

Abstract
Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder resulting from a functional deficiency of arylsulfatase A (ARSA), an enzyme that catalyzes desulfation of 3-O-sulfogalactosylceramide (sulfatide). Lack of active ARSA leads to the accumulation of sulfatide in oligodendrocytes, Schwann cells and some neurons and triggers progressive demyelination, the neuropathological hallmark of MLD. Several therapeutic approaches have been explored, including enzyme replacement, autologous hematopoietic stem cell-based gene therapy, intracerebral gene therapy or cell-based gene delivery into the central nervous system (CNS). However, long-term treatment of the blood-brain-barrier protected CNS remains challenging. Here we used MLD patient-derived induced pluripotent stem cells (iPSCs) to generate long-term self-renewing neuroepithelial stem cells and astroglial progenitors for cell-based ARSA replacement. Following transplantation of ARSA-overexpressing precursors into ARSA-deficient mice we observed a significant reduction of sulfatide storage up to a distance of 300 µm from grafted cells. Our data indicate that neural precursors generated via reprogramming from MLD patients can be engineered to ameliorate sulfatide accumulation and may thus serve as autologous cell-based vehicle for continuous ARSA supply in MLD-affected brain tissue.
Introduction
Metachromatic leukodystrophy (MLD) is an autosomal recessively inherited lysosomal lipid storage disorder resulting from a functional deficiency of arylsulfatase A (ARSA, EC 3.1.6.8).1 The physiological role of this lysosomal enzyme involves desulfation of the galactose moiety of 3-O-sulfogalactosylceramide (sulfatide), being the first step in the lysosomal degradation of this acidic sphingolipid. No other enzyme can compensate for the lack of ARSA activity. Consequently, ARSA deficiency causes accumulation and deposition of sulfatide in lysosomes of various cell types including oligodendrocytes, Schwann cells, microglia, and subpopulations of neurons.2 The accumulating sulfatide is thought to disrupt physiological cell functions eventually leading to a progressive and widespread loss of myelinating cells in the central and peripheral nervous system. The resulting demyelination is associated with rapidly deteriorating neurological symptoms such as ataxia, spastic tetraparesis, optic atrophy, seizures, and dementia leading to premature death.2,3
As with other soluble lysosomal enzymes, lysosomal targeting of newly synthesized ARSA depends on mannose 6-phosphate (M6P) residues that are added to the N-glycans of the enzyme during its passage through the Golgi apparatus.4 In the trans Golgi network, the M6P residues bind to M6P receptors that cycle to the endosomal/lysosomal compartment and separate their ligands from the secretory route. A small fraction of newly synthesized soluble lysosomal enzymes escapes, however, from this biosynthetic sorting pathway and is subsequently released from the cell. Extracellular enzyme can then be endocytosed and lysosomally delivered via M6P receptors that also cycle between the plasma membrane and endosomes. This release-recapture pathway provides the rationale for allogeneic hematopoietic stem cell transplantation as it allows the metabolic correction of ARSA-deficient cells by the transplanted, enzyme competent donor cells. Indeed, hematopoietic stem cell transplantation may prevent the disease progression in milder variants of MLD (juvenile forms), if performed before loss of walking, which typically initiates rapid deterioration.5
Enzyme replacement therapy based on intravenous injection of recombinant enzyme represents another therapeutic approach. It requires repeated and life-long treatment and has been clinically approved for some lysosomal storage diseases without central nervous system (CNS) involvement.6 In mouse models of MLD, intravenous injection of recombinant human ARSA showed some promising effects including improvement of the CNS histopathology and function.7,8 However, due to poor penetration of the blood–brain barrier, repeated applications with high doses of ARSA are required. In an approach to circumvent the blood–brain barrier, MLD mice were treated by intracerebroventricular infusion of ARSA using implantable minipumps.9 Infusion of ARSA into the cerebrospinal fluid of the brain resulted in the complete clearance of sulfatide storage from the infused hemisphere and partial normalization of the ataxic gait. The therapeutic efficacy of a similar approach using an intrathecal application route is presently evaluated in a clinical phase 1/2 trial (NCT01510028).
The peculiarities of the lysosomal sorting process with exchange of soluble lysosomal enzymes between cells make MLD particularly suitable for vector-mediated in vivo and ex vivo gene therapy approaches. Direct delivery of ARSA into the brain using intracerebral injections of lentiviral, adenoviral, or adeno-associated viral vectors resulted in widespread CNS expression of ARSA in rodents and nonhuman primates as well as in improvement of neuropathological and behavioral changes in a mouse MLD model.10,11,12,13,14 Whether these results can be translated to humans is currently not clear, first results from clinical trials in human are expected shortly (NCT01801709, intracerebral administration of AAVrh.10cuARSA 4/2018).
Cell-based delivery of ARSA using hematopoietic stem cells overexpressing ARSA showed ambiguous results in MLD mice ranging from only mild reduction of CNS sulfatide storage14 to almost complete reversion of peripheral and central nervous storage.15 Based on these findings, a phase 1/2 clinical trial was initiated (NCT01560182). First results from these studies indicate that patients receiving treatment before the onset of major cognitive or motor deficits can benefit from this therapy.16
Expression of ARSA by brain cells transplanted into the CNS has emerged as innovative methodology, which bypasses the limitations associated with the blood–brain barrier and permits long-term and continuous delivery of ARSA directly into the brain parenchyma. Indeed, primary mouse neural precursors engineered to overexpress ARSA and transplanted into ARSA-deficient hosts were shown to differentiate into neurons, astrocytes, and oligodendrocytes and to partially reduce sulfatide storage.17,18 Considering the limited access to neural donor tissue in a clinical scenario, we have previously explored embryonic stem (ES) cells as a potential source for cell-mediated ARSA transfer. We could show that mouse ES cell-derived glial precursors overexpressing ARSA and transplanted in a MLD mouse model efficiently differentiate into astrocytes and oligodendrocytes and reduce sulfatide storage in the vicinity of the transplanted cells.19
With the advent of cellular reprogramming of somatic cells toward pluripotency, human pluripotent stem cells directly derived from patients suffering from a particular disease are now available as a possible autologous cell source for translating such results to human. Here we show that panneural and astroglial progenitors derived from induced pluripotent stem cells (iPSCs) generated from a patient with MLD and engineered to overexpress ARSA efficiently engraft into a mouse model of MLD and substantially reduce CNS sulfatide storage within the host brain.
Results
Generation of MLD patient-specific lt-NES cells and astroglial progenitors
iPSCs were generated from skin fibroblasts derived from a 14-year-old female patient suffering from a juvenile variant of MLD using retroviral delivery of the reprogramming factors OCT4, SOX2, KLF4, and c-MYC. The donor carried a homozygous mutation in the ARSA gene resulting in a change of proline to leucine at position 426. The respective P426L allele is the second most frequent MLD allele among European patients.1,20 The mutant enzyme is expressed at slightly reduced levels compared to healthy control cell levels and with an activity in the physiological range, but is rapidly degraded in the lysosome. Thus, the cause of deficiency is decreased intralysosomal stability.20 Five of the primary iPSC clones were expanded and further validated for pluripotency marker expression, transgene silencing, their differentiation potential, and karyotypic integrity. Based on this validation, clone 1 was selected for further experiments. The iPSC line derived from this clone showed expression of the pluripotency-associated markers alkaline phosphatase (AP), OCT4, Tra1-60, and Tra1-81 (Figure 1a–d) and differentiated into cells of all three germ layers in vitro and in vivo (Figure 1e–j). Karyotypic integrity was investigated using genome-wide SNP analysis (Supplementary Figure S1). Sustained silencing of the viral transgenes was confirmed by quantitative PCR analysis of endogenous and transgene expression of the reprogramming factors (Figure 1k). The mutation in the ARSA gene was further confirmed by genomic DNA sequencing (Figure 1l).
