficiently early for this approach to be effective. For MPS I, newborn screening is now being implemented in several states, providing a potential opportunity to conduct first-in-human trials.40
Materials and Methods
Vector production. The AAV8 vectors contained codon-optimized canine or human IDUA cDNA downstream of the thyroid hormone binding globulin promoter. The AAV9 vector contained the chicken β-actin promoter with a cytomegalovirus immediate early enhancer. Both vectors included the rabbit β-globin polyadenylation sequence. Vectors were produced by triple transfection of 293 cells and purified on iodixanol gradients as previously described.41
Study approval. All MPS I dog study protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. All nonhuman primate procedures conformed to the requirements of the Animal Welfare Act and protocols were approved prior to implementation by the Institutional Animal Care and Use Committee at the University of California, Davis.
MPS I dogs. The MPS I dog colony was maintained at the University Of Pennsylvania School Of Veterinary Medicine under NIH and USDA guidelines for the care and use of animals in research. All MPS I dog study protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. For vector injections in neonatal MPS I dogs, the AAV8 vector was diluted in 0.5–1 ml of sterile saline, and injected via the jugular vein. Intrathecal injections of AAV9 vectors and CSF collection were performed via the suboccipital approach. Animals were anesthetized with propofol and intubated, and the suboccipital region was clipped of hair and scrubbed. Suboccipital puncture was performed with a 22-gauge spinal needle using sterile technique. Needle placement was confirmed by CSF flow, and 1–2 ml was collected and immediately frozen. For vector injections, a syringe containing the vector diluted in 1–2 ml phosphate-buffered saline was connected to the spinal needle and slowly injected by hand. Animals were monitored to confirm complete recovery after the procedure. A total of 13 MPS I dogs were included in this study. Genotype was confirmed at birth by PCR and serum enzyme assay. Six dogs were administered an IV injection of the AAV serotype 8 vector (5 × 1012 genome copies per kilogram (GC/kg) body weight) on either the first (N = 3) or seventh (N = 3) day of life. One animal died on postnatal day 3. The remaining five treated animals as well as three naive MPS I dogs were treated with intrathecal AAV9 (1012 GC/kg) at 1 month of age. An additional four animals were treated only with the IV AAV8 vector at 3 months of age. Blood was collected from a peripheral vessel weekly for the first 7 weeks of life then monthly thereafter. CSF (1–2 ml) was collected at the time of intrathecal vector injection (1 month of age), on days 7 and 21 after injection, and monthly thereafter. Euthanasia was performed by administration of sodium pentobarbital (80 mg/kg IV). Five animals (I-549, I-550, I-552, I-553, I-554) were euthanized at 9 months of age; 3 (I-562, I-563, I-564) were euthanized at 11 months of age. Untreated MPS I and controls were euthanized between 6 and 26 months of age (Table 1). Tissues were collected and processed as previously described.10
Rhesus monkeys. All animal procedures conformed to the requirements of the Animal Welfare Act and protocols were approved prior to implementation by the Institutional Animal Care and Use Committee at the University of California, Davis. Activities related to animal care were performed as per California National Primate Research Center standard operating procedures. Normally cycling, adult female rhesus monkeys (Macaca mulatta; N = 4) with a history of prior pregnancy were bred and identified as pregnant, using established methods.42 All dams selected for the study were prescreened to ensure that they were seronegative for AAV antibodies. Fetuses were monitored sonographically during gestation to confirm normal growth and development42 and newborns were delivered by cesarean section at term (160 ± 2 days gestation) according to established protocols.43 Newborns were placed in incubators postdelivery and nursery-reared for the study. Infant health, food intake, and body weights were recorded daily or weekly (dependent on age) in the nursery according to established protocols. At birth, all animals were administered the selected AAV vector IV. At 1 month postnatal age and at subsequent monthly time points (up to 2 months post-transfer, to date) infants were sedated with ketamine (10 mg/kg intramuscularly (IM)) and dexmedetomidine (0.015–0.075 mg/kg IM) in preparation for collection of CSF (~0.5 ml; preinjection then weekly or monthly) and for intrathecal injection via the suboccipital approach (~0.5 ml volume; 1 month and immediately after collection of CSF), all under aseptic conditions. Blood samples were collected at birth then monthly from a peripheral vessel (~3–6 ml) to monitor complete blood counts and clinical chemistry panels, and for collection of serum and plasma. The reversal atipamezole was given IM at a comparable dose to dexmedetomidine when sample collection was completed.
