

Abstract
The adoptive transfer of chimeric antigen receptor (CAR) T cell represents a highly promising strategy to fight against multiple cancers. The clinical outcome of such therapies is intimately linked to the ability of effector cells to engraft, proliferate, and specifically kill tumor cells within patients. When allogeneic CAR T-cell infusion is considered, host versus graft and graft versus host reactions must be avoided to prevent rejection of adoptively transferred cells, host tissue damages and to elicit significant antitumoral outcome. This work proposes to address these three requirements through the development of multidrug-resistant T cell receptor αβ-deficient CAR T cells. We demonstrate that these engineered T cells displayed efficient antitumor activity and proliferated in the presence of purine and pyrimidine nucleoside analogues, currently used in clinic as preconditioning lymphodepleting regimens. The absence of TCRαβ at their cell surface along with their purine nucleotide analogues-resistance properties could prevent their alloreactivity and enable them to resist to lymphodepleting regimens that may be required to avoid their ablation via HvG reaction. By providing a basic framework to develop a universal T cell compatible with allogeneic adoptive transfer, this work is laying the foundation stone of the large-scale utilization of CAR T-cell immunotherapies.
Introduction
The adoptive transfer of chimeric antigen receptor (CAR) T cells represents a highly promising strategy to fight against multiple cancer indications. This strategy relies on the engineering of T cells to redirect their cytolytic activity toward malignant cells via transgenic expression of a tumor antigen-specific receptor at their cell surface. Today, the current protocols of treatment consist in autologous adoptive cell transfer (ACT). In this approach, T lymphocytes recovered from patients, are genetically modified and expanded in vitro before infusion back into patients. This process requires precise logistics, proximity between dedicated production facilities and the bedside and more importantly, delays the availability of genetically engineered T cells for patient treatment.
Recent reports proposed to address these issues by developing a CAR T cell compatible with allogeneic adoptive transfer.1,2,3 This alternative approach consists in generating from a third-party donor, a bulk population of CAR T cells that can be injected into multiple patients, a strategy likely to unleash the full potential of CAR T-cell therapies by bringing them to the industrial level. When allogeneic CAR T-cell adoptive transfer is considered, host versus graft (HvG) and graft versus host (GvH) reactions must be avoided to safely allow effector cells to engraft, proliferate, and specifically kill given tumor cells in patients. While a GvH reaction can be tackled by sequestration of lymphocytes in lymph nodes3 or by targeted gene knockout of T cell receptor (TCRαβ) within CAR T-cell genome,2,4 controlling their rejection via HvG remains a technological hurdle that need to be addressed. It has been proposed that HvG reaction, involving host T-cell activation after direct or indirect allorecognition,5 could be prevented by lymphodepleting regimens. Such regimens, usually consisting of alkylating agents and/or purine nucleotide analogues (PNA) compounds, are known to deplete the host immune system for weeks to month periods, depending on the dose being used.6 They could thus theoretically create a therapeutic window during wich allogeneic CAR T cell could eradicate tumors before being rejected via HvG reaction. If this scenario can be envisionned for the treatment of some hematological tumors reported to be rapidely eradicated by ACT (< 1 month),7,8,9,10,11 it may not be applicable to other type of malignancies including solid tumors that may require an extended period of treatment. Thus, developing strategies to control the extent of therapeutic window for allogeneic ACT treatments is highly desired. One way to address this challenge would be to prolong lymphodepleting regimens during adoptive T-cell transfer. However, because such regimens are also highly likely to deplete adoptively transferred CAR T cells, this strategy requires to use regimen resistant-CAR T cells.
This report describes the genetic engineering and characterization of CAR T cells resistant to three different PNAs currently used in clinic as preconditionning lymphodepleting regimens. Our engineering process includes a lentiviral transduction for CAR expression followed by the simultaneous TALEN-mediated gene processing of TCR constant region (TRAC) and deoxycytidine kinase (dCK) respectively responsible for TCRαβ surface expression and PNA toxicity. It enables expansion as well as recovery of a homogeneous population of engineered CAR T cells that retain their proliferative capacity and cytolitic activity toward tumor cells in the presence of lymphodepleting dose of different PNAs. We envision that these engineered CAR T cells could be generated from third party healthy donors and used in any patients as antitumor allogeneic immunotherapy without generating TCRαβ-dependent GvH reaction. Their drug resistance properties could enable them to resist to simultaneous infusion of lymphodepleting regimens to inhibit the host immune system and control their rate of ablation via HvG reaction.
Results
TALEN-mediated TRAC/dCK dual gene processing is highly efficient in primary T cells
PNAs used as lymphodepleting regimens or as antineoplastic drugs are usually delivered as nucleoside prodrugs. They become toxic after being metabolized to their respective triphosphate forms through sequential phosphorylations catalyzed by deoxycytidine kinase (dCK). To prevent such metabolization from occuring in T cells and to endow them with PNA resistance properties, we sought to inactivate the gene encoding dCK. For this purpose, a TALEN targeting the second exon of the DCK gene (Figure 1a, upper panel) was designed, assembled and its corresponding mRNA was used to electroporate primary T cells. Because our goal was to inactivate TCRαβ concomitantly with dCK, we coelectroporated mRNA encoding TRAC TALEN, reported earlier to efficiently inactivate TCRα gene and prevent TCRαβ surface expression.12 The molecular events generated by both TALEN at their respective locus, were characterized in depth by high-troughput DNA sequencing (Figure 1a). Our sequencing results revealed nucleotide insertions or deletions (Indels) at frequencies of ~80 and 76% at the TRAC and dCK loci respectively. Thus, TALEN-mediated processing of these two genes was highly efficient under our experimental conditions. Deconvolution of sequencing results allowed us to observe that the vast majority of processing events obtained with both TALEN was due to deletions, a pattern consistent with several reports regarding TALEN activity.13 For both loci, deletion events ranged from 2 to 20 bp with a minority of events > 20 bp. Regarding insertions, most of them consisted of two to four nucleotides inserted within the target spacer and generally corresponded to sequence duplication (data not shown).
Figure 1.

TALEN treatment of primary T cells enables simultaneous and highly efficient processing of TRAC and dCK genes. (a) Overall gene architectures of dCK and TRAC open reading frames. Exons (dark blue) and introns (light blue) are indicated. dCK and TRAC TALEN targets are displayed and underlined. The TALEN binding sites and spacer sequences are respectively displayed in upper and lower cases letters. (b) Genotypic characterization of TALEN-mediated inactivation of dCK and TRAC genes by high throughput DNA sequencing. T cells treated with dCK and TRAC TALEN were allowed to grow for 6 days, their genomic DNA was extracted, amplified using TRAC or dCK specific PCR amplicons and analyzed by high throughput DNA sequencing. The frequencies of indels generated at TRAC and dCK locus are indicated.