Figure 1.

Generation and characterization of patient-specific induced pluripotent stem (iPS) cells. (a–d): Metachromatic leukodystrophy (MLD) iPSCs stain positive for pluripotency-associated alkaline phosphatase (a), Oct4 (b), Tra1-60 (c), and Tra1-81 (d). (e–j): Immunocytochemical and histological analysis of in vitro (e,g,i) and in vivo (f,h,j) differentiated MLD iPSCs shows generation of cell types from all three germ layers including βIII-tubulin-positive neurons (e) and neural rosettes (f, both ectoderm), α1-fetoprotein-positive cells (g) and glandular structures (h, both endoderm) or smooth muscle actin-positive cells (i) and cartilage (j, both mesoderm). (k) Silencing of the reprogramming factors OCT4, SOX2, KLF4, and C-MYC was confirmed by quantitative reverse transcription polymerase chain reaction (RT-PCR). Histogram shows relative expression levels. Blue bars represent the iPS cells. Expression of hESC line H9.2 (red bars) was set to 1 for endogenous gene expression. Expression of virus producing PhoenixGP cells (green bars) was set to 1 for transgene expression. Error bars show SD of triplicates. (l) Sequencing of genomic DNA confirmed the P426L missense mutation within the coding region of the arylsulfatase A gene. Scale bars: 100 µm.
From iPSCs, we generated long-term self-renewing neuroepithelial stem cells (lt-NES cells), a robust and homogeneous neural precursor population with strong neurogenic potential in vitro and in vivo.21,22 In the presence of the growth factors FGF2 and EGF, these cells grow in rosette-like structures and express Nestin together with the neuroepithelial stem cell-associated transcription factors SOX2, DACH1, and PLZF (Figure 2a–d). As a correlate of their epithelial nature, the zonula occludens protein 1 (ZO-1) is located apically in these cells (Figure 2d). Upon growth factor withdrawal, lt-NES cells differentiate into a major fraction of neurons expressing βIII-tubulin and MAP2ab (Figure 2e–g) and an only minor fraction of glial fibrillary acid protein (GFAP)-positive astrocytes (Figure 2f).21 To generate a culture of astroglial progenitor cells (APCs), the cells were differentiated for 4 weeks and subsequently trypsinized and replated in the presence of FGF2 and EGF. Under these conditions, the small fraction of APCs proliferate and can be further expanded for multiple passages showing elongated, fibroblast-like morphologies (Figure 2h). Upon growth factor withdrawal, this population gives rise to a virtually pure population of GFAP-positive astrocytes within 3–4 weeks (Figure 2i,j).
Figure 2.

Lt-NES cells and astroglial precursor cells from induced pluripotent stem cells. (a–d) Lt-NES cells grow in rosette-like structures (a) expressing the neural stem cell-associated markers SOX2 (b), Nestin, the dachshund homolog like 1 protein (DACH1; c), the zinc finger and BTB domain containing protein 16 (PLZF) and the apically accentuated protein ZO-1 that is associated with the zona occludens (d). (e,f) Upon growth factor withdrawal, lt-NES cells give rise to a dominant fraction of neurons expressing βIII tubulin (e,f) and only a minor fraction of astrocytes positive for glial fibrillary acidic protein (GFAP; f). (g) Lt-NES cell-derived neurons also express the neuron-specific microtubule associated protein 2 (MAP2ab). (h) Astroglial precursors exhibit a fibroblast-like morphology. (i,j) Upon growth factor withdrawal, they differentiate into cells expressing GFAP. Scale bars: 100 µm.
Lentiviral transduction results in neural cell populations with strong ARSA activity
To generate neural cell populations with high and sustained ARSA activity, we transduced the patient-derived lt-NES cells with a lentiviral vector containing the human ARSA cDNA linked via an internal ribosomal entry site (IRES) to an enhanced green flourescent protein (EGFP) under transcriptional control of the eukaryotic translation elongation factor 1α (EF1α). Progenitor cells transduced with a control vector containing EGFP only served as control (Figure 3a). Cells were selected for Blasticidine S-resistance expressed from an EM7 promoter on the same vector. APCs were subsequently generated from these populations. Overexpression of ARSA was first validated by immunofluorescence. Whereas control lt-NES cells exhibited only a faint ARSA immunoreactivity, strong ARSA expression was observed in lt-NES cells transduced with the ARSA vector (Figure 3b). To assess ARSA enzyme activity in lt-NES cells, APCs and their differentiated progeny, we performed enzymatic ARSA activity assays using the artificial substrate p-nitrocatechol sulfate. As healthy controls we included lt-NES cells and APCs from an unaffected donor.22,23 This assay revealed a strongly reduced ARSA activity in the MLD patient-derived cells compared to healthy control cells (Figure 3c). While lt-NES cells and APCs from healthy control cells exhibited ARSA activities of 0.43 ± 0.05 mU/mg protein and 7.37 ± 0.04 mU/mg, respectively, activities of patient-derived cells were below the limit of detection under the same experimental conditions (assay adjusted to ARSA overexpressing cells). Following differentiation, we observed a slight increase of ARSA activity in patient-derived cells (lt-NES cells: 0.43 ± 0.02 mU/mg; APCs: 0.25 ± 0.01 mU/mg), which was still several fold lower than in controls (lt-NES cells: 1.87 ± 0.17 mU/mg; APCs: 10.25 ± 0.49 mU/mg). In stark contrast, ARSA overexpressing cells showed a strong increase in ARSA activity under all conditions (Figure 3c). Proliferating lt-NES cells and APCs exhibited ARSA activity levels of 140.19 ± 10.59 mU/mg and 612.53 ± 15.15 mU/mg protein, respectively. Remarkably, these levels further increased upon differentiation with differentiated lt-NES cells showing 760.75 ± 15.62 mU/mg and APCs 1792.61 ± 25.52 mU/mg protein ARSA activity.
Figure 3.

Lentiviral gene correction induces high levels of arylsulfatase A (ARSA) activity. (a) Schematic representation of the lentiviral vectors used in the study. (b) Immunofluorescence analysis of ARSA expression in metachromatic leukodystrophy (MLD)-derived lt-NES cells before (left panel) and after lentiviral transduction (right panel). (c) Enzymatic ARSA activity in lt-NES cells and astroglial progenitor cells from unaffected control cells, MLD patient-derived cells and gene corrected MLD cells both, in the proliferating state and following terminal differentiation. Shown are means ± SEM of technical duplicates performed from biological duplicates. (d,e) Intracellular and extracellular ARSA concentrations. Concentrations were measured by enzyme-linked immunosorbent assay (ELISA). To allow for a direct comparison with ARSA levels and to adjust for different cell densities, the calculated total intracellular and extracellular ARSA content was normalized on the total protein content of the cell homogenate of each specific sample (n = 3 wells per cell type). All samples were measured in triplicates. Shown are means ± SD. (d) Intracellular concentrations; (e) extracellular concentrations. (f) Specific activities of ARSA polypeptides expressed by ARSA-transduced cells. Specific ARSA activity was calculated by dividing the total ARSA activity per fraction (mU, measured in the activity assay) through total ARSA mass per fraction (µg, measured in the ELISA) and expressed as mU/µg. Values are means ± SD. APC, astroglial progenitor cells; diff, differentiated cells; prol, proliferating stage. Scale bars: 20 µm.
