

Abstract
Programmed cell death-1 (PD-1) is expressed on activated T cells and represents an attractive target for gene-editing of tumor targeted T cells prior to adoptive cell transfer (ACT). We used zinc finger nucleases (ZFNs) directed against the gene encoding human PD-1 (PDCD-1) to gene-edit melanoma tumor infiltrating lymphocytes (TIL). We show that our clinical scale TIL production process yielded efficient modification of the PD-1 gene locus, with an average modification frequency of 74.8% (n = 3, range 69.9–84.1%) of the alleles in a bulk TIL population, which resulted in a 76% reduction in PD-1 surface-expression. Forty to 48% of PD-1 gene-edited cells had biallelic PD-1 modification. Importantly, the PD-1 gene-edited TIL product showed improved in vitro effector function and a significantly increased polyfunctional cytokine profile (TNFα, GM-CSF, and IFNγ) compared to unmodified TIL in two of the three donors tested. In addition, all donor cells displayed an effector memory phenotype and expanded approximately 500–2,000-fold in vitro. Thus, further study to determine the efficiency and safety of adoptive cell transfer using PD-1 gene-edited TIL for the treatment of metastatic melanoma is warranted.
Introduction
The ability to harness the immune system to induce tumor regression has revolutionized the treatment of patients with metastatic melanoma.1,2,3,4 Administration of high-dose interleukin-2 (IL-2) or checkpoint modulation can induce tumor regression in these patients.2,3,4 In addition, adoptive transfer of autologous tumor infiltrating lymphocytes (TIL) offers an effective therapy for metastatic melanoma with an overall response rate of 49–72% with approximately 20% of patients achieving a durable complete regression.5 Given this success, extensive efforts are underway to develop more effective TIL therapies, designed to increase the response rate and durability of responses.
A promising area of research involves targeting immune checkpoints, which are inhibitory receptors on immune cells that result in inhibition of key effector functions (activation, proliferation, cytokine release, cytoxicity) when they interact with their inhibitory ligand. One of the most studied targets for the induction of checkpoint blockade is Programmed Cell Death-1(PD-1), a member of the CD28 super family of T-cell regulators.6,7,8,9 Its ligands, PD-L1 and PD-L2 are expressed on a variety of tumor cells, including melanoma. The interaction of PD-1 with its ligand PD-L1 inhibits T-cell effector function, results in T-cell exhaustion in the setting of chronic stimulation, and induces T-cell apoptosis in the tumor microenvironment.7,8,10 The significance of this interaction has been demonstrated clinically in a growing number of prospective trials where either an infusion of anti-PD-1 or anti-PD-L1 monoclonal antibody was used to induce tumor regression in approximately 30–40% of patients.11,12,13 However, the durability of the observed responses has yet to be determined.
Consistent with the early clinical success with PD-1 inhibition, we have previously shown that TIL isolated from metastatic melanoma express the PD-1 inhibitory receptor and have impaired effector cytokine production.14 Indeed, the tumor reactive fraction of the TIL population is exclusively found within the PD-1 expressing subset.15 Together, these data suggest that PD-1 inhibition may represent a critical checkpoint in the ability of metastatic melanoma to evade T-cell-mediated elimination. Thus, the ability to permanently silence PD-1 in TIL manipulated ex vivo, prior to ACT, represents an attractive approach to improving the efficacy of TIL, while avoiding the toxicities associated with the long-term administration of a systemic anti-PD-1 antibody.
Multiple technology platforms for research grade gene editing have been developed over the last 10 years including transcription activator-like effector nucleases, the bacterial CRISPR/Cas9 system, and zinc finger nucleases (ZFNs).16,17,18,19,20 Each relies on the recognition of a specific DNA sequence within the genome to target a nuclease domain to this location and mediate the generation of a double-strand break at the target sequence. The repair of this break can result in the introduction of insertion/deletion mutations (indels) by the nonhomologous end-joining pathway (review21). If the nuclease target site resides in the coding sequence, such indels can result in the permanent disruption of the target gene product. While each technology differs with respect to the mechanism used to site-specifically guide the nuclease, all the platforms face a similar challenge for therapeutic use; namely, achieving at clinical scale the cleavage efficiency necessary for biologic impact in the primary human cell/tissue of choice while maintaining sufficient target specificity. To date, only the ZFN platform has been used to treat patients in a clinical trial using ZFNs targeting the HIV coreceptor, CCR5, for the clinical scale production of autologous CD4 T cells refractory to HIV infection for ACT in subjects with HIV.22,23 The results of the initial phase 1 clinical trial showed that the treatment was generally safe and well tolerated, resulted in increased CD4 T-cell levels in all subjects and demonstrated protection of the modified T cells during an antiretroviral drug treatment interruption.23 The implications for this technology to improve patient therapies are numerous, and the ability to harness targeted genome editing in the setting of ACT for the treatment of cancer has yet to be fully explored.
Given the success of ongoing clinical trials using inhibitory antibodies against PD-1, our aim was to determine the feasibility of permanently disrupting the PD-1 gene in human TIL using ZFN-mediated gene editing. Efforts were undertaken to gene edit TIL at clinical scale using flow electroporation with mRNA encoding the PD-1 specific ZFNs. Here we describe the ability of this platform to efficiently and specifically eliminate PD-1 expression within a TIL population at the scale necessary for patient treatment. As a further rationale for exploring this technology in a clinical trial, we characterized the phenotype of the modified TIL and described the effect of PD-1 gene editing on TIL proliferation and effector function.
Results
Efficiency of clinical scale PD-1 gene modification
TIL were harvested from patients with metastatic melanoma as previously described, and induced to rapidly proliferate using a rapid expansion protocol (REP).24 In three separate experiments, TIL from three donors underwent expansion (to a total of approximately 2 × 109 for each donor on day 7 of REP expansion) prior to electroporation with PD-1 ZFN encoding mRNA using MaxCyte's CL-2 flow electroporation processing assembly. At day 7 of the REP, TIL from each donor was resuspended in electroporation buffer to a final concentration of 1 × 108 cells/ml and mixed with PD-1 ZFN RNA at 120 μg/ml (Figure 1b). TIL were then electroporated with RNA encoding a pair of ZFNs targeting the PD-1 gene locus (Figure 1a). The percent indels, or percentage of mutated alleles, was determined by means of the Cel-1 assay. No difference in electroporation efficiency was observed when using either a single small-scale 3 ml processing assembly or the large-scale CL-2 assembly. ZFN-mediated gene editing occurred in 50.9% of alleles when TIL from Donor 1 were electroporated using a small scale cuvette and 47.1% when using the flow electroporation processing assembly. Similarly, gene editing occurred in 44.6% of alleles for Donor 2 and 42.2% for Donor 3 using the CL-2 assembly (Figure 2). Because the Cel-1 nuclease relies on the formation of heteroduplexes of reannealed modified and unmodified alleles during PCR, accuracy is reduced when the efficiency of gene editing increases beyond 50% indels, as modified sequences may reanneal to each other resulting in decreased heteroduplex formation. In order to more accurately determine genetic modification of the intended target site, genomic DNA from the same sample used in the Cel-1 assay was deep-sequenced using the Illumina platform technology (Table 1). The mean % indels was 74.8% (range 69.9–84.1%) for the three donors tested.