Vector biodistribution. At necropsy, tissues for vector biodistribution were quickly dissected and frozen on dry ice. For brain samples, small pieces were dissected from the frontal, temporal, and occipital cortices as well as the medulla and cerebellum of the left hemisphere. The entire left hippocampus was collected. Samples were stored at −80 °C until the time of analysis. DNA was isolated from tissues using the QIAmp DNA Mini Kit and vector genomes quantified by TaqMan PCR as described.41
Enzyme activity assays. Assays for IDUA and Hex activity were performed as described.10
CSF pGAG measurement. CSF pGAG measurement was performed by the Glycotechnology Core at the University of California, San Diego using previously described methods.34 Briefly, GAG was extracted from CSF samples and digested to disaccharides with heparinase I, II, and III. Disaccharides were tagged with aniline 12C by reductive coupling and dried by speed vac. Dried samples were reconstituted in LC-MS grade water and spiked with a known concentration of 12C-aniline tagged standard. Samples were analyzed on a LTQ Orbitrap Discovery electrospray ionization mass spectrometer (Thermo Scientific, Waltham, MA) equipped with Thermo Scientific Ultimate 3000 HPLC system.
Enzyme-linked immunosorbent assay. A C-terminal histidine tag was added to the canine IDUA cDNA by PCR. The tagged cDNA was cloned into an expression cassette driven by a TBG promoter. This plasmid was transfected into 90% confluent 10-cm plates of Huh7 cells using lipofectamine 2000 (Invitrogen, Carlsbad, CA). Supernatant was collected twice at 24-hour intervals; each time the supernatant pH was immediately titrated to pH 5.8 and stored at 4 degrees. The enzyme was purified on a 1 ml HisTrap FF column (GE). The eluted fractions were immediately adjusted to pH 5.8. The fractions containing purified canine IDUA were identified by enzyme assay and SDS–PAGE. The purified protein was incubated at 3 µg/ml in phosphate-buffered saline, pH 5.8 on polystyrene enzyme-linked immunosorbent assay plates overnight at 4 degrees. The plates were washed twice in phosphate-buffered saline, pH 5.8, blocked in 3% BSA, pH 5.8, and then incubated with diluted samples for 1 hour at room temperature. The plates were washed five times, incubated 1 hour with a 1:10,000 dilution of horseradish peroxidase (HRP)-conjugated sheep anti-canine (Pierce, Rockford, IL) in blocking solution, washed five times, and developed using tetramethylbenzidine (TMB) substrate. Titers are based on a standard curve of a serially diluted positive control sample. The assay for antibodies to human IDUA in rhesus monkeys was identical, except that Aldurazyme (Genzyme, Cambridge, MA) 10 µg/ml, was used for coating antigen and the detection antibody was polyclonal goat anti-human (Jackson ImmunoResearch Laboratories, West Grove, PA).