We next analyzed the extracellular and intracellular ARSA concentrations by enzyme-linked immunosorbent assay (ELISA). For the intracellular ARSA content, the levels of proliferating lt-NES cells and APCs as well as differentiated APCs from the patient were below the detection level. Differentiated lt-NES cells contained a normalized value of 0.0006 ± 0.0003 µg ARSA per mg of total protein. For the healthy control, we detected ARSA in all samples with differentiated APCs exhibiting the highest values (proliferating lt-NES cells: 0.003 ± 0.0004; proliferating APCs: 0.01 ± 0.025; differentiated lt-NES cells: 0.006 ± 0.0006; differentiated APCs: 0.34 ± 0.11; all in µg ARSA per mg of total protein). ARSA-transduced samples showed strongly elevated values, again with differentiated APCs exhibiting the highest levels (proliferating lt-NES cells: 1.14 ± 0.12; proliferating APCs: 2.02 ± 0.42; differentiated lt-NES cells: 4.41 ± 0.59; differentiated APCs: 12.03 ± 3.1; all in µg ARSA per mg total protein; Figure 3d).
Similar results were obtained for ARSA concentrations in the supernatant. While proliferating lt-NES cells and both differentiated populations were under the detection level, proliferating APCs showed low levels of secreted ARSA of 0.02 ± 0.02. In contrast, we detected secreted ARSA in all samples derived from the healthy control (proliferating lt-NES cells: 0.05 ± 0.03; proliferating APCs: 0.24 ± 0.008; differentiated lt-NES cells: 0.1 ± 0.03; differentiated APCs: 0.13 ± 0.04; all in µg ARSA per mg of total protein). ARSA secreted from the transduced samples was, again, strongly elevated (proliferating lt-NES cells: 1.54 ± 0.21; proliferating APCs: 2.0 ± 0.38; differentiated lt-NES cells: 0.35 ± 0.06; differentiated APCs: 0.72 ± 0.3; all in µg ARSA per mg of total protein; Figure 3e). Interestingly, and in contrast to the intracellular levels, proliferating cell populations exhibited the highest levels of secreted ARSA in this measurement.
We also calculated the specific activities of ARSA polypeptides, which is the total ARSA activity divided through the total ARSA mass per fraction. For this calculation, ARSA activities were quantified from homogenates and supernatants of the same samples used for the ELISA measurements. As this calculation is error-prone and misleading if ARSA levels are very low, it was only performed for the ARSA-transduced cells. We detected very similar specific activities for all samples, ranging from approximately 60 to 80 mU/µg in the cells and from 30 to 50 mU/µg in the supernatants (Figure 3f). Thus, lentiviral transduction results in robust ARSA expression, activity and secretion in lt-NES cells, astroglial progenitors and their differentiated progeny.
Engrafted neural precursor cell populations survive and differentiate into neurons and glia
We previously demonstrated that mouse embryonic stem cell-derived glial precursors engineered to overexpress human ARSA reduce CNS sulfatide storage in a mouse model of MLD.19 To investigate the potential of gene-corrected ARSA-expressing human neural cell populations to integrate into the brain of ARSA-deficient mice and to prevent the formation of sulfatide deposits in adjacent tissue, we transplanted these cells into the telencephalon of postnatal day 1 ARSA-/- mice.24 While human cells were detectable 4 weeks after transplantation, they were completely depleted by month 3—a time, that was necessary to result in relevant sulfatide deposits—most likely due to rejection of the transplanted cells (data not shown). Hence, we cross-bred ARSA-/- mice with an immunocompromized recombination-activation gene 2 deficient (RAG2-/-) mouse strain25 to obtain ARSA-/-/RAG2-/- mice for further transplantation experiments. Three months following intraventricular injection, donor cell clusters derived from transplanted cells (identified by GFP expression or the human cytoplasma-specific antibody SC121) were observed in various brain regions including cortex, the basal ganglia, fiber tracts, and the hippocampus (see schematic representation of preferential localization in the right panels of Figure 4).
Figure 4.

Engraftment and in vivo differentiation of lt-NES cells and astroglial progenitor cells. (a) Three months after transplantation into the brain of postnatal day 1 immunodeficient arylsulfatase A knockout mice, lt-NES cell-derived transplants differentiated predominantly into neurons expressing human-specific neurofilament. (b) In contrast, astroglial progenitor cell-derived cell clusters exhibited expression of the astrocytic marker glial fibrillary acid protein and only a minor fraction was positive for human neurofilament. Transplanted cells were identified by virtue of their green flourescent protein expression or with the pan-human marker SC121. Schematic drawings on the right depict preferential localization of donor cell clusters. Scale bars: 50 µm.
Differentiated cell types were identified by human-specific antibodies to neurofilament (HO-14; detecting neurons) and GFAP (SC123; detecting astrocytes). Comparable to what we observed in vitro and in analogy to previous transplantation studies,21,26,27 lt-NES cell-derived grafts predominantly differentiated into neurons immunoreactive for the human-specific neurofilament antibody, while human GFAP-positive cells were only observed occasionally (Figure 4a, representative donor cell cluster located in the cortex). In contrast, the majority of transplanted APCs were positive for human GFAP, and only a minor fraction expressed human neurofilament (Figure 4b, representative donor cell cluster located in the anterior–dorsal part of the amygdala). Oligodendrocytes could not be detected in any of the transplanted host brains, which is in line with our previous findings demonstrating that lt-NES cells preferentially differentiate into neurons and astrocytes (data not shown, 21). No teratoma formation or neural overgrowth was observed in any of the animals analyzed.
Engrafted neural precursor cell populations reduce sulfatide storage in a mouse model of MLD
We next investigated whether engrafted neuronal and astroglial cell populations are capable to reduce sulfatide storage in ARSA-/-/RAG2-/- mice brains. Transplanted cells were identified by GFP or ARSA expression. Remarkably, we observed significant reduction of sulfatide deposits (identified with an antibody to sulfatide (Sulph1))28 not only in close vicinity of donor cell clusters but also up to several hundred micrometers next to such clusters. In some grafts, almost complete extinction of sulfatide-positive immunoreactivity was detected in the areas surrounding the transplant-derived clusters (Figure 5a, representative APC donor cell cluster in the septum). Interestingly, ARSA immunoreactivity was not restricted to transplanted cells but also showed a diffuse distribution in the adjacent tissue suggesting transfer of ARSA from the transplanted cells into the adjacent host brain (Figure 5b, representative APC-derived donor cell cluster located in the septum). Strong reduction of sulfatide immunoreactivity was also present in brain regions adjacent to donor cell clusters as for instance in the hilus of the dentate gyrus of animals with donor cell clusters located in the fissure between hippocampus and thalamus (Figure 5c; shown are the ipsilateral and contralateral side of the same animal). Remarkably, at higher magnification individual GFP-positive cells and even cell processes exhibited a deposit-free halo of approximately 10–20 micrometers, suggesting a clearance effect even around single cells or even processes (Figure 5d,e).
Figure 5.

Reduction of sulfatide storage in the vicinity of transplanted arylsulfatase A (ARSA)-expressing cells. (a) Example of an astroglial progenitor donor cell cluster located in the septum. High-magnification inserts depict the density of Sulph1-positive deposits directly next to the grafted cells and in more remote areas. (b) Reduction of Sulph1-positive deposits was also observed in the ARSA “diffusion zone.” In this area, diffuse ARSA-immunoreactivity, but no ARSA-positive cellular structures could be identified. (c) Frequently, reduction of Sulph1-positive deposits was found to extend well into adjacent brain regions abutting the donor cell clusters. Shown are the dentate gyrus in the vicinity of green flourescent protein (GFP)-positive donor cells and the contralateral hippocampus of the same animal. (d,e) Strong reduction of Sulph1-positive deposits was also present in the vicinity of single cells (d) and in the surrounding of GFP-positive neurites (depicted is a merged image from confocal stacks) (e). Scale bars: a–d: 100 µm, e: 20 µm.