Figure 1.

Schematic representation of the modified tumor infiltrating lymphocytes (TIL) process and zinc finger nuclease (ZFN) target site at the PD-1 locus. (a) Schematic of the ZFN target site within the PD-1 gene at the 2q37.3 location. Each ZFN comprises five zinc finger domains that recognize a composite 15bp target site (in red). Each ZFP is coupled to an obligate heterodimer mutant of the FokI endonuclease that mediates dimerization dependent DNA cleavage.46 (b) Work-flow of large clinical scale RNA electroporation with PD-1 targeted ZFN. TIL were induced to rapidly expand (rapid expansion protocol (REP) day 0) and cultured in a G-Rex100 flask at 37 °C and 5% CO2. On day 7 of the REP, TIL were harvested and washed two times with Hyclone Electroporation Buffer. Cells were then counted and resuspended in electroporation buffer at a concentration of 1 × 108/ml. Cells were mixed with 120 μg/ml of PD-1 ZFN RNA and transferred to the CL-2 processing assembly for electroporation on the Maxcyte GT Flow Transfection System.
Figure 2.

Efficiency of PD-1 gene disruption following electroporation with mRNA encoding PD-1-targeted zinc finger nucleases (ZFNs). Cel-1 surveyor nuclease assay performed 3 days following electroporation. ZFN-dependent gene editing occurred in 46.0% of alleles when tumor infiltrating lymphocytes from Donor 1 were electroporated using a smaller scale cuvette and 42.0% when electroporated using the flow electroporation processing assembly. Gene editing occurred in 37.0% of alleles in Donor 2 and 37.0% in Donor 3.
Table 1. Quantification of PD-1 gene modification by deep sequencing using the Illumina MiSeq platform technology.
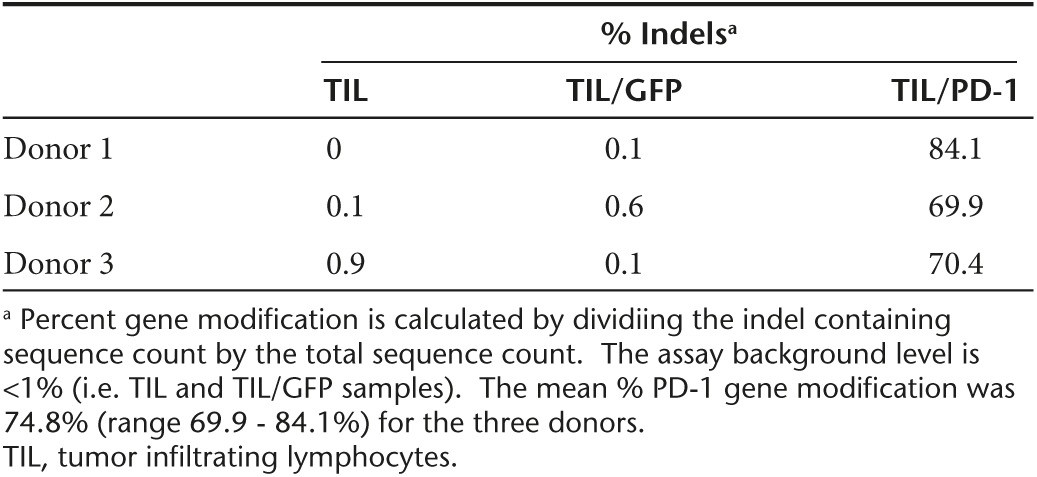
In order to determine the relative frequencies of mono- and bi-allelic target modification, single cell clones were generated from the bulk ZFN modified TIL population. A total of 40 clones from Donor 1 and 40 clones from Donor 3 were successfully isolated, expanded, and sequenced across the PD-1 ZFN target site. The rate of allelic modification across all clones isolated from Donor 1 was 69% (n = 55/80 alleles). Thirty-six of 40 clones from Donor 1 had insertions or deletions at the PD-1 gene locus; with a biallelic modification rate of 48% (n = 19/40 clones). For Donor 3, the rate of allelic modification across all clones isolated was 63% (n = 50/80 alleles). Thirty-four clones from Donor 3 had indels at the PD-1 locus; the percent of these clones with a biallelic modification was 40% (n = 16/40). Taken together, biallelic gene editing occurred in 44% of gene modified TIL (data not shown).
Expression of PD-1 is reduced following zinc finger nuclease-mediated gene editing
To determine the expression of PD-1 in TIL following gene editing, 7 days after electroporation (day 14 of the REP), a portion of TIL were restimulated with anti-CD3/CD28 beads for 48 hours. Both restimulated and unstimulated (no bead restimulation) TIL were assessed for PD-1 surface expression by fluorescence-activated cell sorting analysis (FACS). Figure 3a shows the surface expression of PD-1 on Donor 3 TIL, and is representative of all three donors. For Donor 3, 18 and 16% of TIL and TIL/GFP bulk populations expressed PD-1 on the cell surface, which was reduced to 4% following gene editing, a decrease of nearly 80%. This trend was maintained following anti-CD3/CD28 bead stimulation which increased the fraction of PD-1-positive cells in untreated or mock-electroporated TIL/GFP to 90 and 80%, respectively, while the PD-1 ZFN-treated group achieved only 31.7% PD-1+ TIL. In combining all three experiments, there was a statistically significant decrease in the number of cells expressing PD-1 in the ZFN-treated TIL compared to the untransfected TIL and GFP transduced TIL (Figure 3b). The mean number of CD3+ TIL expressing PD-1 was reduced by 76% on average following ZFN editing (5.6 versus 23.9% in TIL (P = 0.015) and 23.0% in TIL/GFP (P = 0.027)). A similar trend was observed in the bead-stimulated TIL (P = 0.066 and 0.059) (Figure 3b). To further characterize these differences, we performed RT-PCR on the TIL/GFP and TIL/PD-1 KO at sequential time points after anti-CD3/CD28 bead stimulation (Figure 3c). RNA expression of PD-1 in TIL/GFP peaked at 3 hours and subsequently declined, whereas the basal level and the activation-induced upregulation of PD-1 RNA was significantly reduced in PD-1 gene-edited TIL. Taken together, these data demonstrate that the disruption of the PD-1 locus using ZFN in TIL led to frameshift knockouts in a large portion of treated cells (with a 40–48% of biallelic knockout), which in turn significantly reduced expression of both PD-1 mRNA and protein in the ZFN-treated TIL population.