Neutralizing antibody assay. Neutralizing antibodies were evaluated in NHP serum as previously described.44
Histology. Histological analysis of MPS I dog brains was performed as previously described10 with the following modifications for quantifying neurons positive for GM3, cholesterol, and LIMP2 storage: Images of LIMP2- and filipin-stained sections of cerebral cortex were taken with a 10× objective such that the border between layer I (molecular layer) and layer II formed the upper border of the image. A total of 10 images were acquired from each animal. Images of GM3-stained brain sections were taken with a 4× objective from the area directly below the cerebral cortex surface including the cerebral molecular layer. Seven images from each animal were analyzed. All images were processed with ImageJ software (Rasband W. S., National Institutes of Health; http://rsb.info.nih.gov/ij/) using the “Threshold” and “Analyze particles” modules as described previously.10
Statistics. Data are presented as mean ± standard error. Data were evaluated using Mann-Whitney test, Kruskal-Wallis test followed by Dunn's test, or two-way repeated measures ANOVA followed by Holm-Sidak test, as appropriate. P <0.05 was considered statistically significant. All statistical analyses were performed using Prism 6.0 (GraphPad Software).
SUPPLEMENTARY MATERIAL Figure S1. Serum IDUA activity in MPS I dogs. Figure S2. Serum IDUA activity in rhesus monkeys. Table S1. CSF nucleated cell counts in MPS I dogs following vector injection (Cells/μL) Table S2. Vector biodistribution
Acknowledgments
We would like to acknowledge the support of the Vector, Immunology, and Animal Models Cores of the Gene Therapy Program. This work was supported by a grant from REGENXBIO (J.M.W.), National Institutes of Health Grants P40-OD010939 and DK25759 (M.E.H.), and the National Heart, Lung, and Blood Institute (NHLBI) Center for Fetal Monkey Gene Transfer for Heart, Lung, and Blood Diseases grant # HL085794 (A.F.T.), and the Primate Center base operating grant #OD011107 (A.F.T.). J.M.W. is an advisor to REGENXBIO, Dimension Therapeutics, Solid Gene Therapy, and Alexion, and is a founder of, holds equity in, and has a sponsored research agreement with REGENXBIO and Dimension Therapeutics; in addition, he is a consultant to several biopharmaceutical companies and is an inventor on patents licensed to various biopharmaceutical companies.
Supplementary Material
References
- Dahms, NM, Lobel, P and Kornfeld, S (1989). Mannose 6-phosphate receptors and lysosomal-enzyme targeting. J Biol Chem 264: 12115–12118. [PubMed] [Google Scholar]
- Sando, GN and Neufeld, EF (1977). Recognition and receptor-mediated uptake of a lysosomal enzyme, alpha-l-iduronidase, by cultured human fibroblasts. Cell 12: 619–627. [DOI] [PubMed] [Google Scholar]
- Aldenboven, M, Boelens, F and de Koning, TF (2008). The clinical outcome of Hurler syndrome after stem cell transplantation. Biology of Blood and Marrow Transplantation 14: 485–498. [DOI] [PubMed] [Google Scholar]
- Boelens, JJ, Wynn, RF, O'Meara, A, Veys, P, Bertrand, Y, Souillet, G et al. (2007). Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant 40: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, LA, Wraith, JE, Beck, M, Kolodny, EH, Pastores, GM, Muenzer, J et al. (2009). Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 123: 229–240. [DOI] [PubMed] [Google Scholar]
- Langford-Smith, KJ, Mercer, J, Petty, J, Tylee, K, Church, H, Roberts, J et al. (2011). Heparin cofactor II-thrombin complex and dermatan sulphate:chondroitin sulphate ratio are biomarkers of short- and long-term treatment effects in mucopolysaccharide diseases. J Inherit Metab Dis 34: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifuentes, M, Doroshow, R, Hoft, R, Mason, G, Walot, I, Diament, M et al. (2007). A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 90: 171–180. [DOI] [PubMed] [Google Scholar]