To quantify sulfatide load, we determined both the number of Sulph1-positive particles per area and the particle coverage per area using unbiased automatic image analysis software (ImageJ). Donor cells were identified by virtue of their GFP expression, and a ladder of rectangles (each “spoke” 50 µm in height) was placed next to the border of the graft as depicted in Figure 6a. As control, the corresponding contralateral brain region from the same histological specimen not containing any grafted cells was quantified and set to 100. Results were grouped according to donor cell population (lt-NES cells or APCs) and anatomical location (donor cell clusters located in cortical areas, basal ganglia, or the hippocampus). This analysis revealed a direct inverse relationship between sulfatide clearance and distance from the graft.
Figure 6.

Quantification of sulfatide load in different brain region. (a) Schematic demonstrating the approach used for semiquantitative assessment of sulfatide deposits. A grid of rectangles (each 50 µm wide) was placed next to the borders of the grafted cells identified by their green flourescent protein content. (b) Quantification of the average number of Sulph1-positive particles and the area covered by Sulph1-positive particles in the vicinity of donor cell clusters derived from lt-NES cells in percentage compared to the contralateral side. Transplants were grouped according to their localization into cortical transplants, transplants localized in the basal ganglia or hippocampal transplants. The same area of the contralateral ungrafted side of the same animal and the same histological specimen served as control and was set to 100% (indicated as dashed grey line). Numbers (n) given on top of the individual bars represent numbers of areas analyzed for the distance indicated. Graphs were generated and analyzed using Graphpad Prism. Error bars depict SEM. Following analysis of variance test, Fischer's least significant difference test was performed to compute statistics of multiple comparisons to nongrafted contralateral control areas; n.s., not significant, P values * < 0.05, ** < 0.01, *** < 0.001, and **** < 0.0001. (c) Quantification of the average number of Sulph1-positive particles and the area covered by Sulph1-positive particles in the vicinity of transplants derived from astroglial precursor cells. Quantification and statistical analysis was performed analogously to lt-NES cell-derived transplants.
When comparing the number of Sulph1-positive deposits and the total area covered by the deposits directly in donor cell-containing areas, these parameters were reduced to 6.36 ± 10.95 and 4.57 ± 8.86% for lt-NES cell-derived transplants and 6.38 ± 13.20 and 3.07 ± 5.69% for APC-derived transplants, respectively. With increasing distance from donor cells sulfatide deposits became more abundant, but a significant reduction of both parameters was still detectable at distances of up to 300 µm away from transplantation-derived donor cell clusters (Figure 6b,c). Interestingly, we observed no major differences in the sulfatide clearance capacity of both populations (lt-NES cells or APCs). However, in contrast to donor astrocytes, lt-NES cell-derived neurons typically extend widespread axonal projections into the host brain.21,26,27 We therefore asked whether reduction of sulfatide storage could also be observed in neuronal projection areas (defined as areas > 500 µm away from donor cell clusters containing human neurofilament-positive axonal processes but no human cell bodies). Indeed, quantification of the number of Sulph1-positive particles per area and the particle coverage per area revealed a significant reduction of both parameters of up to > 50% in such projection regions such as the corpus callosum (Figure 7a,b). Together, these findings indicate that neuronal and astroglial cell populations derived from iPS cells generated from a MLD patient and engineered to overexpress human ARSA permit an efficient reduction of sulfatide storage across a large perimeter around transplanted donor cells. In case of a neuronal donor cell phenotype, additional and more widespread reduction of sulfatide storage can be achieved in areas containing axonal projections from transplanted ARSA-overexpressing neurons. Thus, autologous transplantation of engineered iPS cell-derived neural cell populations might represent an attractive tool for cell-based delivery of functional ARSA in MLD-affected brain tissue.
Figure 7.

Quantification of sulfatide storage in axonal projection areas. (a) Network of donor-derived axons in the corpus callosum, identified with an antibody to human neurofilament. Sulfatide-positive deposits are stained by an antibody to Sulph1 (left panel). Isolated presentation of human Neurofilaments (black) and Sulph1-positive deposits (red) in false colors (right panel). For sake of visualization, hNeurofilaments and Sulph1-positive deposits have been digitally enlarged in false color image. (b) Quantification of the average number of Sulph1-positive particles and the area covered by Sulph1-positive particles in areas containing donor-derived axonal projections expressed as percentage of the contralateral control region (n = 8; t-test detected significance with P < 0.0001 for both particles/area and particle coverage/area; error bars depict SEM). Scale bar: 40 µm.
Discussion
Lysosomal storage diseases have been discussed as promising candidates for gene therapy, cell transplantation, and enzyme replacement therapies due to the particular characteristics of inter- and intracellular sorting of lysosomal enzymes.4,29 Soluble lysosomal enzymes characteristically carry mannose 6-phosphate (M6P) residues on their N-glycans. This unique modification enables binding of lysosomal enzymes to M6P receptors within the trans Golgi network. From here, these receptors mediate the further vesicular transport of the enzymes to the lysosome. A considerable fraction of M6P bearing enzymes, however, escapes the lysosomal sorting pathway and is released from the cells. M6P receptors on the plasma membrane of neighboring cells can bind the secreted lysosomal enzymes and deliver them to the lysosomal compartment via the endocytotic route. This intricate transfer mechanism opens significant therapeutic options for lysosomal storage disorders.
In gene therapy, cells can be genetically modified to overexpress recombinant enzymes defective in the patient. This genetic modification can either be introduced by direct in vivo injection of viral vectors into the brain or by transplantation of cells, which have been modified ex vivo. The enzyme delivered from such cells can be endocytosed by the resident deficient cells of the patient, a phenomenon referred to as metabolic cross-correction.30 In such a scenario, a single genetically modified cell may suffice to substitute enzyme activity in many cells of the patient. Based on this rationale we previously transplanted mouse embryonic stem cell-derived glial precursors genetically engineered to overexpress ARSA into the brain of neonatal ARSA-deficient mice.19 We showed that sulfatide storage was reduced in the vicinity of ARSA delivering engrafted cells. Here, we aimed at extending this approach to human cells and to a scenario enabling autologous transplantation, which would bypass the need of immunosuppression. To that end, we employed iPS cells, which represent a versatile autologous donor source for a cell-mediated gene therapy. Generated from somatic cells of a patient and modified in vitro, they provide an inexhaustible source of “healthy cells” for transplantation without the risk of transplant rejection. In a proof of principle experiment, iPSCs generated from a mouse model of sickle cell anemia were genetically corrected and, following differentiation into hematopoietic progenitors, transplanted back into the mouse, resulting in phenotypic recovery.31 In another pioneering study, human iPSCs from limb-girdle muscular dystrophy Type 2D (LGMD2D) patients were differentiated into mesangioblast-like mesodermal progenitor cells that were lentivirally engineered to express wild type α-sarcoglycan. Following transplantation into a mouse model of LGMD2D, these cells differentiated into α-sarcoglycan-positive muscle fibers resulting in improved muscle function compared to nontransplanted animals.32 We extended this autologous transplantation approach to a cell-mediated enzyme replacement therapy in MLD.
Lentiviral overexpression of ARSA in neural progenitors derived from patient-specific iPS cells resulted in strongly elevated levels of intracellular and secreted ARSA compared to GFP-only transduced cells or cells derived from healthy controls. Importantly, the specific activity of ARSA from the supernatant of the lentivirally transduced populations ranged between ~30 to 50 mU/µg. For comparison, enzyme that has been purified from the supernatant of ARSA-overexpressing Chinese hamster ovary cells has a specific activity of ~40 mU/µg.8 Thus, lentivirally transduced iPSC-derived cells generate ARSA with a physiological specific activity, indicating correct modification of the newly expressed ARSA by the formyl glycine generating enzyme (FGE).33 The fact that FGE becomes not limiting under the condition of high ARSA overexpression renders it unlikely that ARSA depletes FGE and thereby lowers the specific activity of other cellular sulfatases, also requiring this modification for their activation.