Figure 3.

Expression of PD-1 is reduced following zinc finger nuclease-mediated gene editing. (a) PD-1 cell surface expression by FACs analysis is shown for Donor 2. Results are representative of all three donors. (b) Cell surface expression of PD-1 is reduced at baseline and following anti-CD3/CD28 bead stimulation in tumor infiltrating lymphocytes (TIL) following PD-1-targeted gene editing (TIL/PD-1) compared to untransfected TIL (TIL) and GFP-transfected TIL (TIL/GFP). Percent CD3+/PD-1+ cells for each condition is presented as the mean ± SEM for all three donors. (c) Time course of PD-1 mRNA expression in GFP transfected (TIL/GFP) and gene edited TIL (TIL/PD-1 KO) for Donor 2 during anti-CD3/CD28 bead stimulation analyzed by RT-PCR.
Effect of electroporation and gene editing on T-cell proliferation, phenotype, and effector function
Next we sought to determine what effect the gene editing had on T-cell proliferation, phenotype and effector function. Both TIL/GFP (mock) and TIL/PD-1 showed a modest decrease in cell proliferation during the initial REP (REP1) when rapid expansion was interrupted by the electroporation process (Figure 4a). However, a second rapid expansion revealed no consistent differences between TIL, TIL/GFP, and TIL/PD-1 KO with respect to proliferative capacity following REP2 (Figure 4b).
Figure 4.

The effect of the flow electroporation protocol and PD-1 gene editing on tumor infiltrating lymphocytes (TIL) proliferation during rapid expansion protocol (REP). (a) Both GFP-transfected (TIL/GFP) and gene-edited TIL (TIL/PD-1) sustained a reduction in fold proliferation when rapid expansion was interrupted by the electroporation process (day 7). (b) A second REP revealed no consistent difference between TIL, TIL/GFP, and TIL/PD-1 KO with respect to fold expansion/proliferative capacity.
The phenotype of the gene-edited TIL was assessed by FACS staining using anti-CD45RA and anti-CD62L antibodies to determine the fractions of naive (CD45RA+/CD62L+), central memory (CD45RA-/CD62L+), effector memory (CD45RA-/CD62L-) and EMRA (CD45RA+/CD62L-) T cell subsets. Although slight differences in the distribution of T-cell subsets were observed across TIL from different subjects (i.e., donor variability), the gene-edited TIL displayed a similar phenotype compared with unmodified cells and GFP transfected TIL. In all cases, the majority of the cells had an effector memory-like phenotype (CD45RA-/CD62L-) with slight differences in the number of cells expressing higher levels of CD45RA (Figure 5).
Figure 5.

Gene edited tumor infiltrating lymphocytes (TIL) display a similar phenotype as unmodified cells and GFP-transfected TIL. FACs analysis was performed of each condition for all three donors. The majority of T cells within the bulk TIL had an effector memory-like phenotype (CD45RA-/CD62L-) with slight differences in number of cells expressing higher levels of CD45RA that were independent of gene editing.
Tumor-specific effector function of TIL was evaluated by coculturing the TIL with tumor target cells and measuring cytokine release (Figure 6) or cytolytic activity against HLA-matched antigen-positive target cells. Tumor target cell lines used in our study were isolated from patients as previously described25 and included both HLA-unmatched (HLA-A2-; mel888 and mel938) and HLA-matched (HLA-A2+; mel526, mel624, and mel624/PD-L1) targets that express the melanoma-associated antigen MART-1. TILs from Donors 1 and 2 showed recognition of the shared melanoma antigen, MART-1. PD-1 gene-edited TIL showed a significantly improved polyfunctional cytokine profile, relative to TIL/GFP controls, with release of TNFα, GM-CSF, and IFNγ (P < 0.0001) when cocultured with mel526. The same was true when cocultured with mel624, with the exception of IFNγ release for Donor 2, which no significant difference was observed. To further characterize the role of the PD-1/PD-L1 axis in TIL effector function, a mel624 tumor cell line was engineered to overexpress PD-L1 (mel624/PD-L1; Supplementary Figure S3) and was used in coculture. The increased expression of PD-L1 in the tumor cell line mel624/PD-L1 decreased the cytokine levels to those comparable with HLA-unmatched tumor cell lines in unmodified TIL from Donor 1. Importantly, while TIL/PD-1 KO cells also exhibited reduced cytokine secretion when cocultured with the mel624/PD-L1, the reduction was to a much lesser extent and significantly higher than unmodified TIL (Figure 6). TIL/PD-1 KO from Donor 2 also showed significantly enhanced cytokine secretion levels with the exception of IFNγ levels following coculture with mel624 and mel624/PD-L1. Donor 3, recognizing a known mutated antigen expressed by an autologous tumor cell line generated from the patient's tumor with no recognition of MART-1. TIL/PD-1 KO cells from Donor 3 showed no increase in cytokine secretion following coculture with the autologous tumor cell line (data not shown).26 From these results, we conclude that gene editing of PD-1 in TIL significantly enhanced T-cell effector function in vitro as measured by the ability to secrete TNFα, GM-CSF, and IFNγ following stimulation with antigen-specific tumor targets even when the targets overexpressed PD-L1. Of note, while increased IFNγ secretion was observed for all PD-1 gene-edited TIL following coculture with mel624/PD-L1, no additional increase in cytokine secretion was observed when the PD-1/PD-L1 axis was targeted with a blocking anti-PD-1 antibody (Supplementary Figure S2).
Figure 6.