TALEN-mediated TRAC/dCK dual gene processing does not promote genetic adverse event
Potential off-site targets were carefully evaluated in silico and in vitro for both loci. We defined off-site targets as genomic sequences bearing any combinations of TALEN half binding sites containing ≤ 4 mismatches with respect to the sequence to target and separated form one another by 9 to 30 bp. Because we intended to perform two simultaneous gene inactivations, all combinations of TRAC and dCK TALEN half binding sites were considered (Table 1) as well as how close they were to coding sequences. The vast majority of potential offsite targets identified in silico contained a total ≥ 5 mismatches when combining the two half-sites, a number reported to abrogate TALEN activity.14,15 From this list, 16 different off-sites located inside or less than 30 bp away from exonic regions were selected and studied by high-through DNA sequencing (Supplementary Material and Supplementary Figure S1). As expected, we were not able to detect indels events above the threshold set by the control experiment performed with mock transfected primary T cells (Table 1). In addition, the presence and stability of translocations induced by two simultaneous TALEN treatment were also checked by nested polymerase chain reaction (PCR). Our results showed that TALEN treatment induced translocations that were selected against during a prolonged 30-day expansion (Supplementary Materials and Supplementary Figure S2). Together these results are consistent with the high specificity of TALEN and their suitability for therapeutic applications.14,15
Table 1. Assessment of targeting specificity of TRAC and dCK TALEN in primary T cells.

TALEN-mediated TRAC/dCK dual gene processing disrupts TCRαβ expression at the surface of primary T cells and increased their resistance to clofarabine
To verify that TRAC gene processing prevented surface expression of the functional TCRαβ receptor in primary T cells, cells were analyzed by flow cytometry, 6 days postelectroporation with mRNAs encoding TRAC and dCK TALEN. As expected from previous results obtained in jurkat cells,12 we found that 81% of TALEN-treated T cells had lost TCRαβ surface expression (Figure 2a, lower left panel). TCRαβ-deficient T cells were purified using anti-CD3 magnetic beads (Figure 2a, lower right panel), enabling the recovery of a highly homogenous preparation of TCRαβ-deficient T cells (>99% of the bulk population). Further analysis of their genomic DNA allowed us to determine that they displayed 89 and 93% of indels at TRAC and dCK loci respectively (Figure 2b).
Figure 2.

Multiplex TALEN treatment of primary T cells enables efficient depletion of TCR membrane expression. (a) Purification of TRAC KO T cells from the bulk population of cell transfected with mRNA encoding TRAC and dCK TALEN. Labeling control experiment performed with T cells in the presence or in the absence of anti TCR mAb-PE (upper panel) and monitoring of TCRαβ-deficient cells gathered before and after TRAC KO T cells purification using anti-CD3 magnetic beads and MACS LD-column (lower panel). (b) Genotypic characterization of TALEN-mediated inactivation of TRAC and dCK genes by high throughput DNA sequencing before and after TCR-cells purification.
To determine if dCK gene inactivation resulted in PNA resistance, T cells treated with dCK and TRAC TALEN were analyzed for their in vitro resistance to the purine nucleotide analogue clofarabine. T cells were treated with dCK and TRAC TALEN and allowed to grow for 13 days before assessing their resistance property to clofarabine (Figure 3a, upper part). Resistance was determined by comparing the viability of wild-type (wt) and TALEN-treated T cells after 48 hours incubation in the presence of increasing amounts of clofarabine. Our results showed that the viability of wt T cells dropped at low doses of clofarabine with an IC50~100 nmol/l, while engineered T cells viability remained constant up to 10 µmol/l of clofarabine and displayed IC50 values of about 100 µmol/l (Figure 3b open symbols and Table 2). Such IC50 differences, spanning about 3 logs of clofarabine concentration, indicated that TALEN-mediated processing of the dCK gene enabled increased resistance of T cells to clofarabine. In addition, this resistance property was not impaired by the simultaneous inactivation of TRAC, as indicated by similar IC50 values obtained for single and double knock-out T cells (Table 1 and Supplementary Figure S3, KO-D and KO-D/T, compare open triangles and open squares).
Figure 3.

TALEN-mediated dCK processing increased primary T cells resistance to clofarabine without needing time consuming and potentially harmfull drug selection process. (a) Scheme of the experimental workflow used to engineer and characterize T cells. Primary T cells were electroporated with mRNA encoding dCK and TRAC TALEN to generate double KO T cells called T KO D/T in the legend. Cells were allow to grow for 3 days (D0-D3) and then cultured for 5 days in the presence or in the absence of 1 µmol/l clofarabine (D3-D8). After 5 additionnal days of culture (D8-D13), cells were plated in a 96-well plate at 105 cells per well in presence of increasing concentration of clofarabine (from 0 to 100 µmol/l) and in a total volume of 100 µl. Two days after plating (D15), viability of cells was determined by flow cytometry. (b) Clofarabine resistance properties of untreated (T) and TALEN-treated T cells (T KO D/T) cultured in the absence or in the presence of 1 µmol/l clofarabine (+Clo). Solid lines are best fits for decreasing exponential function (see Material and Methods) obtained for experiments performed in the absence of clofarabine selection.
Table 2. Assessment of IC50 values of different PNAs toward wt and engineered T cells.
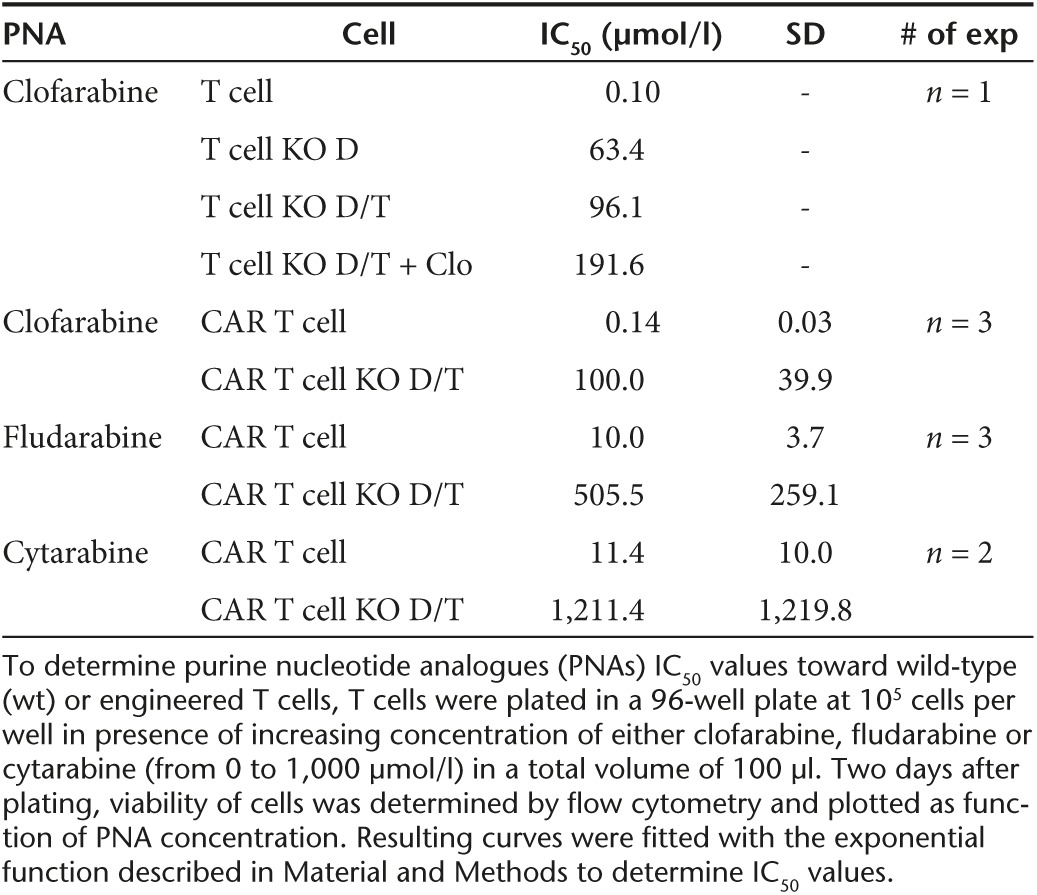
Because TALEN-mediated dCK gene inactivation was not total (Figure 1b), we hypothesized that depleting the remaining wt T cells bearing active dCK from the bulk T-cell population would improve the overall resistance properties the cells. To test this assumption, we implemented a 5-day selection process in the presence of 1 µmol/l clofarabine, a dose previously determined to be lethal for wt T cells (1 µmol/l, Figure 3a, bottom). These T cells were then cultured for five additional days in the absence of clofarabine before testing their clofarabine resistance at D13 as described above (Figure 3b, closed symbols). Our results showed that the 5-day selection barely increased IC50 values (Table 2, Figure 3b, compare close and open symbols), indicating that such a selection process did not markedly improve clofarabine resistance of the TALEN-treated T-cell population. Therefore, our results indicated that TALEN-mediated inactivation of dCK gene, even when combined with TRAC inactivation, is efficient enough to generate clofarabine resistant T cells without requiring any chemical selection or purification process.