The ratio between intracellular and extracellular ARSA varied substantially between the different cultured cell types (see Figure 3d,e). The rate by which lentivirally transduced patient-derived cells deliver ARSA is of particular interest, as they are supposed to serve as a local enzyme donor following implantation into the brain parenchyma. Notably, ARSA-overexpressing patient-derived cells (APCs and lt-NES cells) secrete substantial amounts of enzyme into the medium as long as they are in the proliferating state. As a consequence, ARSA is almost equally distributed between the medium and the cellular fraction. However, as the cells differentiate and total ARSA increases, this ratio drops and less ARSA is secreted, both on a percentage basis and in absolute terms. Compared to the extracellular ARSA concentration of proliferating APCs and lt-NES cell cultures, the extracellular levels decrease to 35.8 and 22.8% after differentiation, respectively. Still, ARSA overexpressing differentiated patient-derived cells secrete considerable amounts of ARSA. Compared to healthy control cells, the extracellular levels are 5.6-fold (APCs) and 3.7-fold (lt-NES cells) higher, respectively.
Consequently, we detected a significant reduction of sulfatide storage when transplanting these cells into the brain of ARSA-deficient mice. Depending on the brain region, significant reduction of sulfatide storage could be observed up to a distance of 300 µm away from transplanted cells, with both, lt-NES cell- and APC-derived transplants exhibiting comparable efficiencies in reducing sulfatide storage in the vicinity of donor cell clusters. In addition, we observed a significant reduction of sulfatide deposits around processes of donor neurons, implying that neuron-mediated gene transfer might, in the end, permit ARSA delivery to larger and more remote host brain regions than glial cells. In line with this notion, axonal transport and release has previously been demonstrated in primary mouse neurons lentivirally transduced with a tagged version of human ARSA.34 On the other hand, it is currently not clear to what extent neuronal innervation of transplanted cells might lead to pathological innervation and disturbance of endogenous networks. Until addressed in detail, glial cells as a more “inert” cell population might be considered as a safer alternative for cell-mediated gene transfer in MLD. Importantly, our quantification was only performed using coronal sections and in one plane, which does not permit extrapolation of sulfatide reduction across 3D space. Another limitation of the current study is that we used bulk infection of the cells resulting in a polyclonal cell population. Translation toward a clinical setting would require clonal lines subjected to stringent quality control including viral copy numbers and their integration potential. In addition, cell delivery to far larger areas would be required. It is difficult to extrapolate how many cells might be required to achieve widespread distribution of ARSA secreting cells throughout human CNS. While some of the transplanted precursor cells continue to proliferate, others die. Results from our previous studies involving transplantation of pluripotent stem cell-derived neural stem cells indicate that only <20% of the initially grafted cells survive on long-term.26 However, surviving neurons extent very elaborate axonal projections, which may be sufficient to reach large parts of the human brain. The proliferative potential of astroglial progenitors, on the other hand, seems to be more extensive so that one might speculate that lower initial numbers in combination with multiple injection sites might suffice for proper distribution throughout the brain. In line with this notion, recent studies showed an impressive distribution of human astrocytes derived from human fetal glial cell progenitors after multifocal injection into the postnatal brain.35 How these findings can be translated to the human brain need to be determined.
Comparing our transplantation model to the clinical scenario it is important to note that the immunodeficient host animals used in this study cannot reflect the immunological situation of a MLD patient brain where neuroinflammation is a prominent component of the disease. Whether and to what extent neuroinflammatory changes may impede such a transplantation approach cannot be predicted from our preclinical study.
Compared to other methods of enzyme delivery, our cell-mediated approach comes with distinct advantages. First, direct cell delivery to the CNS bypasses problems associated with poor penetration of recombinant enzyme or peripherally administered cells across the blood–brain barrier. Second, integration of enzyme-releasing cells into the host CNS can enable stable and perhaps life-long supply, rendering repetitive invasive interventions unnecessary. On the other hand, the peripheral nervous system and non-neural tissues will not benefit from such transplants at the same extent. Thus, combinatorial methodologies including efficient delivery of ARSA to the periphery might be required to affect ARSA deficiency in both systems.
It is important to note that neurological symptoms in MLD are considered to derive to a large extent from demyelination resulting from the demise of oligodendrocytes accumulating sulfatide. While lt-NES cells represent a highly standardized cell population suitable for neural transplantation,21,26 they show notoriously low oligodendroglial differentiation potential.21,22 Although in case of early intervention demyelination might be prevented, more advanced cases would benefit from enzyme delivery from a cell population more amenable to oligodendroglial differentiation, thereby tackling both, enzyme deficiency and myelin loss.
Materials and Methods
Human material. The study was approved by the local ethics committee. All subjects (MLD patient and healthy control) gave written informed consent.
Generation and validation of iPS cells. Retroviral plasmids for OCT4, SOX2, KLF4, and c-MYC (plasmids 17217, 17218, 17219, and 17220)36 were obtained from Addgene (Cambridge, MA). PhoenixGP cells (ATCC no. 3514; Wesel, Germany) cultured in 10-cm plates were cotransfected with the individual retroviral vectors and the VSV-G vector (plasmid 12259, Addgene) using the CaPO4 method. Viral supernatants were harvested on day 2 and 3 after transfection, filtered through a 0.45 µm cellulose acetate filter and pelleted at 5 × 104 average g force for 120 minutes. Dermal fibroblasts were plated at a density of 5 × 104 cells per well of a gelatin-coated 12-well plate and infected overnight with a combination of all four retroviruses in the presence of 6 mg/ml polybrene and 4 ng/ml FGF2. Four days after transduction, cells were split into plates preseeded with mouse embryonic fibroblasts (MEFs). Medium was switched to human iPSC medium containing DMEM/F12 culture medium supplemented with 20% KnockOut serum replacement, 0.1 mmol/l nonessential amino acids, 1 mmol/l L-glutamine, 0.1 mmol/l β-mercaptoethanol and 8 ng/ml basic fibroblast growth factor (bFGF; all from Life Technologies, Darmstadt, Germany). Four weeks after transduction, colonies with human ES cell-like morphology were manually isolated, carefully triturated and expanded to establish human iPSC lines. iPSCs were maintained on irradiated MEFs in iPSC media and split at a 1:3 to 1:4 ratio every 4–5 days. For teratoma formation, 1 × 106 cells were resuspended as clumps in iPSC medium and injected into the testis of immune-compromised SCID beige mice. After 5–8 weeks, tumors were isolated, fixed overnight in 4% paraformaldehyde, and subjected to histological examination using hematoxylin and eosin staining. For transgene silencing, total RNA was extracted using TRIzol reagent (Life Technologies). To generate cDNA, RNA was incubated with DNase I (Promega, Mannheim, Germany) and reverse transcribed with BioRad iScript Kit (BioRad, Munich, Germany) according to the manufacturer's manual. Quantitative RT–PCR was performed using the iCycler (BioRad) platform following the manufacturer's instruction. All results were normalized to GAPDH. Primers used for the analysis of transgene silencing and expression of pluripotency-associated genes were:
OCT4 endo forward: GACAGGGGGAGGGGAGGAGCTAG
OCT4 endo reverse: GTTCCCTCCAACCAGTTGCCCCAAAC
SOX2 endo forward: GTATCAGGAGTTGTCAAGGCAGAG
SOX2 endo reverse: TCCTAGTCTTAAAGAGGCAGCAAAC
c-MYC endo, forward: TTCGGGTAGTGGAAAACCAG
c-MYC endo reverse: CCTCCTCGTCGCAGTAGAAA
KLF4 endo, forward: GACCAGGCACTACCGTAAACA
KLF4 endo reverse: CTGGCAGTGTGGGTCATATC
OCT4 trans forward: GTACTCCTCGGTCCCTTTCC
SOX2 trans forward: CATGTCCCAGCACTACCAGA
c-MYC trans forward: GGAAACGACGAGAACAGTTGA
KLF4 trans forward: GACCACCTCGCCTTACACAT
ALL trans reverse: TGACACCAGACCAACTGGTAA
“Endo” refers to primers for amplification of endogenous genes, “trans” to primer pairs specific for the amplification of the viral transgenes.