Enhanced polyfunctional effector cytokine profile of PD-1 knockout tumor infiltrating lymphocytes (TIL) in vitro. Coculture with melanoma cell lines was performed on day 12 of the rapid expansion protocol, 5 days after electroporation, and cytokine (CXCL8/IL-8, GM-CSF, IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 p70, TNFα, and VEGF) levels were measured using the Luminex Performance Human High Sensitivity Cytokine Panel. Donor 1 showed a polyfunctional effector phenotype and a significant increase in TNFα, GM-CSF, and IFNγ secretion following PD-1 gene editing (black bars) as compared to the unmodified TIL (P < 0.0001, hatched bars). Donor 1 PD-1 KO TIL also showed significantly less inhibition following coculture with mel624/PD-L1, a PD-L1-overexpressing cell line. Similar results seen for Donor 2; however, no significant difference in IFNγ secretion was observed when compared to unmodified TIL with the exception of Donor 2 PD-1 KO TIL cocultured with mel526. Shown are representative data from Donors 1 and 2, one-way analysis of variance, P ≤ 0.0001).
To further test the effector function of ZFN treated TIL, specific cytolytic activity was measured by chromium release assay (Supplementary Figure S4) with cocultures of mel624 and mel624/PD-L1. PD-1 ZFN gene edited TIL from Donor 1 showed a modest improvement in target-specific cytolysis against mel624/PD-L1 as compared to untransfected TIL or TIL/GFP. For Donors 2 and 3, there were no significant differences observed between the three TIL preparations (TIL, TIL/GFP, TIL/PD-1).
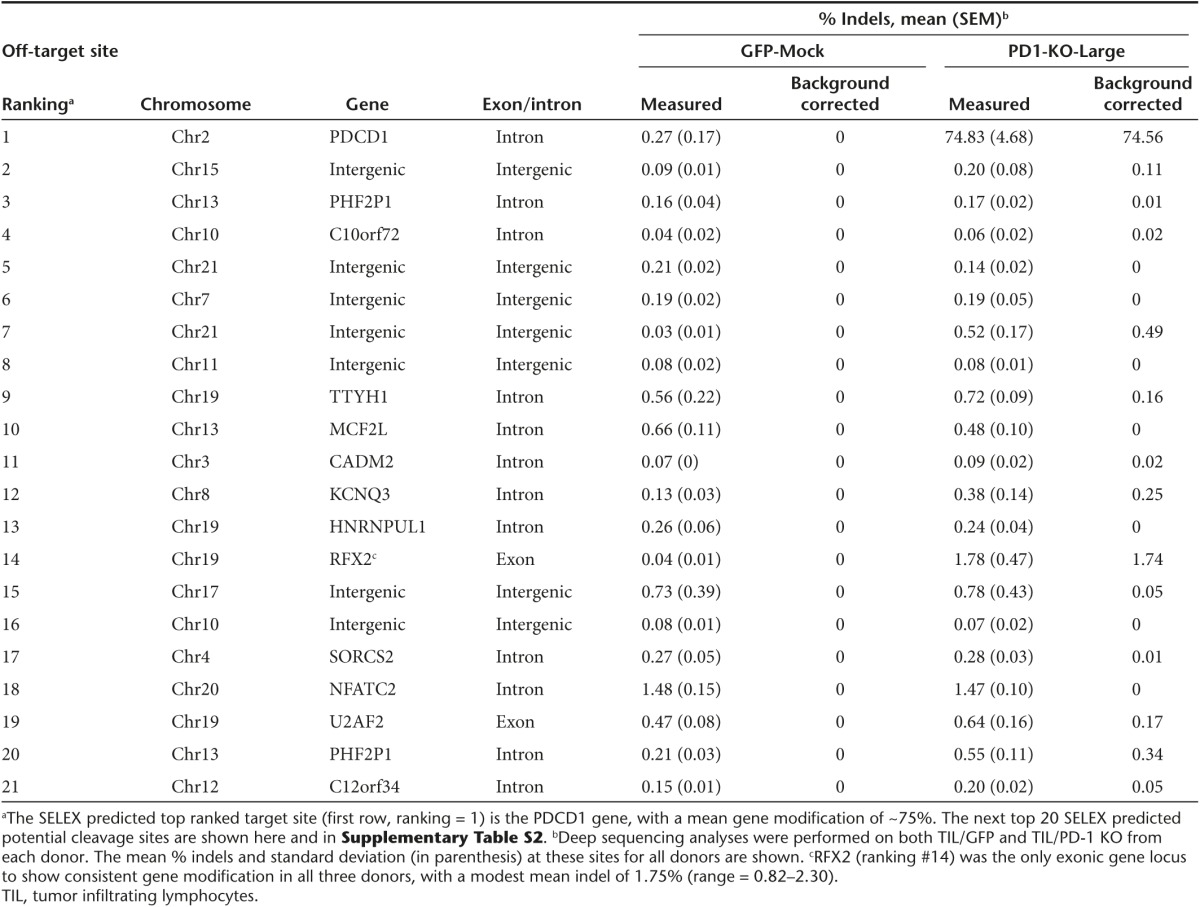
Target specificity of PD-1 ZFNs
The consensus PD-1 ZFN binding sites were determined by SELEX as previously described20,27,28 revealing highly specific binding by both ZFN monomers. To identify candidate off target cleavage sites, the SELEX determined consensus-binding sites were scanned against the human genome and potential heterodimer cleavage targets were scored according to their degree of match to the SELEX-derived base frequency matrices (Supplementary Tables S1 and S2). Sequencing of the 20 top-scoring potential cleavage sites was performed on both TIL/GFP and TIL/PD-1 KO from each donor and the % indel at these sites are shown in Table 2. Only two of the top twenty SELEX based potential off-target sites are in exonic regions. RFX2, is a gene in the regulatory factor X gene family, and with a mean rate of modification of 1.75% indels (0.82–2.30), was the only exonic target to show consistent, but relatively modest gene modification. In contrast, the mean on target PD1 gene modification was 75% (P = 0.004).
Table 2. PD-1 gene editing by ZFNs is target specific.