TALEN-treated CAR T cells are resistant to multiple PNAs and are able to proliferate in the presence of their clinically relevant dose
Having demonstrated that treatment of primary T cells by TRAC and dCK TALEN could eliminate cell surface expression of TCRαβ and endow them with clofarabine resistance, we next evaluated their potential as an antitumor immunotherapy agent toward Daudi cells, a CD19+ lymphoblast cell line used here as an convenient tumor model. To do so, we transduced T cells with a lentiviral vector containing a polycistronic DNA expression cassette encoding a membrane exposed rituximab-dependent depletion system (RQR8 (ref. 16)), followed by a 2A cis-acting hydrolase element and a CD19-specific CAR7 (Figure 4a). Such a polycistronic coding sequence was designed to endow T cells with cytolytic properties toward the CD19+ tumor cells and to allow their selective ablation in an event of uncontroled acute cytotoxicity using the complement-dependent RQR8-specific monoclonal antibody (mAb) named rituximab.16
Figure 4.

TALEN-treated CAR T cells are resistant to lethal dose of different purine and pyrimidine nucleotide analogues. (a) Scheme of the polycistronic DNA expression cassette encoding the membrane exposed rituximab-dependent depletion system (RQR8), followed by a 2A cis-acting hydrolase element and the anti-CD19 CAR. (b) Scheme of the experimental workflow used to engineer and characterize T cells proliferative capacity and resistance properties toward clofarabine, fludarabine and Cytarabine. (c,d,e) Clofarabine, fludarabine, and cytarabine IC50 determination. 105 CAR T cells were incubated in the presence of increasing concentration of clofarabine, fludarabine or cytarabine for 48 hours before being analyzed by flow cytometry to determine their viability. Experimental viability values obtained were normalized with respect to the one obtained in the absence of drug and then plotted as a function of drug concentration. The resistance properties of untreated (CAR T cell) and TALEN-treated CAR T cells (CAR T cell KO D/T) are displayed. (f–h) Proliferation capacity of CAR T cells in the presence or in the absence of clofarabine, fludarabine and cytarabine. 106 CAR T cells were grown for a total of 9 days in the absence or in the presence of 1, 5, and 10 µmol/l of clofarabine, fludarabine and cytarabine respectively. The number of viable untreated (CAR T cell) and TALEN-treated CAR T cells (CAR T cell KO D/T) counted at each passage are plotted as a function of time. Vertical arrows indicate each passage step where cells were diluted to 106 cell/ml in the presence or in the absence of drug.
Efficient generation of RQR8-positive CAR T cells (Supplementary Figure S4) enabled us to begin our characterization by evaluating their resistance toward clofarabine. We made the most of this opportunity to test other purine and pyrimidine nucleotide analogues currently used in clinic as lymphodepleting regimens or antineoplastic agents. We first determined the IC50 values of clofarabine, fludarabine, and cytarabine with respect to untreated and TALEN-treated CAR T cells using the protocol described earlier (Figure 4b). Consistent with our previous results, our data showed that clofarabine IC50 value obtained for TALEN-treated CAR T cells was about 3 logs higher than the ones obtained for untreated CAR T cells (100 µmol/l and 140 nmol/l respectively, Figure 4c and Table 2). Interestingly, similar differences in IC50 values were obtained with fludarabine and cytarabine (Figure 4d,e respectively and Table 2). Together, these data first indicated that the lentiviral transduction of a RQR8-CAR construction did not impair the ability of engineered T cells to resist to clofarabine. In addition, they provided clear evidence that these cells were resistant not only to purine (i.e., clofarabine and fludarabine) but also to pyrimidine nucleotide analogues (i.e., cytarabine), thus demonstrating their broad spectrum of PNA resistance.
The ability to proliferate in the presence of clinically relevant doses of PNA was then evaluated. Toward this aim, untreated and TALEN-treated CAR T cells were grown for 10 days in the presence or in the absence of 1 µmol/l clofarabine, 5 µmol/l fludarabine, or 10 µmol/l cytarabine, the doses previously reported as their respective average Cmax after their uptake in human patients.17,18,19 Viable cell numbers were determined every 2–3 days and plotted as a function of time (Figure 4f,g,h). Our results demonstrated that the proliferation of untreated and TALEN-treated CAR T cells were similar in the absence of any PNA. Thus, the dual inactivation of TCR and dCK did not impair the rate of T cells division. In addition, the presence of PNAs in the culture media, strongly impaired or completely abolished proliferation of untreated CAR T cells while being harmless with respect to TALEN-treated CAR T cells (Figure 4f,g,h). Similar conclusions could be drawn out of identical experiments performed with three other blood donors (Supplementary Figure S5). Such difference between the proliferation pattern of untreated and TALEN-treated CAR T cells were consitent with their respective drug resistance curves. Indeed, the presence of either 1 µmol/l clofarabine, 5 µmol/l fludarabine, and 10 µmol/l cytarabine decreased the cellular viability of untreated CAR T cells by about 80, 30, and 50% respectively while only a minimal impact was observed with TALEN-treated CAR T cells (Figure 4c–e). Noteworthy, TALEN-treated CAR T cells were also able to proliferate in the presence of a combination of clofarabine and cytarabine (1 and 10 µmol/l respectively, Supplementary Materials, Supplementary Figure S6). Together, these results indicated that TALEN-treated CAR T cells were able to resist and proliferate in the presence of clinically relevant doses of different PNAs.
Clofarabine prevents alloreactivity of PBMC toward CAR T cells in vitro
To evaluate the ability of PNA to prevent alloreactivity of peripheral blood mononuclear cells (PBMCs) toward CAR T cells, we performed allogeneic mixed lymphocyte reaction in the presence or in the absence of clofarabine. Briefly, PBMC freshly purified from a donor B (PBMC B) and labeled with carboxy fluorescein succinimidyl ester (CFSE) were incubated with either T cells purified from the same blood donor (T cells B) or with CAR T cells engineered out of a different blood donnor (CAR T cell A). After 6 days of incubation, we evaluated the extent of PBMC viability, activation, and proliferation by flow cytometry and anti IFNγ ELISA (Supplementary Figure S7). Our results showed that when mixed with CAR T cells A, PBMC B showed a typical pattern of activation/proliferation that was similar to the one observed in the positive control experiment performed with phytohaemagglutinin (PHA) (Supplementary Figure S7a). This pattern consisted in a significant dilution of CFSE dye, an increase of cellular viability and size along with a high level of IFNγ release (Supplementary Figure S7b), typically associated with T cells activation and blasting. As expected, this phenomenom was not observed in the presence of T cells B. Interestingly addition of 1 µmol/l clofarabine to the reaction media, was able to deplete about than 90 % of PBMC (Supplementary Figure S7c) and impair their alloreactivity toward CAR T cells, as illustrated by the low level of IFNγ release (Figure 7b). Together, our results indicated that clinically relevant dose of clofarabine prevents the alloreaction between PBMC and CAR T cells in vitro.