SNP analysis. Genomic DNA was prepared using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Whole-genome single nucleotide polymorphism (SNP) genotyping was performed at the Institute of Human Genetics at the University of Bonn. Genomic DNA at a concentration of 50 ng/µl was used for whole-genome amplification. Afterwards, the amplified DNA was fragmented and hybridized to sequence-specific oligomers bound to beads on an Illumina HumanOmniExpress BeadChip 12V1.0 chip (Illumina, San Diego, CA). Data were analyzed using Illumina GenomeStudio V2011.1 (Illumina).
Genotype analysis. Genomic DNA was extracted using the Viagen DirectPCR DNA Extraction System (Viagen Biotech, Los Angeles, CA). PCR amplification of exon 9 including parts of the 3′ UTR was performed using Phusion polymerase (Finnzymes; primers: MLDfw, CTAGATCCCTGGCCCCTCCTC; MLDrv, CCCAGTGAGGAGCCATCACATG). PCR included a preincubation at 98 °C for 30 seconds followed by 35 cycles of denaturation at 98 °C for 10 seconds, annealing at 60 °C for 30 seconds and extension at 72 °C for 30 seconds with a final extension step at 72 °C for 10 minutes. The 423 bp fragment was extracted from the gel and subsequently sequenced (Seqlab, Göttingen, Germany).
Neural differentiation. Lt-NES cells were generated as described previously21 and continuously propagated on poly-L-ornithine/laminin-coated plates in N2 medium supplemented with B27 (1 µl/ml, Life Technologies), 10 ng/ml FGF2, and 10 ng/ml EGF (both from R&D systems, Minneapolis, MN). Neuronal differentiation was induced by removing the growth factors FGF2 and EGF from the media and culturing the cells in Neurobasal medium supplemented with B27 (1:50, Life Technologies) and DMEM/F12 supplemented with N2 (1:100) mixed at a 1:1 ratio. 300 ng/ml cAMP was added to the differentiation media. Astroglial progenitors were generated by withdrawal of growth factors from lt-NES cells for 4 weeks. Following dissociation by trypsin and further cultivation in the presence of the growth factors FGF2 and EGF, a proliferating cell population exhibiting a fibroblast-like morphology was established. Differentiation was induced by omitting the growth factors for 3–4 weeks in the presence of 10% fetal calf serum (FCS).
Vector design and lentiviral transduction. For constitutive overexpression, a modified version of the pLenti6/V5-D-TOPO system was employed in which the cytomegalovirus promoter of the pLenti6/V5-D-TOPO plasmid had been replaced with the truncated elongation factor 1α promoter and a woodchuck postregulatory element (WPRE) added before the SV40/EM7 promoter controlling the Blasticidine resistance gene (pLentiWE). The cDNA sequence of the EGFP was amplified by PCR and cloned into the pLentiWE plasmid, resulting in pLentiWE-EGFP. The cDNA sequence of human ARSA followed by an IRES was cloned into the pLentiWE-EGFP plasmid just before the EGFP sequence, resulting in the plasmid pLentiWE-hARSA-IRES-EGFP. Lentiviral particles were generated and concentrated as described previously.37,38 Briefly, 293FT cells were transduced with the lentiviral plasmids pLentiWE-EGFP or pLentiWE-hARSA-IRES-EGFP, the packaging plasmid psPAX2, and the envelope plasmid pMD2.G, harvested daily for 2 days and enriched by centrifugation. Lt-NES cells were transduced with pLentiWE-EGFP or pLentiWE-hARSA-IRES-EGFP vectors and passaged in the presence of 20 µg/ml Blasticidine S (Life Technologies) for 1 week to generate stable cell lines. From selected lt-NES cell lines, astroglial progenitor cells were generated as outlined above.
Enzymatic activity assay. Cells were grown to confluency in six-well tissue culture plates. Cell extracts were obtained by cell lysis in ice-cold Tris-buffered saline/0.5% Triton X-100 supplemented with protease inhibitors (1 mmol/l iodoacetamide, 1 mmol/l phenylmethanesulfonyl fluoride, and 5 mmol/l ethylene-diaminetetraacetic acid), followed by a 5-minute centrifugation at 11,000 g. ARSA activity in cell lysates was determined with the artificial substrate p-nitrochatechol sulfate.39 Fifty microliters of each sample were incubated for 30 minutes at 37 °C with 10 mmol/l p-nitrochatechol sulfate in 0.5 M sodium citrate-phosphate buffer (pH 5) containing 10% NaCl (w/v) and 0.2% bovine serum albumin (w/v). The reaction was stopped by addition of 500–750 µl of 1 M NaOH and absorbance was measured at 515 nm on a Beckman DU640 spectrophotometer (Beckman-Coulter, Pasadena, CA). ARSA activities were related to protein concentrations determined by the DC protein assay (BioRad).
Determination of intra- and extracellular ARSA levels by ELISA. Transduced and nontransduced iPSC-derived neuronal and APCs were cultured to confluency in six-well tissue culture plates and 2 ml serum-free culture medium. After a culture period of 48 hours without change of medium, cells and conditioned media were harvested. Conditioned media were centrifuged at 11,000 g for 5 minutes to remove floating cells. The supernatant was transferred to a new vial and supplemented with bovine serum albumin to a final concentration of 1% (w/v) in order to stabilize ARSA. Cells were harvested by trypsinization and homogenized in 100 µl ice-cold Tris-buffered saline/0.5% Triton X-100 supplemented with protease inhibitors (Halt Protease Inhibitor Cocktail, Thermo Scientific, Sankt Leon-Rot, Germany). The homogenates were centrifuged at 11,000 g for 5 minutes, and aliquots of the supernatant were used for the determination of the ARSA content and protein concentration (DC protein assay, Bio-Rad). ARSA concentrations and activities were determined in triplicates using an indirect sandwich ELISA40 and the artificial substrate p-nitrochatechol sulfate,39 respectively. Recombinant human ARSA expressed from Chinese hamster ovary cells was utilized as a standard.8 Optical densities were measured with a Genios Spectra Fluor Plus 96-well plate reader from Tecan (Groedig, Austria).
Statement about animal studies. Rag2-/--immunodeficient animals were used for teratoma assays validating multi-germ layer differentiation of iPSCs as previously described.41 Neonatal ARSA-deficient Rag2-/- mice were used for the assessment of lt-NES cells or astroglial progenitors to reduce sulfatide storage. The studies are in accordance with the legal requirements of the local authorities (permit numbers: 8.87-50.10.37.09.290 and 87.51.04.2010.A022).
Transplantation of neonatal animals. Neonatal animals (postnatal day 1) were anesthetized by hypothermia. Using transcranial injection with a glass capillary, 100,000 lt-NES cells or astroglial progenitors suspended in a total volume of 2 µl were injected into the lateral ventricle. Transplanted animals were placed on an electrical thermal blanket at 37 °C for recovery and relocated to their mothers.
Immunocytochemical analysis. Cells were fixed in 4% paraformaldehyde for 10 minutes at room temperature, washed with phosphate-buffered saline (PBS) and blocked with 10% fetal calf serum in the presence of 0.1% Triton X-100 (blocking solution). Primary antibodies were applied in blocking solution for 16 hours at 4 °C. Secondary antibodies were added for 1 hour at room temperature. 4′,6-diamidino-2-phenylindole solution was used for 5 minutes for nuclear counterstaining (Life Technologies) and cells were mounted with Mowiol/DABCO before visualization by a Zeiss Axiovision microscope with Apotome technology (Zeiss, Jena, Germany). Antibodies applied and concentrations used are outlined in Table 1.