- Souillet, G, Guffon, N, Maire, I, Pujol, M, Taylor, P, Sevin, F et al. (2003). Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant 31: 1105–1117. [DOI] [PubMed] [Google Scholar]
- Whitley, CB, Belani, KG, Chang, PN, Summers, CG, Blazar, BR, Tsai, MY et al. (1993). Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet 46: 209–218. [DOI] [PubMed] [Google Scholar]
- Hinderer, C, Bell, P, Gurda, BL, Wang, Q, Louboutin, JP, Zhu, Y et al. (2014). Intrathecal gene therapy corrects CNS pathology in a feline model of mucopolysaccharidosis I. Mol Ther 22: 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith, JE, Beck, M, Lane, R, van der Ploeg, A, Shapiro, E, Xue, Y et al. (2007). Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 120: e37–e46. [DOI] [PubMed] [Google Scholar]
- Langereis, EJ, van Vlies, N, Church, HJ, Geskus, RB, Hollak, CE, Jones, SA et al. (2015). Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long-term enzyme replacement therapy. Mol Genet Metab 114: 129–137. [DOI] [PubMed] [Google Scholar]
- Fan, X, Ang, A, Pollock-Barziv, SM, Dipchand, AI, Ruiz, P, Wilson, G et al. (2004). Donor-specific B-cell tolerance after ABO-incompatible infant heart transplantation. Nat Med 10: 1227–1233. [DOI] [PubMed] [Google Scholar]
- McCarthy, SA and Bach, FH (1983). The cellular mechanism of maintenance of neonatally induced tolerance to H-2 class I antigens. J Immunol 131: 1676–1682. [PubMed] [Google Scholar]
- Billingham, RE, Brent, L and Medawar, PB (1953). Actively acquired tolerance of foreign cells. Nature 172: 603–606. [DOI] [PubMed] [Google Scholar]
- Adkins, B (1999). T-cell function in newborn mice and humans. Immunol Today 20: 330–335. [DOI] [PubMed] [Google Scholar]
- Wang, G, Miyahara, Y, Guo, Z, Khattar, M, Stepkowski, SM and Chen, W (2010). “Default” generation of neonatal regulatory T cells. J Immunol 185: 71–78. [DOI] [PubMed] [Google Scholar]
- Hu, C, Cela, RG, Suzuki, M, Lee, B and Lipshutz, GS (2011). Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci USA 108: 2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traas, AM, Wang, P, Ma, X, Tittiger, M, Schaller, L, O'donnell, P et al. (2007). Correction of clinical manifestations of canine mucopolysaccharidosis I with neonatal retroviral vector gene therapy. Mol Ther 15: 1423–1431. [DOI] [PubMed] [Google Scholar]
- Xu, L, Mei, M, Haskins, ME, Nichols, TC, O'donnell, P, Cullen, K et al. (2007). Immune response after neonatal transfer of a human factor IX-expressing retroviral vector in dogs, cats, and mice. Thromb Res 120: 269–280. [DOI] [PubMed] [Google Scholar]
- Xu, L, Gao, C, Sands, MS, Cai, SR, Nichols, TC, Bellinger, DA et al. (2003). Neonatal or hepatocyte growth factor-potentiated adult gene therapy with a retroviral vector results in therapeutic levels of canine factor IX for hemophilia B. Blood 101: 3924–3932. [DOI] [PubMed] [Google Scholar]
- Kakkis, E, McEntee, M, Vogler, C, Le, S, Levy, B, Belichenko, P et al. (2004). Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab 83: 163–174. [DOI] [PubMed] [Google Scholar]
- Shull, RM, Helman, RG, Spellacy, E, Constantopoulos, G, Munger, RJ and Neufeld, EF (1984). Morphologic and biochemical studies of canine mucopolysaccharidosis I. Am J Pathol 114: 487–495. [PMC free article] [PubMed] [Google Scholar]
- Menon, KP, Tieu, PT and Neufeld, EF (1992). Architecture of the canine IDUA gene and mutation underlying canine mucopolysaccharidosis I. Genomics 14: 763–768. [DOI] [PubMed] [Google Scholar]
- Terlato, NJ and Cox, GF (2003). Can mucopolysaccharidosis type I disease severity be predicted based on a patient's genotype? A comprehensive review of the literature. Genet Med 5: 286–294. [DOI] [PubMed] [Google Scholar]