TALEN-treated CAR T cells display similar antitumor cytotoxicity than wt CAR T cells
The cytolytic properties of TALEN-treated CAR T cells toward CD19+ tumor cells were then evaluated. For that purpose, we performed a flow cytometry-based cytotoxicity assay20 to assess their killing potency and evaluate their level of specificity toward CD19+ cells. Our results showed that untreated and TALEN-treated CAR T cell displayed similar specific cell lysis activity toward Daudi cells (35% ± 2 and 39% ± 2 respectivelly, Figure 5 and Supplementary Figure S8). These results indicated that dual inactivation of TRAC and dCK genes did not markedly affect CAR T cells cytolytic activity and specificity.
Figure 5.

CAR T-cell cytolytic activity is not affected by dual inactivation of dCK and TCRαβ. To assess the impact of TALEN treatment on the cytolytic activity and specificity of CAR T cells toward Daudi cells the flow-based cytotoxicity assay reported in ref. 20 was used. 104 Daudi (CD19+) and 104 K562 (CD19- negative control) cells labeled respectively with CellTrace CFSE and CellTrace violet were coincubated with 105 effector CAR T cells (E/T ratio of 10:1), for 5 hours at 37 °C. Cells were then recovered and their viability was determined by flow cytometry. Viability of Daudi and K562 cells was used to calculate the frequencies of targeted cytotoxicity using the formula decribed in ref. 20. Untransduced T cells, untreated or treated by dCK and TRAC TALEN (Lanes 1 and 2), were used as negative controls. Error bars represent the mean of two experiments + SD.
We then investigated the ability of TALEN-treated CAR T cells to retain their antitumor activity after being expanded in the presence of lethal dose of PNA. To this end, cells were cultured for 2–9 days in the presence of 1 µmol/l clofarabine and their cytolytic activity was assessed. Using the flow cytometry-based cytotoxicity assay described earlier, the specific cell lysis activities of TALEN-treated CAR T cells cultured with or without clofarabine were compared to those obtained for untreated CAR T cells or T cells cultured without clofarabine (Figure 6 and Supplementary Figure S9). Our results showed that the presence of 1 µmol/l clofarabine, while lethal for untreated CAR T cells (Figure 4c, closed circles and Figure 4f, open triangles), did not impair the cytolytic properties of TALEN-treated CAR T cells even after 2–9 days of culture (Figure 6, compare lanes 1 and 2 with 3 and 4). Indeed, they display similar activity to those cultured in the absence of drug, whatever the length of culture of our experimental conditions. In addition, our results confirmed that the dual inactivation of dCK and TRAC did not impair T-cell cytolytic activity as demonstrated by the similar specific cell lysis activites observed for the TALEN-treated and untreated CAR T cells (Figure 6, compare lanes 2 with lane 6).
Figure 6.

TALEN-treated CAR T cells retain their cytolytic activity after 2 to 9 days of culture in the presence of clinically relevant dose of clofarabine. 104 Daudi (CD19+) and 104 K562 (CD19- negative control) cells labeled respectively with CellTrace CFSE and CellTrace violet were coincubated for 5 hours at 37 °C with 105 of TALEN-treated CAR T cells grown for 2 or 9 days in the presence or in the absence of 1 µmol/l clofarabine. Cells were then recovered and their viability was determined by flow cytometry. Viability of Daudi and K562 cells were used to calculate the frequencies of specific cell lysis using the formula decribed in ref. 20 (lanes 1 to 4). T cells and CAR T cells were cultivated in the absence of clofarabine for 9 days were respectively used as negative and positive controls of specific cell lysis activity (lanes 5 and 6). Results are displayed as specific cell lysis activities relative to the control experiment performed with CAR T cells. Error bars represent the mean of two experiments + SD.
TALEN-treated CAR T cells can pair up with PNA chemotherapy for better antitumor activity
Clofarabine is currently used in clinic as cytotoxic agent to treat patients with relapsed or refractory acute lymphoblastic leukemia and displays potent cellular toxicity toward multiple tumor cell lines,21,22,23 including Daudi cells.24 We thus sought to investigate whether a combination of CAR T-cell immunotherapy with clofarabine chemotherapy would enhance the antitumor activity toward Daudi cells. To do so, TALEN-treated CAR T cells were used to perform a flow cytometry based cytotoxicity assay with CFSE-labeled Daudi cells preincubated for 24 hours in the presence of 1 µmol/l clofarabine (Cmax). To effectively assess the potential benefit of combination therapy, two control experiments were performed to determine the cytotoxicity elicited by clofarabine or CAR T cell alone toward Daudi cells. An additional experiment performed in the absence of both treatments was performed and used as reference for Daudi cells maximal viability. Our results showed that combination therapy led to more than a 50% drop of Daudi cells viability (Figure 7a, lane 4 and Supplementary Figure S10a). Such decrease was greater than those obtained with TALEN-treated CAR T cells alone (Figure 7a, lane 3) or with clofarabine alone (Figure 7a, lane 2). Similar results were obtained when clofarabine was replaced by 10 µmol/l cytarabine (Cmax, Figure 7b and Supplementary Figure S10b). Therefore, altogether our results indicated that when provided simultaneously, both treatments elicited a better antitumor activity compared with either treatment alone.
Figure 7.

TALEN-treated CAR T cells can pair up with chemotherapy for better antitumor activity. 104 Daudi (CD19+) cells preincubated in the presence or in the absence of purine nucleotide analogues (1 µmol/l clofarabine or 10 µmol/l cytarabine) for 24 hours, were labeled with CellTrace CFSE and coincubated for 5 hours at 37 °C in the absence or the presence of 105 TALEN-treated CAR T cells (CAR T KO D/T). Cells were then recovered and their viability was determined by flow cytometry. The residual viability of tumor cells (Daudi) obtained in the presence or in the absence of clofarabine (a) or cytarabine (b) are displayed. p values numbers, calculated according to the procedure described in material and methods, are indicated. Error bars represent the mean of 2 or 3 experiments + SD performed in the presence of clofarabine or cytarabine respectively.
Discussion
The clinical outcome of CAR T-cell adoptive therapies is intimately linked to the ability of effector cells to engraft, proliferate, and specifically kill given tumor cells. When allogeneic CAR T-cell infusion is considered, HvG and GvH reactions must be avoided to prevent, (i) rejection of adoptively transferred cells, (ii) host tissue damage, and (iii), to elicit significant antitumor activity. In this study, we have engineered primary T cells so that they could potentially address these three requirements. Using the TALEN-mediated gene editing method, we generated a homogeneous population of TCRαβ-deficient CAR T cells resistant toward clofarabine, fludarabine, and cytarabine, three nucleotide analogues used in the clinic as lymphodepleting preconditionning regimens for ACT. TALEN-treated CAR T cells were able to proliferate in vitro in the presence of drugs and displayed similar antitumor properties compared to untreated CAR T cells. In addition, we showed that when combined, chemotherapy and CAR T cells immunotherapy resulted in a better antitumor activity compared with either treatment alone. Due to the absence of TCRαβ at the surface, such engineered T cells could be generated from third party donors and used in patients as an antitumor allogeneic immunotherapy treatment without generating a GvH reaction. In addition, their broad PNAs resistance properties could enable them to resist to multiple lymphodepleting regimens used to inhibit the host immune system and prevent their ablation via HvG reaction.