Table 1. Antibodies and concentrations used.
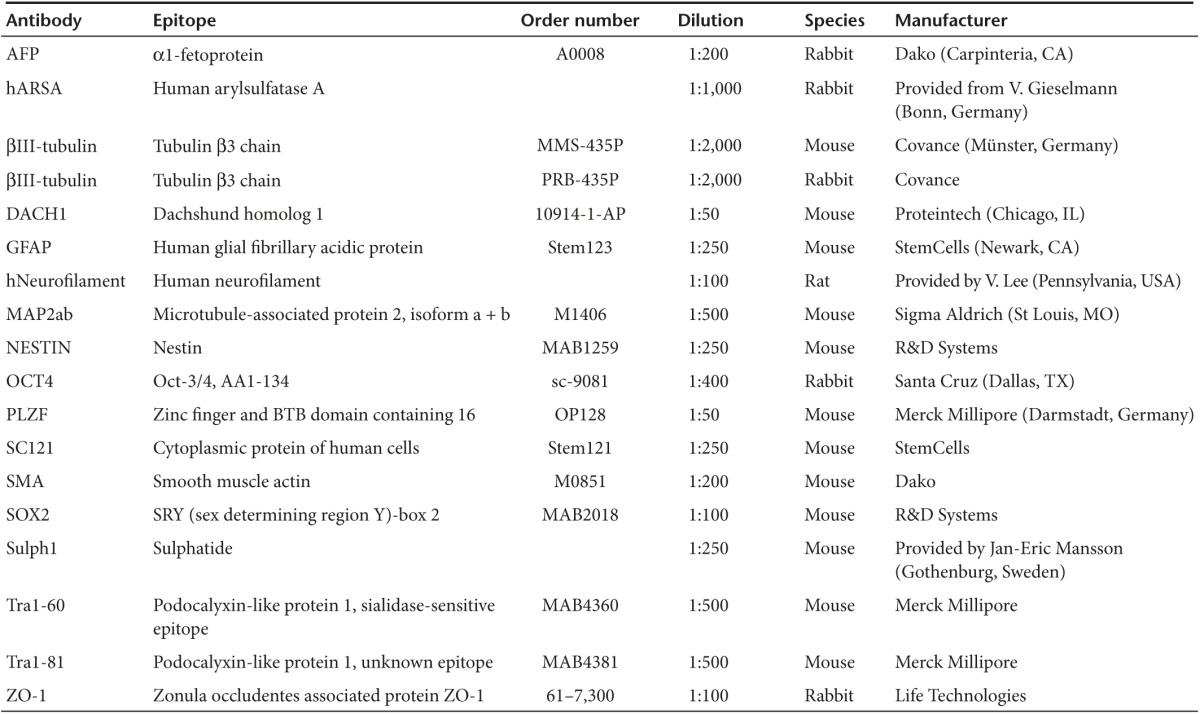
Immunohistochemical analysis. One to three months following transplantation, animals were sacrificed and perfused with paraformaldehyde (4%). Thirty-µm thick coronal sections were generated using a cryostat, dried, reconstituted, and washed with PBS for 30 minutes at room temperature. For analysis of sulfatide deposits, the histological specimen then was treated with a 0.05% Glycine/PBS solution for 30 minutes at room temperature, washed three times with PBS and blocked overnight at 4 °C with 1% bovine serum albumin and 0.05% Tween 20 in PBS. Specimens were washed additional three times (PBS, 10 minutes) and primary antibodies were applied overnight at 4 °C in PBS supplemented with 0.01% Tween 20 and 1% bovine serum albumin. Following washing with PBS (3×, 10 minutes), secondary antibodies were applied for 2 hours at room temperature. The expression of all other epitopes was examined by blocking the sections in 10% FCS, 0.1% Triton X-100 and 0.1% sodium azide and applying primary and secondary antibodies in the same solution for the same times as indicated above. Primary antibodies and concentrations used are outlined in Table 1.
Quantification of sulfatide storage. For quantification of sulfatide storage, 30-µm thick sections were processed and images were acquired under standardized acquisition times using a Zeiss Axiovision microscope. Grafted cells were identified by virtue of their GFP expression, and a ladder of rectangular sectors (each “spoke” 50 µm in height) was tangentially aligned to the border of the graft in a way, which (i) allowed continuous quantification into the adjacent host tissue within the same anatomical brain region for at least 50 µm and (ii) enabled a maximal width of tangential apposition of the bar-ladder to the graft. The number and area of sulfatide-positive deposits was quantified using unbiased automatic image analysis software (ImageJ; http://imagej.nih.gov/ij/) and correlated to the total analyzed area. Quantification of sulfatide deposits in contralateral areas devoid of donor cells was used as control (set to 100%). The number of sulfatide deposits within sectors with an identical distance to the donor cell clusters was averaged across multiple animals and sites within the same anatomical compartment. Measurements were conducted dependent on the feasibility to stay within the same anatomical compartment, resulting in different numbers (n) for different regions.
For quantification of sulfatide storage in axonal projection areas, host brain regions >500 µm away from transplanted donor cells containing human neurofilament-positive donor cell-derived axons but no human cell bodies were selected. Corresponding regions from the contralateral side were used as control, and values of the grafted side were expressed as percentage of the control (which was set to 100%).
SUPPLEMENTARY MATERIAL Figure S1. Single nucleotide polymorphism-based karyotyping.
Acknowledgments
We are grateful to Anke Leinhaas and Svenja Auel for excellent technical support, to the Institute of Human Genetics (LIFE & BRAIN Center) for single nucleotide polymorphism analysis, to Virginia Lee for providing the human neurofilament (Ho14) antibody and to Jan Eric Mansson for providing the Sulph1 antibody. This study was supported by the German Federal Ministry for Education and Research (grant 01GN0813), BONFOR and the Hertie Foundation. J.D. conceived and designed the study; collected, analyzed, and interpreted data; and wrote the manuscript. A.B., B.E., and J.L. collected, analyzed, and interpreted data. U.M. collected, analyzed, and interpreted data; provided material and wrote the manuscript. M.E. and V.G. interpreted data, provided material, and wrote the manuscript. O.B. conceived the study, interpreted data, provided financial support, and wrote the manuscript. P.K. conceived and designed the study; collected, analyzed, and interpreted data; and wrote the manuscript.
Supplementary Material
Single nucleotide polymorphism-based karyotyping.
References
- von Figura, K, Gieselmann, V, and Jaeken, J (2001). The metabolic and molecular bases of inherited disease. In: Scriver, CR, Beaudet, AL, Sly, WS, Valle, D, Childs, B, Kinzler, KW, Vogelstein, B, (eds.). Metachromatic Leukodystrophy. McGraw-Hill: New York. pp. 3695–3724.