In vivo toxicity studies in NSG mice
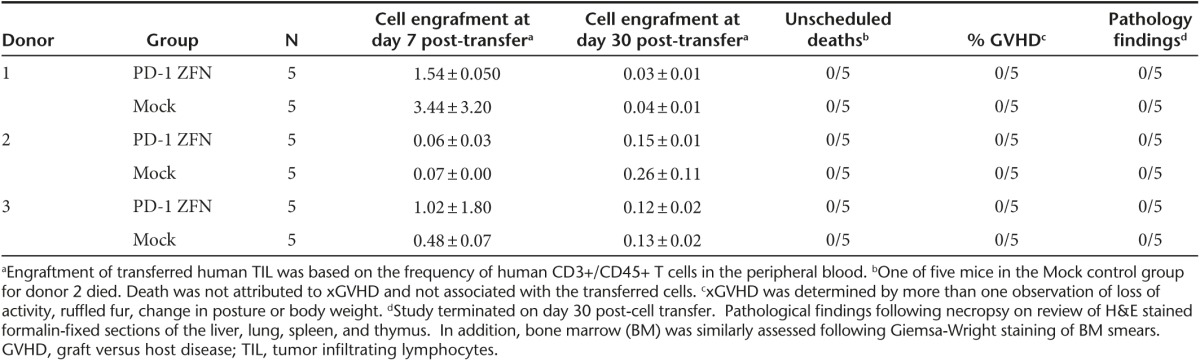
PD-1 gene-edited and mock transfected TIL were transferred into NSG mice to evaluate whether ex vivo knockout of PD-1 in T lymphocytes may result in proliferative abnormalities or tumor formation. In this study, 5 million TIL/PD-1 or TIL/GFP from three separate donors were transferred into NSG mice (five mice per group, Table 3) without IL-2 supplementation. Engraftment was assessed post-cell transfer by measuring the frequency of human CD3+/CD45+ cells circulating in the peripheral blood. Low-level engraftment was detected 1-week postinfusion (0.06–3.44%), with no observable difference between the GFP transfected or PD-1 gene-edited TIL. Engraftment of TIL at 1 month post-cell transfer reduced to near background levels, at which point the experiment was terminated and euthanized mice were evaluated at necropsy. No tissue abnormalities or tumor formation was observed following H&E staining of formalin-fixed tissues (liver, lung, spleen, thymus, and bone marrow). Based on the lack of long-term circulating human T cells in the periphery and no evidence of tissue abnormalities, PD-1-ZFN-related carcinogenicity was not observed (Table 3).
Table 3. In-vivo toxicity assessment of engrafted PD-1 knockout melanoma TIL.
Discussion
With a response rate of 50%, adoptive cell transfer (ACT) using autologous TIL offers an effective therapy for metastatic melanoma.5 Building on this success, efforts are ongoing in to further improve response rates by developing better methods to manufacture TIL, better selection of tumor reactive T cells within the bulk TIL population,15,29 and by preventing the downregulation of T-cell effector functions mediated by the immunosuppressive tumor microenvironment. To that end, we exploited a gene editing technology, which involves the disruption of the human PD-1 gene locus using ZFNs. Herein, we demonstrate that delivery of ZFNs via a high flow-through electroporation system provides an efficient platform to decrease PD-1 expression in TIL at a scale and efficiency suitable for patient treatment. Our results indicate that gene modification was highly efficient (70–84% disruption at the PD-1 locus) and resulted in a significant decrease in both PD-1 mRNA and cell surface expression on treated T cells. We demonstrate how a pair of ZFNs directed against exon 1 of the PD-1 gene can result in bi-allelic disruption of the gene locus with high efficiency, i.e., approaching 50% of the TIL population without selection for the desired effect. While there was a decrease in the cell expansion during the initial electroporation/REP cycle (seen in both the TIL/GFP and TIL/PD-1 samples), the difference was transient and the T cells retained a proliferative capacity similar to untransfected TIL following a second REP. The TIL (mock or PD-1 KO) were not adversely affected by the electroporation process and were mostly of the effector memory phenotype. TIL/PD-1 KO exhibited a polyfunctional cytokine secretion profile and released significantly higher levels of TNFα, GM-CSF, and IFNγ for donors 1 and 2, with the exception of IFNγ for donor 2, in an antigen-specific manner as compared to TIL/GFP. In addition, TIL/PD-1 maintained their capacity to secrete significantly higher levels of cytokine when cocultured with a tumor cell line that overexpresses PD-1 inhibitory ligand, mel624/PD-L1. Taken together, these results suggest that ZFNs can be used to create PD-1-deficient TIL with significantly enhanced effector function at a scale necessary for clinical study in subjects with metastatic melanoma.
For our preclinical study, it was important to determine if a reduction in PD-1 expression on TIL was important for tumor-specific effector function or target-mediated cell lysis. An important function of PD-1 is the decrease in functional capacity when its ligand, PD-L1, is engaged.30 As such, PD-1 blockade should release the cells from PD-L1-induced suppression. While some have shown that blocking the PD-1/PD-L1 axis in vitro and in some murine models resulted in increased in vitro effector function (increased IFNγ secretion or cytolysis),31 this is not a general finding. Our findings showed a significant increase in in vitro effector function based on a polyfunctional cytokine profile following coculture with antigen-specific target cells. Comparison of PD-1 gene knockout versus human anti-PD-1 monclonal antibody blockade was attempted, but our efforts to demonstrate a positive effect on TIL effector function following PD-1 blockade were inconclusive (Supplementary Figure S2).32 Data to support the complete blockade of the PD-1/PD-L1 axis using the anti-PD-1 monoclonal antibody (clone EH12.2H7) are lacking. In addition, only modest benefits in cell proliferation have been reported following PD-1 blockade on T lymphocytes, which does not necessarily directly translate into a more tumor-reactive population of TIL.33,34
There is a growing body of evidence suggesting that TIL cause tumor regression through the recognition of mutated antigens, making it even more difficult to extrapolate murine tumor models or humanized mouse models to the human TIL setting.29,35 The most compelling evidence for the role of the PD-1/PD-L1 axis in evading the host immune response to cancer comes from a growing body of clinical trials where blocking the PD-1/PD-L1 interaction with a monoclonal antibody has been shown to induce tumor regression in patients with metastatic melanoma, renal cell, colonic adenocarcinoma, and non–small-cell lung cancer.13,36,37 Treatment with an anti-PD-1 antibody can result in an objective response as high as 38% in patients with metastatic melanoma.13 However, there are adverse events related to inflammation and autoimmunity with the systemic therapy that include pneumonitis, renal dysfunction, and elevation in aminotransferase levels.12 Our proposed strategy to silence PD-1 ex vivo in TIL prior to adoptive cell transfer could confer the advantages of blocking the PD-1/PD-L1 axis while avoiding the toxicities associated with the administration of a systemic antibody.
The heterodimer-dependent cleavage by the Fok-I nuclease domain of the ZFN mandates binding of both ZFPs to a targeted region prior to DNA cleavage. As part of our preclinical evaluation, we performed off target analysis by sequencing the top 20 SELEX predicted potential cleavage sites from ZFN-treated cells. Only two of these sites were in an exonic region. RFX2 was the only exonic target to show consistent, but relatively modest gene modification, with a measured rate of modification of 1.75% indels. RFX2 is a transcriptional activator and can bind to cis elements in the promoter of the IL-5 receptor alpha gene, yet the full biological role remains unknown.38 Based on the previously established safety profile of ZFNs, the minimal off-target gene modification in our analysis, continued in vitro dependence on IL-2 following electroporation (Supplementary Figure S5), no proliferative abnormalities and no observed tumorgenicity in an NSG engraftment model (Table 3), this platform appears safe for further evaluation.