A haute couture editing method to process primary T-cell genome in a highly precise and efficient manner: a prerequisite for therapeutic applications
The efficiency of TALEN-mediated dual inactivation of dCK and TCRα gene in primary T cells was unprecedented. Both loci were shown to be processed at Indels frequencies of ~90%, a value exceeding former studies describing gene inactivation in primary T cells.2,4 Such high indels frequencies are likely due to multiple parameters that were chosen in an educated manner.25 Among the most important ones are the choice of TALEN target and the method used to vectorize TALEN into T cells.
Regarding the choice of TRAC and dCK TALEN targets, we made sure to carefully consider their epigenetic status in T cells, their nucleotide composition, and predict their potential off-site targets using the TALEN specificity matrices reported in ref. 14. Regarding their position, both TALEN targets were chosen to be at, or within the immediate vicinity of an exon/intro border (Figure 1a). Processing of this type of region is expected to disrupt donor or acceptor splice sites in addition to the exonic regions, a strategy highly likely to promote aberrant splice variants and result in the production of inactive truncated proteins.25 Concerning the vectorization method, we chose to use RNA encoding TALEN and the Agilpulse electroporation method. This combination was shown to have two main advantages to engineer primary T cells. On the one hand, using mRNA instead of DNA prevents any unexpected random insertion of TALEN encoding sequence and improves the gene processing efficiency (Poirot et al., submitted). On the other hand, this electroporation method, optimized in-house for T cells transfection, was shown to decrease cellular stress and mortality usually associated with this step.12
Highly efficient on-target processing did not imply off-site target activity and downstream genetic adverse events. Indeed, we showed that while about 90% of both TRAC and dCK loci were processed, no off-site activity could be detected above the background measured with mock transfected T cells, at the most probable off-site targets predicted in silico (see Table 1 and Supplementary Material). Regarding translocation events, they could be detected in the double TALEN-treated CAR T cells 10 days after being treated with TALEN. However, after an additional 20 days of culture, corresponding to the cellular expansion step needed for a standard CAR T-cell production process,11 most of these events were selected against. This likely indicated that the translocations were unstable and did not confer any proliferative advantages to engineer CAR T cells. Untreated and TALEN-treated CAR T cells consistently displayed similar proliferation rates (Figure 4f–h), indicating that simultaneous processing of TRAC and dCK genes did not generate durable adverse events in T-cell genome under our experimental conditions. Regarding that matter, it is important to consider that the frequency of translocation induced by two simultaneous TALEN treatments are loci-dependent.26,27 When clear evidence of a stable translocation is observed, one may consider sequential gene inactivation using short lifespan mRNA encoding engineered nuclease.4 However, although it has yet to be demonstrated, such sequential process could impact the antitumor efficacy of CAR T cell by increasing the cellular stress associated with consecutive transfection steps and by extending the length of the production process.
Engineered CAR T cells display stable, robust and multi drug resistant phenotype allowing prevention of HvG alloreaction
We have demonstrated that TALEN-mediated inactivation of dCK translated into stable and efficient purine and pyrimidine nucleotide analogue resistance phenotype of CAR T cells. We were able to recover PNA-resistant CAR T cells without requiring any PNA-dependent selection process. In addition, inactivation of dCK neither altered CAR T cells cytotoxicity toward tumor cells nor their capacity to proliferate. This latter property was expected because rather than being involved in the pivotal de novo synthesis of dNTP, dCK is implicated in the nonessential nucleotide salvage pathway.28 Thus, TALEN-mediated inactivation of dCK represents two major advantages for the production of PNA-resistant CAR T cells: (i) it allows to expand and recover large quantities of CAR T cells in a short time frame and (ii) it does not expose them to genotoxic selection agents, a harmfull procedure that would jeopardize their utilization in adoptive transfer therapies.
The PNA resistance properties of CAR T cells allowed them to resist to lymphodepleting treatment (Figure 4) necessary to prevent HvG reaction in an allogeneic context (Supplementary Figure S7). Thus, we envisioned that this treatment could modulate the extent of the therapeutic window during which allogeneic CAR T cells could engraft and eradicate tumors before being rejected by the patient immune system. The benefit of sustained engraftment of CAR T cells is still unclear. Indeed, previous autologous ACT studies showed that 15 days was sufficient to elicit a complete response in multiple instances.9 Nonetheless, a longer persistence could benefit other patients. Thus, ideally, controlling the duration of lymphodepletion in allogeneic settings could provide CAR T cells with enough time to eliminate tumor cells while minimizing risks associated with prolonged lymphodepletion.
The broad PNA resistance of engineered CAR T cells generated in this work offers different options for lymphodepletion regimen. Today, fludarabine alone or in combination with cyclophosphamide, is frequently used as lymphodepleting agent prior to adoptive T-cell transfer.8,9,29 Utilization of either one or both chemotherapies was shown to improve antitumor efficacy by removing endogenous cellular elements (cytokine sinks) that compete with transferred T cells for supportive cytokines.30 Because of their similar chemical structure and physiological properties, clofarabine and cytarabine could also be used for the same purpose as demonstrated recently for adoptive CAR T-cell immunotherapy of acute lymphoblastic leukemia.8
A multidrug resistant CAR T cell allowing combination therapy for better therapeutic outcomes
Endowing CAR T cell with resistance to PNA is not the only functionality allowed by our gene editing approach as it could also enable combination therapy. Combination therapy using chemo- and cell-based therapies represents an appealing strategy to improve the efficiency and persistence of treatment as demonstrated for stem cell adoptive therapy.31,32,33 The potential advantage of this approach for CAR T-cell immunotherapies relies on the well-known ability of certain chemotherapies to increase the persistence and activity of effector T cells,30 increase their tumor trafficking,34,35 modulate immunosupressive factors36 and for some of them including PNAs, directly affect tumor cell viability.6 While highly promising, the development of combination therapies have been dampened by the cytotoxic properties of chemotherapy toward immunotherapeutic cellular agents. However, during the past ten years, development of genome engineering techniques through transgene expression or shRNA transfection have enabled development of cells resistant to methotrexate,37 trimetrexate and temolozide,38 cyclophosphamide derivative,37 micofenolate mofetil,39 6-thioguanine,31 and rapamycin40 as nonexhaustive examples. While our work extended the portfolio of genome engineering approaches enabling permanent and stable cellular drug resistance without relying on transgene expression, it also established the in vitro proof of principle that combining CAR T cells with PNAs lead to better antitumor outcome than either treatment alone. Noteworthy, because all the experiments delineated in this work were performed in vitro, we could not assess the therapeutic benefit of PNA-dependent immunomodulation including the depletion of regulatory T cells (Treg)41 and myeloid-derived suppressor cells (MDSC),42 two main cellular protagonists of T-cell inhibtion. We envision that inhibition of such cellular subsets including others, could potentialize CAR T-cell cytolytic activity in vivo and thus significantly improve the overall therapeutic outcome. It will be now interesting to challenge this system in a more complex environment such as humanized xenograft mouse models.
