

Abstract
Objective:
To identify the molecular genetic basis of a syndrome characterized by rapidly progressing cerebral atrophy, intractable seizures, and intellectual disability.
Methods:
We performed exome sequencing in the proband and whole-genome single nucleotide polymorphism genotyping (copy number variant analysis) in the proband-parent trio. We used heterologous expression systems to study the functional consequences of identified mutations.
Results:
The search for potentially deleterious recessive or de novo variants yielded compound heterozygous missense (c.1202G>A, p.Cys401Tyr) and frameshift deletion (c.2396delG, p.Ser799IlefsTer96) mutations in ADAM22, which encodes a postsynaptic receptor for LGI1. The deleterious effect of the mutations was observed in cell surface binding and immunoprecipitation assays, which revealed that both mutant proteins failed to bind to LGI1. Furthermore, immunoprecipitation assays showed that the frameshift mutant ADAM22 also did not bind to the postsynaptic scaffolding protein PSD-95.
Conclusions:
The mutations identified abolish the LGI1-ADAM22 ligand-receptor complex and are thus a likely primary cause of the proband's epilepsy syndrome, which is characterized by unusually rapidly progressing cortical atrophy starting at 3–4 months of age. These findings are in line with the implicated role of the LGI1-ADAM22 complex as a key player in nervous system development, specifically in functional maturation of postnatal synapses. Because the frameshift mutation affects an alternatively spliced exon with highest expression in postnatal brain, the combined effect of the mutations is likely to be hypomorphic rather than complete loss of function. This is compatible with the longer survival of the patient compared to Lgi1−/− and Adam22−/− mice, which develop lethal seizures during the first postnatal weeks.
Many types of epilepsies are caused by genetic defects in ion channel or neurotransmitter genes, but an alternative epileptogenic mechanism is revealed by dysfunction of the complexes formed by the secreted neuronal glycoprotein LGI1 and its pre- and postsynaptic receptors ADAM23 and ADAM22, respectively.1,2 ADAM22 and ADAM23 are members of the ADAM (A Disintegrin And Metalloproteinase) family of transmembrane proteins implicated in proteolysis and cell adhesion.3 Unlike many ADAM proteins, neuronally expressed ADAM22 and ADAM23 are catalytically inactive and act by binding to other proteins. LGI1-ADAM22 and LGI1-ADAM23 complexes have recently been suggested to function as transsynaptic players forming macromolecular complexes regulating synapse maturation and function, particularly in postnatal brain1,2,4,5 (reviewed in reference 6). In addition to the postsynaptic membrane, ADAM22 also functions in axons in the CNS7 and peripheral nervous system (PNS)8 and in Schwann cells.8
LGI1, ADAM22, and ADAM23 are all genetically linked to epilepsy. Heterozygous mutations in LGI1, which impair LGI1 secretion or binding to its receptors,9 cause autosomal dominant lateral temporal lobe epilepsy (ADLTE; OMIM #600512).10 Although ADAM22 and ADAM23 mutations have not been reported in human diseases, knockout mice for Lgi1,2,11,12 Adam22,13 and Adam2314 manifest with lethal seizures during the first postnatal weeks. Furthermore, ADAM23 has been implicated in canine epilepsy.15
Advances in high-throughput sequencing methods have enhanced identification of genes for rare disorders, including epilepsy syndromes.16–18 We used whole-exome sequencing to characterize the genetic cause of a severe infantile-onset progressive encephalopathy with intractable seizures. We discovered compound heterozygous mutations in ADAM22 that compromise the protein function.
METHODS
Study patient.
The proband was ascertained from a series of 30 Finnish patients with severe epilepsy syndromes who underwent whole-exome sequencing. The clinical data were reviewed from hospital records. H.P. and M.S. personally examined the patient. The original CT and MRI images obtained prior to the age of 11 years were not available for re-review.
Standard protocol approvals, registrations, and patient consents.
An institutional review board at the Helsinki University Hospital approved the study. Informed consent for DNA analysis was obtained from the parents.
Exome sequencing and variant calling.