The implications of PD-1 gene editing may reach beyond the current application of TIL for the treatment of metastatic melanoma. We have previously shown that PD-1 expression on CD8+ TIL accurately identifies the repertoire of clonally expanded tumor-reactive cells.15 Adoptively transferred T cells, enriched by sorting for tumor-reactive markers like PD-1, may benefit from gene editing of the PD-1 locus prior to ACT. This would effectively create a tumor-specific population of T cells less susceptible to the immunosuppressive effects of tumor PD-L1 expression. Another application is that of T-cell receptor (TCR) or chimeric antigen receptor (CAR)-engineered lymphocytes which can induce cancer regression in patients not amenable to TIL therapy.39,40,41,42,43 Using expression profiling, we have previously reported the overexpression of multiple inhibitory receptors, including PD-1, in TCR-engineered lymphocytes that persist in vivo following ACT.44 Our present study validates the use of a platform that could be used for gene editing of PD-1 in TCR or CAR-transduced T cells prior to ACT.
In summary, ZFNs provide an efficient way to knock out PD-1 expression in TIL at clinical scale without adversely affecting T-cell effector function or altering the TIL phenotype. Gene modification is specific while supporting on-target cleavage levels that result in bi-allelic disruption of PD-1 in approximately half of the TIL population. Using this approach to improve the efficacy of TIL and other T-cell-based therapies in patients with cancer warrant further clinical investigation.
Materials and Methods
PD-1 ZFN construct and ZFN messenger RNA. ZFNs targeting the human PDCD1 (PD-1) gene were designed by modular assembly using an archive of one- and two-finger modules and optimized for binding as previously described (Figure 1a).45 DNA segments encoding the two ZFNs were then linked into a single gene that expresses both ZFNs separated by a 2A self-processing polypeptide, and the resulting gene was subsequently cloned into a T7 expression cassette (Invitrogen, Carlsbad, CA) for in vitro transcription of ZFN encoding mRNA. mRNA was made using a standard in vitro transcription method by a commercial vendor (Trilink Biotechnologies, San Diego, CA). The full amino acid sequence of the PD-1 ZFNs (PD1-ZFN-L and PD1-ZFN-R) is disclosed in the supporting supplementary information (Supplementary Figure S1).
Cell culture and cell lines. All human cells and tissues were obtained under Institutional Review Board approved protocols (National Cancer Institute, Bethesda, MD). Melanoma tumor cell lines HLA-A2+/MART-1+ (526, 624, 624/PD-L1) and HLA-A2−/MART-1+ (888, 938) were generated at the Surgery Branch (National Cancer Institute, National Institutes of Health, Bethesda, MD), as previously described.25 Adherent tumor cell lines were grown under standard conditions in Roswell Park Memorial Institute (RPMI) 1640 with 10% fetal bovine serum medium (Sigma Aldrich, St. Louis, MO), 25 mmol/l HEPES (Invitrogen) and penicillin/streptomycin (Invitrogen) at 37 °C with 5% CO2. TIL were cultured in G-Rex100 flasks (Wilson-Wolf Manufacturing, New Brighton, MN) with T-cell medium (AIM V, Invitrogen) plus 5% human AB serum, 25 mmol/l HEPES, penicillin/streptomycin, and 500 IU/ml interleukin-2 (IL-2, Aldesleukin, Prometheus, San Diego, CA).
REP and TIL electroporation. As shown in Figure 1b, TIL electroporation was performed using the MaxCyte GT Flow Transfection System (MaxCyte, Gaithersburg, MD). In order to generate a sufficient number of transduced T cells for adoptive cell transfer, the TIL were induced to proliferate using a REP.46 Briefly, 1 × 107 TIL were combined with 1 × 109 allogeneic, irradiated (5,000 rad) peripheral blood mononuclear cells (PBMC), and these cells were suspended in 400 ml of T-cell media containing 30 ng/ml of OKT3. The cells were cultured in a G-Rex100 flask at 37 °C and 5% CO2. Five days later, 200 ml of media was aspirated and replaced. Seven days after the start of the REP, TIL were harvested and washed two times with Hyclone Electroporation Buffer (Hyclone Laboratories, Logan, UT). Cells were then counted and resuspended in electroporation buffer at a concentration of 1 × 108/ml. Cells were then transferred to the MaxCyte CL-2 processing assembly and mixed with 120 μg/ml of PD-1 ZFN mRNA (or GFP mRNA for GFP transfected TIL/GFP). Electroporation was performed as per MaxCyte's protocol. Following electroporation, TIL were transferred from the processing assembly to a T-175 flask and placed in an incubator at 37 °C for 20 minutes. Following this incubation step, TIL were resuspended in AIM-V media at a concentration of 1 × 106/ml. Cells were then placed in an incubator set at 30 °C for an overnight low temperature incubation as previously described.47 The following day, TIL were transferred to a 37 °C incubator and left undisturbed until REP day 10 (3 days following electroporation).
Cel-1 and RT-PCR assay. The level of ZFN-induced PD-1 gene modification was quantified by the Cel-1 assay as previously described.48 Cel-1 assay was performed using the forward primer 5′- ACCTGGGACAGTTTCCCTTC and the reverse primer 5′- CTGGCTCTGGGACACCTG to target PD-1 exon 1 region.
To evaluate the expression timecourse of PD-1 mRNA, TILs were electroporated with either GFP mRNA or PD-1 ZFN mRNA. Following electroporation, cells were stimulated with anti-CD3/CD28 beads. Transduced cells were then harvested at 0, 3, 6, 9, and 18 hours following stimulation after which total RNA was isolated from treated cells. RT-PCR assay was performed with 1 µg total RNA using SuperScript III (Life Tech, CA) along with the human PD-1 forward primer, ACCTGGGACAGTTTCCCTTC, and reverse primer, CTGGCTCTGGGACACCTG. For PCR control reactions, human β-actin was amplified at the same time with forward, ACACTGTGCCCATCTACGAGG, and reverse, AGGGGCCGGACTCGTCATACT, primers, respectively.