A dual activity lock for better safety of adotive CAR T-cell therapy
CAR T cells therapy has been reported to be highly efficient at killing tumor cells. However, one of the downsides of this therapeutic approach is its potential to induce the cytokine release syndrome (CRS), an acute adverse event thought to be linked to their capacity to proliferate and depending on the tumor burden, to release toxic levels of cytokines.43 Such phenomenon was associated with multiple symptoms ranging from mild nausea and fever to life threatening organ faillures. Another downside relies on their capacity to stably engraft into patient for long periods ranging from 6 months44 up to 10 years.45,46 While such a long-term engraftment can be considered as a clear advantage to maintain on-target on-tumor activity and eradicate tumor cells, it also extends the on-target off-tumor activity of T cells in vivo. In the instance of anti-CD19+ CAR T-cell adoptive therapies, such activity leads to a longstanding aplasia of normal B cells that compromise normal immune system functions. Albeit not life threatening in that case, when other type of malignancies are considered, on-target off-tumor activity could occur on essential organs that express tumor antigen at lower levels and in that case, long-term persistence of CAR T cells could represent a greater threat.
Thus, avoiding the aformentionned acute and long-term adverse events appears to be mandatory to improve the safety of adoptive cell immunotherapies. For two main reasons, we believe that the cellular engineering method delineated in this work provides a basic frame work to address these points. First, because of the RQR8 depleting system expression at the surface of CAR T cells (Supplementary Figure S4), one could rapidly trigger their ablation as comprehensively demonstrated in ref. 16. Noteworthy, similar ablation could also be efficiently mediated by other surface exposed depleting systems including ICasp9/AP1903 (ref. 47) and in that sense, the CAR T cell developped in this work could be considered as a modular immunotherapeutic platform. Second, as cells are intended to be infused in an allogeneic context, they are prone to be naturally depleted by the host immune system recovery, a physiological phenomenon likely to follow PNAs clearance and to occur within few weeks through homeostatic peripheral expansion.48 Thus, although it has yet to be proven in vivo, we envision that by stopping the PNAs-based lymphodepletion regimen, one could promote ablation of CAR T cells and prevent the outbreak of unwanted long-term adverse events.
Conclusion
In summary, we report the development of multidrug resistant and TCRαβ-deficient CAR T cells compatible with allogeneic immunotherapy. Engineered CAR T cells resistance towards PNA chemotherapies enabled them to proliferate in their presence while retaining their antitumor activity and specificity. Such PNA resistance provides two main advantages. First, it could enable allogeneically transferred CAR T cells to survive lymphodepleting regimens used to maintain the host immune system in check and control their rate of ablation via a HvG reaction. Second, it could allow combination therapy, an approach likely to improve clinical outcomes by potentializing CAR T cell antitumor activity in vivo. In addition, due to their lack of surface exposed-TCR, these cells could be theoretically generated from third party healthy donors and used as allogeneic immunotherapy without generating GvH reaction. Finally, the methodology delineated here could also be applied to different cellular subtypes and thus improve outcomes of multiple cell-based therapies including those performed in autologous settings. In conclusion, we believe that the cellular engineering strategy described in this work will provide a basic frame work to generate a universal T cell that will foster large scale utilization of ACT and thus benefit a broader range of patients.
Materials and Methods
Materials. Clofarabine, fludarabine (fludarabine-phosphate), and cytarabine were obtained from Sigma (St Louis, MO, cat #C7495,# F9813, and #C3350000 respectively), diluted according to the manufacturer protocol. The concentrations of diluted PNA solutions were accurately determined by spectrophotometry. dCK and TRAC TALEN were produced in-house using the solid phase assembly method described in ref. 49. Regarding cell culture reagents, X-vivo-15 was obtained from Lonza (Basel, Switzerland cat#BE04-418Q), IL-2 from Miltenyi Biotech (Bergisch Gladbach, Germany, cat#130-097-748), human serum AB from Seralab (West Sussex, UK cat#GEM-100–318), human T activator CD3/CD28 from Life Technologies (Beverly, MA, cat#11132D), MACS-LD column from Miltenyi Biotech (cat#130-042-901), fixable viability dye eFluor780 from eBioscience (San Diego, CA, cat#65-0865-14). CFSE dye was obtained from Life Technologies (cat#C34554) and anti-IFNγ ELISA kit was obtained from R&D systems (Minneapolis, MN, cat#DIF50).
Generation of TCR/dCK-deficient primary T cells. To generate TCRαβ/dCK-deficient T cells, primary T cells were first purified from buffy-coats, activated and transfected according to the procedure described in ref. 12. Briefly regarding transfection, 4 days after their activation by Dynabeads human T activator CD3/CD28, 5 million of T cells were simultaneously transfected with 5 µg of each mRNA encoding left and right arms of TALEN targeting the TCRα constant chain and dCK exon 2 (see Supplementary Material). Transfection was performed using Agilpulse technology, by applying two 0.1 ms pulses at 3,000 V/cm followed by four 0.2 ms pulses at 325 V/cm in 0.4 cm gap cuvettes and a final volume of 200 µl of Cytoporation buffer T (BTX Harvard Apparatus, Holliston, MA). Cells were then immediately diluted in X-Vivo-15 media supplemented by 20 ng/ml IL-2 (final concentration) and 5% human serum AB. Transfected T cells were eventually diluted at 1 × 106/ml and kept in culture at 37 °C in the presence of 5% CO2 and 20 ng/ml IL-2 (final concentration) and 5% human AB serum for further characterization.
A similar procedure was used to generate TCRαβ/dCK-deficient primary T cells expressing anti-CD19 CAR (FMC63 construct), given the few difference described in the following. Freshly purified primary T cells were activated for 3 days according to the procedure described above and in ref. 12, and then transduced with lentiviral vectors harboring a RQR8-2A-FMC63 CAR expression cassette under the control of the Ef1α promoter (see Supplementary Material), at the multiplicity of infection (MOI) of 5. Two days after transduction, T cells were transfected with mRNA encoding TRAC and dCK TALEN using the procedure described above.
Isolation of TCRαβ-deficient T cells using magnetic separation. TCRαβ-deficient cells were purified according to the protocol described in refs. 12,25. Briefly, about 107 T cells recovered 6 days after TALEN treatment were labeled with biotin conjugated anti-TCRαβ antibody MicroBeads before being loaded onto a MACS LD-Column placed in the magnetic field of a MACS Separator. Using this procedure, the magnetically labeled CD3-positive cells were retained in the column while the unlabeled TCRαβ-deficient cells could be recovered in the flow through. One round of purification was usually necessary to obtain a homogeneous population of TCRαβ-deficient cells (purity > 99%).
Detection of translocation event by nested-PCR. The nested-PCR protocol used to detect translocation event after treating T cells with TRAC and dCK TALEN has been adapted from the protocol described in ref. 26. Briefly, it consists of amplifying the genomic DNA, extracted from a bulk population of TALEN-treated T cells, via a first PCR specific for the four translocations named TR1, TR2, TR3, and TR4 expected to occur between TRAC and dCK loci. This PCR is then amplified using a second set of oligonucleotides designed to anneal inside the first PCR amplicons. The first and second PCR were performed with the Herculase II Fusion DNA Polymerases kit (Agilent Technologies) using 25 cycles of amplification and a Tm = 50 °C. A comprehensive list of oligonucleotides and translocation matrix sequences used to set up and optimize the nested-PCR protocol is documented in the Supplementary Material section.