Genomic DNA extracted from peripheral blood of the proband was exome-sequenced at the Wellcome Trust Sanger Institute (Hinxton, Cambridge, United Kingdom) using methods described previously.19 Briefly, SureSelect Human All Exon 50 Mb V3 RNA baits (Agilent Technologies, Santa Clara, CA) were used to enrich exonic targets, and sequencing was carried out using HiSeq 2000 platform (Illumina, San Diego, CA). Alignment of the sequence reads to Human Reference Genome hs37d5 (based on GRCh37) and their further processing were performed as described previously.19 Single nucleotide variants and indels of the proband exome were called jointly with 84 previously published exomes19 and 42 unpublished in-house exomes using GATK HaplotypeCaller (v. 3.3; https://www.broadinstitute.org/gatk/).20–22
Variant analysis under recessive and de novo inheritance models.
Reflecting the different possible inheritance patterns of the underlying mutation(s) in the study patient with no affected relatives, rare autosomal or sex-linked recessive and novel heterozygous de novo mutations were explored. Mutations in mitochondrial genome were also analyzed. The variant filtering process is described in detail in appendix e-1 and figure e-1 at Neurology.org/ng.
Variant validation and segregation analysis.
Candidate variants were confirmed, and segregation of the variants was analyzed by bidirectional Sanger sequencing. Primers are available from the authors upon request.
Analysis of copy number variants.
Illumina Human CoreExome single nucleotide polymorphism array with 548K markers enriched to genic regions was used to detect copy number variants (CNVs) from genomic DNA isolated from peripheral blood of the proband and parents and to confirm correct relatedness of DNA samples in the proband-parent trio. PennCNV23 was used to call CNVs (http://www.openbioinformatics.org/penncnv/).
Functional studies.
Plasmid construction.
The complementary DNA (cDNA) of human LGI1 was purchased from Thermo Scientific (clone ID: 4811956) and subcloned into pCAGGS with FLAG tag at its C-terminus. The cDNA of human ADAM22 (same sequence as in NM_021723 except the c.242C>G, p.Pro81Arg polymorphism in the Pro domain, which is cleaved from the mature ADAM22) was subcloned into pCAGGS. ADAM22 mutants Cys401Tyr and Ser799IlefsTer96 were generated by using site-directed mutagenesis. pGW1-PSD-95-FLAG was constructed by replacing a green fluorescent protein (GFP) fragment of pGW1-PSD-95-GFP with a synthetic DNA fragment encoding FLAG.24 All PCR products were verified by DNA sequencing.
Antibodies.
The following antibodies were used: rabbit polyclonal antibodies to LGI1 (ab30868; Abcam, San Francisco, CA), mouse monoclonal antibodies to KDEL (10C3; Enzo Life Sciences, New York, NY), PSD-95 (MA1-046; Thermo Scientific, Waltham, MA), and FLAG (M2; Sigma-Aldrich, St. Louis, MO). Rabbit polyclonal antibodies to ADAM22 were raised against GST-mouse ADAM22 (aa 444-526), corresponding to the extracellular disintegrin domain. Antibodies were affinity-purified on a cyanogen bromide–activated Sepharose 4B (GE Healthcare, Piscataway, NJ) column containing the immunizing antigen.
Cell surface binding assay.
The assay was performed in COS7 cells with minor modifications to a previously reported method.1 Details of the assay are presented in appendix e-1.
Immunoprecipitation.
HEK293T cells were cotransfected with wild-type LGI1-FLAG or PSD-95-FLAG together with either wild-type ADAM22 or one of the ADAM22 mutants, Cys401Tyr or Ser799IlefsTer96. The immune complexes were precipitated with FLAG-M2 agarose and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Gels were subjected to Western blotting. A more detailed description of the method is presented in appendix e-1.
Accession codes.
The raw aligned sequence reads were submitted to the European Genome-phenome Archive (https://www.ebi.ac.uk/ega/home) by Wellcome Trust Sanger Institute under study accession ID EGAS00001000190.
RESULTS
Clinical features.
The patient, currently 26 years of age, was born at term after an uncomplicated pregnancy. She is the second child of healthy nonconsanguineous parents of Finnish ancestry, with no family history of encephalopathy or epilepsy. During pregnancy, fetal movements were normal. Birth measurements were 3,700 g/51 cm (+0.5 SD)/33.5 cm (−1 SD). She had an Apgar score of 10 and was discharged from the hospital normally. The first signs of disease were brief episodes of apnea at 2 months of age, and soon thereafter she lost her ability to smile and make eye contact. At 3 months, she was hospitalized for repeated brief seizures. The seizures started with focal symptoms and later generalized. EEG was severely abnormal, and generalized spikes initiating from the left side with abnormal background activity were seen. The seizures proved to be intractable; treatment with barbiturate anesthesia and phenobarbital, phenytoin, carbamazepine, and valproic acid in different combinations was ineffective. The ammonium levels were mildly and briefly elevated during hospitalization, but metabolic screening tests remained normal. At 3.5 and 4 months, the brain CT was normal, but 6 weeks later, marked supratentorial atrophy was present. From 6 to 9 months the seizures were better controlled, but thereafter she started to have short focal clonic seizures or myoclonias.