Generation of gene-edited TIL clones. Using a modified FACS Aria instrument (BD Biosciences, San Jose, CA), single cells were sorted into individual wells of a 96-well plate containing 100 allogeneic, irradiated (5,000 rad) PBMC, in 150 μl of T-cell media with 30 ng/ml of OKT3. Plates were covered with aluminum foil to prevent desiccation and incubated at 37 °C with 5% CO2. Seven days later, 100 μl of T-cell media (containing OKT3) was added to each well and plates were returned to the incubator. On day 14 of the culture, the T-cell clones were harvested and genomic DNA was isolated for clonal assessment of PD-1 gene modification.
Indel quantification via next-generation sequencing/MiSeq. Loci of interest (both on target PD1 as well as potential off-target sites) were PCR amplified, and the levels of modification at each locus were determined by paired-end deep sequencing on an Illumina MiSeq sequencer. Paired sequences were merged via SeqPrep (John St. John, https://github.com/jstjohn/SeqPrep, unpublished). A Needleman-Wunsch alignment was performed between the target amplicon genome region and the obtained Illumina sequence to map indels.49
Flow cytometry and PD-1 induction. FACS was performed using an APC conjugated anti-PD1 antibody (eBioscience, San Diego, CA, clone MIH-4) according to manufacturer's recommendations. FACS analysis was performed on day 14 of the REP, 7 days following electroporation. To induce PD-1 expression, TIL were activated with anti-CD3/CD28 Dynabeads (Life Technologies, Grand Island, NY). Cells were plated in a 24-well tissue culture plate at 1 × 106/ml at a ratio of four beads/cell for 48 hours prior to FACS analysis. Cells were analyzed using a FACScan flow cytometer with FlowJo software (Tree Star, Ashland, OR).
Coculture and cytokine assays. 1 × 105 TIL were combined with an equal number of target cells in the indicated well of a 96-well plate containing T-cell media without IL-2. The cells were incubated at 37 °C overnight. The following day, the supernatant from each well was collected and an IFNγ ELISA assay (Pierce, Rockford, IL) was performed according to manufacturer's protocol. In addition, secreted cytokine (CXCL8/IL-8, GM-CSF, IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 p70, TNFα, and vascular endothelial growth factor (VEGF)) levels were also measured using the Luminex Performance Human High Sensitivity Cytokine Panel (R&D systems, Minneapolis, MN) according to manufacturer's protocol. Cocultures were also performed using a PD-1 blocking antibody as a treatment condition for comparison (Supplementary Figure S2). For these experiments, 1 × 105 TIL were incubated for 1 hour at 37 °C in T-cell media without IL-2 along with either 5 or 30 μg/ml of Ultra-LEAF (low endotoxin, azide-free) purified anti-human PD-1 blocking antibody (clone EH12.2h7, BioLegend, San Diego, CA) prior to plating an equal number of target tumor cells.
Cytolytic assay. Melanoma tumor cell lines were incubated for 120 minutes with 100 μCi chromium 51 (51Cr) (PerkinElmer, Boston, MA) and washed three times with culture media. Chromium-labeled melanoma cell lines were plated at 1 × 104 cells/well of a 96-well U-bottom plate along with either TIL, TIL/GFP, or TIL/PD-1 KO cells at the indicated effector/target ratios. The coculture was incubated for 4.5 hours at 37 °C. The supernatant was harvested, and the amount of 51Cr released was determined by γ-counting. Tumor specific lysis was calculated from triplicate samples using the following equation ((experimental cpm – spontaneous cpm)/(maximal cpm – spontaneous cpm)) and expressed as a percentage.25
Off-target analysis. Binding sites recognized by PD1-L and PD1-R were determined using a SELEX approach as described.20 This study yielded the base frequency matrices summarized in Supplementary Table S1. The human genome (hg19) was then scanned for potential heterodimer cleavage targets using the approximate string-matching algorithm and core consensus sequences derived from Supplementary Table S1 (GTCGTYMGGGYGGTG for PD1-ZFN-R and AGGGCGGCTGTGGGA for PD1-ZFN-L).27 Candidate targets were then scored and ranked using full base frequency matrices essentially as described to yield the ranked list provided in Supplementary Table S2.28 For identifying and ranking candidate cleavage sites, a separation distance of 5 or 6 bp was allowed between ZFN binding sites, reflecting the ability of ZFNs to efficiently dimerize and cleave in either configuration.
In vivo toxicity studies in NSG mice. Four- to 6-week-old NOD.Cg-PrkdcscidIl2rgtm1Wj1/SzJ (NSG) female mice (the Jackon Laboratory, Bar Harbor, ME) were used for adoptive cell transfer experiments. All procedures were approved by the National Cancer Institute animal care and use committee. Briefly, each mouse received an intravenous injection of 1–5 × 106 unmodified or PD-1 KO TIL resuspend in 500 μl of HBSS. Peripheral blood was collected at 1 week and 1 month post-cell transfer and stained for CD3, CD4, CD8, and CD45. Daily cage observations were made to assess gross animal health and signs of xGVHD. Following termination of the experiments, mice were subjected gross visual assessment at necropsy. In addition, multiple organs (spleen, liver, lung, thymus, bone marrow, bone) were collected. Samples were split for DNA isolation (snap freeze) and formalin fixation followed by sectioning and H & E staining and subsequent analysis. Any gross pathological abnormalities were analyzed further by immunohistochemical staining for T cell infiltration.
Statistical analysis. Continuous variables were expressed as median with range or mean ± standard error of mean or standard deviation and compared using analysis of variance (ANOVA). For significant differences, a post hoc analysis was performed using the Student's t-test for 2 × 2 comparison. Statistical significance was set at P ≤ 0.05. Analysis was performed using Graphpad Prism software v6.0b (La Jolla, CA).
SUPPLEMENTARY MATERIAL Figure S1. Amino acid sequence of PD1-ZFN-L and PD1-ZFN-R. Figure S2. IFNγ secretion by TIL when cocultured with mel624/PD-L1 (donors 1 and 2) or an autologous tumor cell line (donor 3). Figure S3. PDL-1 expression by FACs analysis on target melanoma tumor cells lines: HLA-A2−/MART-1+ (888, 938) and HLA-A2+/MART-1+ (526, 624/BSR, 624/PDL-1). Figure S4. PD-1 knockout TIL retain lytic potential in vitro. Figure S5. Growth curves of each TIL preparation (TIL, TIL/GFP, TIL/PD-1) for donors 2 and 3 following IL-2 withdrawal from culture media. Table S1. Base frequency matrices for PD1-L and PD1-R, as derived from an alignment of sequences recovered via SELEX analysis. Table S2. Top ranking candidate PD1 ZFN cleavage sites in the human genome.