IC50 determination. IC50 of a given drug is defined as the concentration of drug need to decrease the cellular viability by 50%. To determine IC50 of a clofarabine, fludarabine and cytarabine, 100 103 T cells were incubated in the presence of increasing concentration of drugs (from 0 to 100 µmol/l typicially) for 48 hours and in a total volume of 100 µl X-Vivo-15 media supplemented by 20 ng/ml IL-2 and 5% human AB serum. Cells were washed with 100 µl of phosphate buffer saline (PBS) and then labeled by eFluor 780 for 15 minutes at 4 °C according to the manufacturer protocol. Labeling was stopped by addition of PBS 2% fetal veal serum (SVF) and cells were eventually fixed by 4% paraformaldehyde (PFA) before being analyzed by flow cytometry to determine their viability. Evolution of cell viability as a function of PNA concentration was fitted with the drfit R package software using the formula: y = 1/(1 + EXP((LOG10(x) − LOG10(IC50))/z))*100 with y, x, and z corresponding to viability frequency, the concentration of drug, and the scale parameter.
Flow-based cytotoxicity assay. The cytolytic activity and specificity of CAR T cell was assessed according to the flow cytometry-based cytotoxicity assay described in ref. 20. This assay consisted of labeling 104 Daudi (CD19+) and 104 K562 (CD19- negative control) cells with 1 µmol/l CellTrace CFSE and 1 µmol/l CellTrace violet respectively (Life Technology) and coincubate them with 105 effector CAR T cells (E/T ratio of 10:1) in a final volume of 100 µl X-Vivo-15 media, for 5 hours at 37 °C. Cells were then recovered and labeled with eFluor780 viability marker before being fixed by 4% PFA as described above. Fixed cells were then analyzed by flow cytometry to determine their viability. The frequency of specific cell lysis was calculated using the formula described in ref. 20 and displayed in the following:
Frequency of specific cell lysis = (Via Daudi-T/ViaK562-T)/(Via Daudi/Via K562)
where Via Daudi-T and ViaK562-T correspond respectively to the % of viable Daudi and K562 cells obtained after 5 H in the presence of CAR T cells and where Via Daudi and Via K562 correspond respectively to the % of Daudi and K562 cells obtained after 5 H in the absence of CAR T cells.
Allogeneic mixed lymphocyte reaction. 20 × 106 PBMC freshly purified from a donor B (PBMC B) were labeled by a 10-minute incubation with 2 nmol/l CFSE in the dark, at 37 °C in a final volume of 5 ml. CFSE-labeled PBMC B were then diluted two times in fetal bovine serum, and washed with X-Vivo-15 media supplemented by 20 ng/ml IL-2 and 5% human AB serum. Cell number and viability was then determined and their concentration was adjusted to 1 × 106 viable cells/ml. To perform mixed lymphocyte reaction, CFSE-labeled PBMC B were incubated with either T cells purified from the same blood donor (T cells B) or with CAR T cells engineered out of a different blood donnor (CAR T cell A) in X-Vivo-15 media supplemented by 20 ng/ml IL-2 and 5% human AB serum. CFSE labeled PBMC viability, activation and proliferation were determined after 6 days of incubation, by flow cytometry and anti IFNγ ELISA.
Statistical analysis. Viability values obtained on Daudi cells were averaged for each condition in each experiment (n = 2 with two different donors and n = 3 with three different donors for experiments performed with cytarabine and clofarabine respectively), and these averages were used in a paired t-test (pairing by experiment) to compare two different conditions and determine P values indicated in Figure 7.
SUPPLEMENTARY MATERIAL Figure S1. High-throughput DNA sequencing analysis method Figure S2. Simultaneous treatment of primary T cells with dCK and TRAC TALEN does not generate long-standing genetic adverse events. Figure S3. TALEN-mediated dCK processing increased primary T cells resistance to clofarabine without needing time consuming and potentially harmfull drug selection process. Figure S4. Efficient expression of RQR8 depleting system at the surface CAR T. Figure S5. TALEN-treated CAR T cells can proliferate in the presence of lethal dose of different purine and pyrimidine nucleotide analogues (PNAs). Figure S6. TALEN-treated CAR T cells can proliferate in the presence of a combination of purine and pyrimidine nucleotide analogues (PNAs). Figure S7. PNA treatment prevents alloreactivity of PBMC toward CAR T cells in vitro. Figure S8. CAR T cell cytolytic activity is not affected by dual inactivation of dCK and TCRαβ. Figure S9. TALEN-treated CAR T cells retain their cytolytic activity after 2 to 9 days of culture in the presence of clinically relevant dose of clofarabine. Figure S10. TALEN-treated CAR T cells can pair up with chemotherapy for better antitumor activity.
Acknowledgments
All authors are Cellectis employees. J.V. and L.P. conceived the work; V.G., A.M., and J.M.F. performed experiments; A.D. and A.J. contributed significantly to the interpretation of results; and J.V. wrote the manuscript with support from all authors. We would like to thank Stephanie Langevin for bioinformatic analysis, Julianne Smith, Roman Galetto, Cecile Schiffer-Mannioui, and Jean-Pierre Cabaniols for fruitful discussions, Ramon Martinez, and Sylvain Goyat for technical assistance.