- Kachur, E and Del Maestro, R (2000). Mucopolysaccharidoses and spinal cord compression: case report and review of the literature with implications of bone marrow transplantation. Neurosurgery 47: 223–8; discussion 228. [DOI] [PubMed] [Google Scholar]
- Taccone, A, Tortori Donati, P, Marzoli, A, Dell'Acqua, A, Gatti, R and Leone, D (1993). Mucopolysaccharidosis: thickening of dura mater at the craniocervical junction and other CT/MRI findings. Pediatr Radiol 23: 349–352. [DOI] [PubMed] [Google Scholar]
- Vijay, S and Wraith, JE (2005). Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr 94: 872–877. [DOI] [PubMed] [Google Scholar]
- Wang, L, Wang, H, Bell, P, McMenamin, D and Wilson, JM (2012). Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum Gene Ther 23: 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoDuca, PA, Hoffman, BE and Herzog, RW (2009). Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther 9: 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudele, JM, Finn, JD, Siner, JI, Martin, NB, Niemeyer, GP, Zhou, S et al. (2015). AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 125: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, JD, Ozelo, MC, Sabatino, DE, Franck, HW, Merricks, EP, Crudele, JM et al. (2010). Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood 116: 5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, B, Kulis, MD, Young, SP, Hobeika, AC, Li, S, Bird, A et al. (2010). Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther 18: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, R, Brown, JR, Al-Mafraji, K, Lamanna, WC, Beitel, JR, Boons, GJ et al. (2012). Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat Chem Biol 8: 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietupski, JB, Hurlbut, GD, Ziegler, RJ, Chu, Q, Hodges, BL, Ashe, KM et al. (2011). Systemic administration of AAV8-a-galactosidase A induces humoral tolerance in nonhuman primates despite low hepatic expression. Mol Ther 19: 1999–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurigot, V, Marco, S, Ribera, A, Garcia, M, Ruzo, A, Villacampa, P, et al. (2013). Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest 123: 3254–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer, C, Bell, P, Vite, CH, Louboutin, J-P, Grant, R, Bote, E, et al. (2014). Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol Ther — Methods & Clinical Development 1: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, P, Gao, G, Haskins, ME, Wang, L, Sleeper, M, Wang, H et al. (2011). Evaluation of adeno-associated viral vectors for liver-directed gene transfer in dogs. Hum Gene Ther 22: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer, C, Bell, P, Gurda, BL, Wang, Q, Louboutin, JP, Zhu, Y et al. (2014). Liver-directed gene therapy corrects cardiovascular lesions in feline mucopolysaccharidosis type I. Proc Natl Acad Sci USA 111: 14894–14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins, PV, Campbell, C, Klug, T, Rogers, S, Raburn-Miller, J and Kiesling, J (2015). Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr 166: 172–177. [DOI] [PubMed] [Google Scholar]
- Wang, L, Calcedo, R, Bell, P, Lin, J, Grant, RL, Siegel, DL et al. (2011). Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther 22: 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantal, AF. Ultrasound Imaging in Rhesus (Macaca Mulatta) and Long-Tailed (Macaca fascicularis) Macaques: Reproductive and Research Applications. In: The Laboratory Primate. Academic Press, London, UK, 2005. pp 317–352. [Google Scholar]
- Tarantal, AF, McDonald, RJ, Jimenez, DF, Lee, CC, O'Shea, CE, Leapley, AC et al. (2005). Intrapulmonary and intramyocardial gene transfer in rhesus monkeys (Macaca mulatta): safety and efficiency of HIV-1-derived lentiviral vectors for fetal gene delivery. Mol Ther 12: 87–98. [DOI] [PubMed] [Google Scholar]
- Calcedo, R, Vandenberghe, LH, Gao, G, Lin, J and Wilson, JM (2009). Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.