Acknowledgments
The authors would like to thank David Shivak (Sangamo BioSciences) for his assistance in defining the ZFN off-target specificities, Arnold Mixon and Shawn Farid (Surgery Branch) for their technical support with FACS analysis, and Sid Kerkar (Department of Pathology, NCI) for his pathology assessment in the NSG engraftment study. Sangamo BioSciences provided the ZFNs and electroporation reagents necessary to complete this project. In addition, Sangamo BioSciences has not selected or edited the data presented. The Surgery Branch/NCI coauthors have no financial interests to disclose.
Supplementary Material
References
- Restifo, NP, Dudley, ME and Rosenberg, SA (2012). Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins, MB, Lotze, MT, Dutcher, JP, Fisher, RI, Weiss, G, Margolin, K et al. (1999). High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17: 2105–2116. [DOI] [PubMed] [Google Scholar]
- Pardoll, DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12: 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, SA (2014). IL-2: the first effective immunotherapy for human cancer. J Immunol 192: 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, SA, Yang, JC, Sherry, RM, Kammula, US, Hughes, MS, Phan, GQ et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, T, Akiba, H, Iwai, H, Matsuda, H, Aoki, M, Tanno, Y et al. (2002). Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169: 5538–5545. [DOI] [PubMed] [Google Scholar]
- Barber, DL, Wherry, EJ, Masopust, D, Zhu, B, Allison, JP, Sharpe, AH et al. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439: 682–687. [DOI] [PubMed] [Google Scholar]
- Dong, H, Strome, SE, Salomao, DR, Tamura, H, Hirano, F, Flies, DB et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8: 793–800. [DOI] [PubMed] [Google Scholar]
- Dong, H, Zhu, G, Tamada, K and Chen, L (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 5: 1365–1369. [DOI] [PubMed] [Google Scholar]
- Freeman, GJ, Long, AJ, Iwai, Y, Bourque, K, Chernova, T, Nishimura, H et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192: 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer, JR, Tykodi, SS, Chow, LQ, Hwu, WJ, Topalian, SL, Hwu, P et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, SL, Hodi, FS, Brahmer, JR, Gettinger, SN, Smith, DC, McDermott, DF et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid, O, Robert, C, Daud, A, Hodi, FS, Hwu, WJ, Kefford, R et al. (2013). Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369: 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh, M, Johnson, LA, Heemskerk, B, Wunderlich, JR, Dudley, ME, White, DE et al. (2009). Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114: 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros, A, Robbins, PF, Yao, X, Li, YF, Turcotte, S, Tran, E et al. (2014). PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest 124: 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung, JK and Sander, JD (2013). TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. (2013). RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D et al. (2014). Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov, FD, Rebar, EJ, Holmes, MC, Zhang, HS and Gregory, PD (2010). Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646. [DOI] [PubMed] [Google Scholar]
- Miller, JC, Tan, S, Qiao, G, Barlow, KA, Wang, J, Xia, DF et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148. [DOI] [PubMed] [Google Scholar]
- Cai, M and Yang, Y (2014). Targeted genome editing tools for disease modeling and gene therapy. Curr Gene Ther 14: 2–9. [DOI] [PubMed] [Google Scholar]
- Maier, DA, Brennan, AL, Jiang, S, Binder-Scholl, GK, Lee, G, Plesa, G et al. (2013). Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther 24: 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, SA, Yang, JC, Sherry, RM, Kammula, US, Hughes, MS, Phan, GQ et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, SL, Solomon, D and Rosenberg, SA (1989). Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol 142: 3714–3725. [PubMed] [Google Scholar]
- Huang, J, El-Gamil, M, Dudley, ME, Li, YF, Rosenberg, SA and Robbins, PF (2004). T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol 172: 6057–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S and Manber, U (1992). Fast text searching: allowing errors. Commun ACM 35: 83–91. [Google Scholar]
- Perez, EE, Wang, J, Miller, JC, Jouvenot, Y, Kim, KA, Liu, O et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, E, Turcotte, S, Gros, A, Robbins, PF, Lu, YC, Dudley, ME et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedoeem, A, Azoulay-Alfaguter, I, Strazza, M, Silverman, GJ and Mor, A (2014). Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol 153: 145–152. [DOI] [PubMed] [Google Scholar]
- Iwai, Y, Ishida, M, Tanaka, Y, Okazaki, T, Honjo, T and Minato, N (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA 99: 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, F, Kaneko, K, Tamura, H, Dong, H, Wang, S, Ichikawa, M et al. (2005). Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 65: 1089–1096. [PubMed] [Google Scholar]
- Velu, V, Kannanganat, S, Ibegbu, C, Chennareddi, L, Villinger, F, Freeman, GJ et al. (2007). Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol 81: 5819–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziewicz, H, Ibegbu, CC, Fernandez, ML, Workowski, KA, Obideen, K, Wehbi, M et al. (2007). Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol 81: 2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Lu, YC, El-Gamil, M, Li, YF, Gross, C, Gartner, J et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian, SL, Drake, CG and Pardoll, DM (2012). Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 24: 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson, EJ, Sharfman, WH, Drake, CG, Wollner, I, Taube, JM, Anders, RA et al. (2013). Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res 19: 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama, A, Pan, J, Zhang, P, Reith, W, Mach, B, Tenen, DG et al. (1999). Dimeric RFX proteins contribute to the activity and lineage specificity of the interleukin-5 receptor alpha promoter through activation and repression domains. Mol Cell Biol 19: 3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Dudley, ME, Wunderlich, JR, Hughes, MS, Yang, JC, Sherry, RM et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314: 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Morgan, RA, Feldman, SA, Yang, JC, Sherry, RM, Dudley, ME et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Feldman, SA, Wilson, WH, Spaner, DE, Maric, I et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule, MA, Savoldo, B, Myers, GD, Rossig, C, Russell, HV, Dotti, G et al. (2008). Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14: 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abate-Daga, D, Hanada, K, Davis, JL, Yang, JC, Rosenberg, SA and Morgan, RA (2013). Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood 122: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651. [DOI] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon, Y, Choi, VM, Xia, DF, Vo, TD, Gregory, PD and Holmes, MC (2010). Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods 7: 459–460. [DOI] [PubMed] [Google Scholar]
- Guschin, DY, Waite, AJ, Katibah, GE, Miller, JC, Holmes, MC and Rebar, EJ (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256. [DOI] [PubMed] [Google Scholar]
- Needleman, SB and Wunsch, CD (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.