Supplementary Material
References
- Marcus, A and Eshhar, Z (2014). Allogeneic chimeric antigen receptor-modified cells for adoptive cell therapy of cancer. Expert Opin Biol Ther 14: 947–954. [DOI] [PubMed] [Google Scholar]
- Torikai, H, Reik, A, Liu, PQ, Zhou, Y, Zhang, L, Maiti, S et al. (2012). A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119: 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus, A, Waks, T and Eshhar, Z (2011). Redirected tumor-specific allogeneic T cells for universal treatment of cancer. Blood 118: 975–983. [DOI] [PubMed] [Google Scholar]
- Berdien, B, Mock, U, Atanackovic, D and Fehse, B (2014). TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther 21: 539–548. [DOI] [PubMed] [Google Scholar]
- Benichou, G, Yamada, Y, Yun, SH, Lin, C, Fray, M and Tocco, G (2011). Immune recognition and rejection of allogeneic skin grafts. Immunotherapy 3: 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordheim, LP, Durantel, D, Zoulim, F and Dumontet, C (2013). Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 12: 447–464. [DOI] [PubMed] [Google Scholar]
- Porter, DL, Levine, BL, Kalos, M, Bagg, A and June, CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude, SL, Frey, N, Shaw, PA, Aplenc, R, Barrett, DM, Bunin, NJ et al. (2014). Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Kassim, SH, Somerville, RP, Carpenter, RO, Stetler-Stevenson, M et al. (2015). Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer, JN, Dudley, ME, Feldman, SA, Wilson, WH, Spaner, DE, Maric, I et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119: 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galetto, R, Lebuhotel, C, Poirot, L, Gouble, A, Toribio, ML, Smith, J et al. (2014). Pre-TCRα supports CD3-dependent reactivation and expansion of TCRα-deficient primary human T-cells. Mol Ther Methods Clin Dev 1: 14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y, Kweon, J and Kim, JS (2013). TALENs and ZFNs are associated with different mutation signatures. Nat Methods 10: 185. [DOI] [PubMed] [Google Scholar]
- Juillerat, A, Dubois, G, Valton, J, Thomas, S, Stella, S, Maréchal, A et al. (2014). Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res 42: 5390–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger, JP, Pattanayak, V, Reyon, D, Tsai, SQ, Sander, JD, Joung, JK et al. (2014). Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods 11: 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip, B, Kokalaki, E, Mekkaoui, L, Thomas, S, Straathof, K, Flutter, B et al. (2014). A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 124: 1277–1287. [DOI] [PubMed] [Google Scholar]
- Gandhi, V, Kantarjian, H, Faderl, S, Bonate, P, Du, M, Ayres, M et al. (2003). Pharmacokinetics and pharmacodynamics of plasma clofarabine and cellular clofarabine triphosphate in patients with acute leukemias. Clin Cancer Res 9: 6335–6342. [PubMed] [Google Scholar]
- Long-Boyle, JR, Green, KG, Brunstein, CG, Cao, Q, Rogosheske, J, Weisdorf, DJ et al. (2011). High fludarabine exposure and relationship with treatment-related mortality after nonmyeloablative hematopoietic cell transplantation. Bone Marrow Transplant 46: 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramis, VI, Champagne, J, Sato, J, Krailo, M, Ettinger, LJ, Poplack, DG et al. (1990). Pharmacology of fludarabine phosphate after a phase I/II trial by a loading bolus and continuous infusion in pediatric patients. Cancer Res 50: 7226–7231. [PubMed] [Google Scholar]
- Zhao, Y, Moon, E, Carpenito, C, Paulos, CM, Liu, X, Brennan, AL et al. (2010). Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70: 9053–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley, RG, Roth, S, Kurtzberg, LS, Rouleau, C, Yao, M, Crawford, J et al. (2009). Bone marrow CFU-GM and human tumor xenograft efficacy of three antitumor nucleoside analogs. Int J Oncol 34: 1329–1340. [PubMed] [Google Scholar]
- Waud, WR, Schmid, SM, Montgomery, JA and Secrist, JA 3rd (2000). Preclinical antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D- arabinofuranosyl)adenine (Cl-F-ara-A). Nucleosides Nucleotides Nucleic Acids 19: 447–460. [DOI] [PubMed] [Google Scholar]
- Takahashi, T, Kanazawa, J, Akinaga, S, Tamaoki, T and Okabe, M (1999). Antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine, a novel deoxyadenosine analog, against human colon tumor xenografts by oral administration. Cancer Chemother Pharmacol 43: 233–240. [DOI] [PubMed] [Google Scholar]
- Valdez, BC, Murray, D, Nieto, Y, Li, Y, Wang, G, Champlin, RE et al. (2012). Synergistic cytotoxicity of the DNA alkylating agent busulfan, nucleoside analogs and suberoylanilide hydroxamic acid in lymphoma cell lines. Leuk Lymphoma 53: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton, J, Cabaniols, JP, Galetto, R, Delacote, F, Duhamel, M, Paris, S et al. (2014). Efficient strategies for TALEN-mediated genome editing in mammalian cell lines. Methods 69: 151–170. [DOI] [PubMed] [Google Scholar]
- Piganeau, M, Ghezraoui, H, De Cian, A, Guittat, L, Tomishima, M, Perrouault, L et al. (2013). Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res 23: 1182–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui, H, Piganeau, M, Renouf, B, Renaud, JB, Sallmyr, A, Ruis, B et al. (2014). Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell 55: 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard, P (1988). Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem 57: 349–374. [DOI] [PubMed] [Google Scholar]
- Wallen, H, Thompson, JA, Reilly, JZ, Rodmyre, RM, Cao, J and Yee, C (2009). Fludarabine modulates immune response and extends in vivo survival of adoptively transferred CD8 T cells in patients with metastatic melanoma. PLoS One 4: e4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni, L, Finkelstein, SE, Klebanoff, CA, Antony, PA, Palmer, DC, Spiess, PJ et al. (2005). Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202: 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary, R, Baturin, D, Fosmire, S, Freed, B and Porter, CC (2013). Knockdown of HPRT for selection of genetically modified human hematopoietic progenitor cells. PLoS One 8: e59594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gori, JL, Tian, X, Swanson, D, Gunther, R, Shultz, LD, McIvor, RS et al. (2010). In vivo selection of human embryonic stem cell-derived cells expressing methotrexate-resistant dihydrofolate reductase. Gene Ther 17: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, CC and DeGregori, J (2008). Interfering RNA-mediated purine analog resistance for in vitro and in vivo cell selection. Blood 112: 4466–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Yang, JC, Sherry, RM, Topalian, SL, Restifo, NP et al. (2005). Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23: 2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nars, MS and Kaneno, R (2013). Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy. Int J Cancer 132: 2471–2478. [DOI] [PubMed] [Google Scholar]
- Takebe, N, Zhao, SC, Adhikari, D, Mineishi, S, Sadelain, M, Hilton, J et al. (2001). Generation of dual resistance to 4-hydroperoxycyclophosphamide and methotrexate by retroviral transfer of the human aldehyde dehydrogenase class 1 gene and a mutated dihydrofolate reductase gene. Mol Ther 3: 88–96. [DOI] [PubMed] [Google Scholar]
- Dasgupta, A, Shields, JE and Spencer, HT (2012). Treatment of a solid tumor using engineered drug-resistant immunocompetent cells and cytotoxic chemotherapy. Hum Gene Ther 23: 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonnalagadda, M, Brown, CE, Chang, WC, Ostberg, JR, Forman, SJ and Jensen, MC (2013). Engineering human T cells for resistance to methotrexate and mycophenolate mofetil as an in vivo cell selection strategy. PLoS One 8: e65519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huye, LE, Nakazawa, Y, Patel, MP, Yvon, E, Sun, J, Savoldo, B et al. (2011). Combining mTor inhibitors with rapamycin-resistant T cells: a two-pronged approach to tumor elimination. Mol Ther 19: 2239–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer, M, Kochanek, M, Darabi, K, Popov, A, Jensen, M, Endl, E et al. (2005). Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 106: 2018–2025. [DOI] [PubMed] [Google Scholar]
- Gabrilovich, DI, Ostrand-Rosenberg, S and Bronte, V (2012). Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, ML, Riviere, I, Wang, X, Bartido, S, Park, J, Curran, K et al. (2014). Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6: 224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M, Levine, BL, Porter, DL, Katz, S, Grupp, SA, Bagg, A et al. (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colovos, C, Villena-Vargas, J and Adusumilli, PS (2012). Safety and stability of retrovirally transduced chimeric antigen receptor T cells. Immunotherapy 4: 899–902. [DOI] [PubMed] [Google Scholar]
- Scholler, J, Brady, TL, Binder-Scholl, G, Hwang, WT, Plesa, G, Hege, KM et al. (2012). Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4: 132ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi, A, Tey, SK, Dotti, G, Fujita, Y, Kennedy-Nasser, A, Martinez, C et al. (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365: 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, KM, Hakim, FT and Gress, RE (2007). T cell immune reconstitution following lymphodepletion. Semin Immunol 19: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daboussi, F, Leduc, S, Maréchal, A, Dubois, G, Guyot, V, Perez-Michaut, C et al. (2014). Genome engineering empowers the diatom Phaeodactylum tricornutum for biotechnology. Nat Commun 5: 3831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.