- Gieselmann, V and Krägeloh-Mann, I (2010). Metachromatic leukodystrophy–an update. Neuropediatrics 41: 1–6. [DOI] [PubMed] [Google Scholar]
- Birkholz, T, Irouschek, A, Knorr, C and Schmidt, J (2009). Alternative anesthetic management of a child with spastic quadriplegia due to metachromatic leukodystrophy using total intravenous anesthesia. Paediatr Anaesth 19: 551–552. [DOI] [PubMed] [Google Scholar]
- Coutinho, MF, Prata, MJ and Alves, S (2012). Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab 105: 542–550. [DOI] [PubMed] [Google Scholar]
- Krägeloh-Mann, I, Groeschel, S, Kehrer, C, Opherk, K, Nägele, T, Handgretinger, R et al. (2013). Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow Transplant 48: 369–375. [DOI] [PubMed] [Google Scholar]
- Laney, DJ, White, AL, Rhead, WJ and Fernhoff, P (2008). Creating genetics-based infusion centers: a case study of two models. Genet Med 10: 626–632. [DOI] [PubMed] [Google Scholar]
- Matzner, U, Lüllmann-Rauch, R, Stroobants, S, Andersson, C, Weigelt, C, Eistrup, C et al. (2009). Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol Ther 17: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes, F, Stroobants, S, Gerlach, D, Wohlenberg, C, Wessig, C, Fogh, J et al. (2012). Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum Mol Genet 21: 2599–2609. [DOI] [PubMed] [Google Scholar]
- Stroobants, S, Gerlach, D, Matthes, F, Hartmann, D, Fogh, J, Gieselmann, V et al. (2011). Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum Mol Genet 20: 2760–2769. [DOI] [PubMed] [Google Scholar]
- Consiglio, A, Quattrini, A, Martino, S, Bensadoun, JC, Dolcetta, D, Trojani, A et al. (2001). In vivo gene therapy of metachromatic leukodystrophy by lentiviral vectors: correction of neuropathology and protection against learning impairments in affected mice. Nat Med 7: 310–316. [DOI] [PubMed] [Google Scholar]
- Kurai, T, Hisayasu, S, Kitagawa, R, Migita, M, Suzuki, H, Hirai, Y et al. (2007). AAV1 mediated co-expression of formylglycine-generating enzyme and arylsulfatase a efficiently corrects sulfatide storage in a mouse model of metachromatic leukodystrophy. Mol Ther 15: 38–43. [DOI] [PubMed] [Google Scholar]
- Colle, MA, Piguet, F, Bertrand, L, Raoul, S, Bieche, I, Dubreil, L et al. (2010). Efficient intracerebral delivery of AAV5 vector encoding human ARSA in non-human primate. Hum Mol Genet 19: 147–158. [DOI] [PubMed] [Google Scholar]
- Piguet, F, Sondhi, D, Piraud, M, Fouquet, F, Hackett, NR, Ahouansou, O et al. (2012). Correction of brain oligodendrocytes by AAVrh.10 intracerebral gene therapy in metachromatic leukodystrophy mice. Hum Gene Ther 23: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzner, U and Gieselmann, V (2005). Gene therapy of metachromatic leukodystrophy. Expert Opin Biol Ther 5: 55–65. [DOI] [PubMed] [Google Scholar]
- Biffi, A, Capotondo, A, Fasano, S, del Carro, U, Marchesini, S, Azuma, H et al. (2006). Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest 116: 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Kawabata, K, Migita, M, Mochizuki, H, Miyake, K, Igarashi, T, Fukunaga, Y et al. (2006). Ex vivo cell-mediated gene therapy for metachromatic leukodystrophy using neurospheres. Brain Res 1094: 13–23. [DOI] [PubMed] [Google Scholar]
- Givogri, MI, Galbiati, F, Fasano, S, Amadio, S, Perani, L, Superchi, D et al. (2006). Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy. J Neurosci 26: 3109–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, D, Schmandt, T, Muth-Köhne, E, Perez-Bouza, A, Segschneider, M, Gieselmann, V et al. (2006). Embryonic stem cell-based reduction of central nervous system sulfatide storage in an animal model of metachromatic leukodystrophy. Gene Ther 13: 1686–1695. [DOI] [PubMed] [Google Scholar]
- Polten, A, Fluharty, AL, Fluharty, CB, Kappler, J, von Figura, K and Gieselmann, V (1991). Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med 324: 18–22. [DOI] [PubMed] [Google Scholar]
- Koch, P, Opitz, T, Steinbeck, JA, Ladewig, J and Brüstle, O (2009). A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci USA 106: 3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk, A, Koch, P, Kesavan, J, Takashima, Y, Ladewig, J, Alexander, M et al. (2012). Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One 7: e29597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens, J, Stüber, K, Wunderlich, P, Ladewig, J, Kesavan, JC, Vandenberghe, R et al. (2013). APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based γ-secretase modulation. Stem Cell Reports 1: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, B, Saftig, P, Hartmann, D, Coenen, R, Lüllmann-Rauch, R, Goebel, HH et al. (1996). Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc Natl Acad Sci USA 93: 14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai, Y, Rathbun, G, Lam, KP, Oltz, EM, Stewart, V, Mendelsohn, M et al. (1992). RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68: 855–867. [DOI] [PubMed] [Google Scholar]
- Steinbeck, JA, Koch, P, Derouiche, A and Brüstle, O (2012). Human embryonic stem cell-derived neurons establish region-specific, long-range projections in the adult brain. Cell Mol Life Sci 69: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki, K, Tatarishvili, J, Wood, J, Koch, P, Wattananit, S, Mine, Y et al. (2012). Human-induced pluripotent stem cells form functional neurons and improve recovery after grafting in stroke-damaged brain. Stem Cells 30: 1120–1133. [DOI] [PubMed] [Google Scholar]
- Fredman, P, Mattsson, L, Andersson, K, Davidsson, P, Ishizuka, I, Jeansson, S et al. (1988). Characterization of the binding epitope of a monoclonal antibody to sulphatide. Biochem J 251: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, P, Dahms, NM and Kornfeld, S (2003). Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4: 202–212. [DOI] [PubMed] [Google Scholar]
- Fratantoni, JC, Hall, CW and Neufeld, EF (1969). The defect in Hurler and Hunter syndromes. II. Deficiency of specific factors involved in mucopolysaccharide degradation. Proc Natl Acad Sci USA 64: 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna, J, Wernig, M, Markoulaki, S, Sun, CW, Meissner, A, Cassady, JP et al. (2007). Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318: 1920–1923. [DOI] [PubMed] [Google Scholar]
- Tedesco, FS, Gerli, MF, Perani, L, Benedetti, S, Ungaro, F, Cassano, M et al. (2012). Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Transl Med 4: 140ra89. [DOI] [PubMed] [Google Scholar]
- Dierks, T, Schlotawa, L, Frese, MA, Radhakrishnan, K, von Figura, K and Schmidt, B (2009). Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease - Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim Biophys Acta 1793: 710–725. [DOI] [PubMed] [Google Scholar]
- Luca, T, Givogri, MI, Perani, L, Galbiati, F, Follenzi, A, Naldini, L et al. (2005). Axons mediate the distribution of arylsulfatase A within the mouse hippocampus upon gene delivery. Mol Ther 12: 669–679. [DOI] [PubMed] [Google Scholar]
- Windrem, MS, Schanz, SJ, Guo, M, Tian, GF, Washco, V, Stanwood, N et al. (2008). Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2: 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K, Tanabe, K, Ohnuki, M, Narita, M, Ichisaka, T, Tomoda, K et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872. [DOI] [PubMed] [Google Scholar]
- Mertens, J, Stüber, K, Poppe, D, Doerr, J, Ladewig, J, Brüstle, O et al. (2013). Embryonic stem cell-based modeling of tau pathology in human neurons. Am J Pathol 182: 1769–1779. [DOI] [PubMed] [Google Scholar]
- Koch, P, Siemen, H, Biegler, A, Itskovitz-Eldor, J and Brüstle, O (2006). Transduction of human embryonic stem cells by ecotropic retroviral vectors. Nucleic Acids Res 34: e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, H, Dodgson, KS and Spencer, B (1959). The assay of arylsulphatases A and B in human urine. Clin Chim Acta 4: 453–455. [DOI] [PubMed] [Google Scholar]
- Matzner, U, Harzer, K, Learish, RD, Barranger, JA and Gieselmann, V (2000). Long-term expression and transfer of arylsulfatase A into brain of arylsulfatase A-deficient mice transplanted with bone marrow expressing the arylsulfatase A cDNA from a retroviral vector. Gene Ther 7: 1250–1257. [DOI] [PubMed] [Google Scholar]
- Koch, P, Breuer, P, Peitz, M, Jungverdorben, J, Kesavan, J, Poppe, D et al. (2011). Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480: 543–546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Single nucleotide polymorphism-based karyotyping.