At 1 year of age, she was very hypotonic, with no active movement, and had brisk reflexes in the lower extremities and rigidity in both the upper and lower extremities. She continued to have short tonic seizures, sometimes with focal clonic jerks in her left eye and in the corner of the mouth. The occipital frontal circumference (OFC) was 43 cm (−2.5 SD). She had a narrow forehead, open mouth, full lips, and a high and narrow palate. The nose was short and the ear lobules were outward-turning, and she had intermittent swelling of extremities and face. Electroretinography was normal. On brain MRI, the supratentorial atrophy had progressed, and there was marked subdural effusion. Cerebellum and brain stem were normal. In morphologic assessment of muscle biopsy, nicotinamide adenine dinucleotide and cytochrome c stainings were normal, but frequent lipid vacuoles were seen. Electron microscopic analysis of the sural nerve was normal, and electroneuromyography also showed normal results. The liver enzymes were moderately elevated, serum lactate was slightly above normal range, and respiratory chain analysis revealed unspecific changes (low consumption of oxygen and high complex IV activity with cytochrome c) not exclusively pointing toward mitochondrial defect. Neuropathy, ataxia, retinitis pigmentosa syndrome point mutation analysis and mitochondrial DNA Southern blotting gave normal results.
At 2 years, the OFC was 44.5 cm (−3 SD) and the CT showed marked supratentorial atrophy and subdural effusion. The cerebellum was normal. She experienced brief generalized seizures up to 4 times per day and was treated with phenobarbital, valproic acid, and diazepam. No voluntary movements were seen, and microcephaly together with profound intellectual disability was present. The ophthalmologic examination was normal at 2 years and showed mild atrophy of the optic nerve at 4 years 10 months. No signs of hearing loss were noted, and sometimes an auditory stimulus preceded a seizure. After the rapid progression during the first year of life, her epilepsy has remained stable with focal spikes and decreased background activity. The MRI images at 11 years showed severe cortical atrophy, widening of both central and cortical CSF spaces, and skull thickening (figure 1). She is currently nonambulatory, has intractable epilepsy, and is living in institutional care.
Figure 1. Brain imaging findings in the patient.
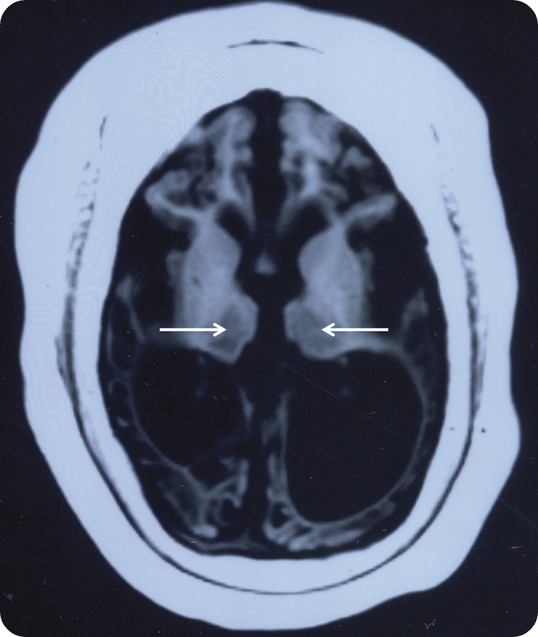
An axial T1-weighted MRI image of the patient at the age of 11 years. There is extensive loss of both gray and white matter with corresponding widening of the CSF spaces and thickening of the skull. Both thalami (arrows) show shrinking and signal change.
Genetic analysis reveals compound heterozygous mutations in ADAM22.
We analyzed the exome variant data of the proband (sequencing metrics in appendix e-1) for rare and potentially deleterious autosomal or X-linked recessive mutations (figure e-1). Variants in 2 genes, ADAM22 and CPA4, passed the recessive filtering (figure e-1, table e-1), but only the mutations in ADAM22 segregated in an autosomal recessive manner in the family (figure 2A and figure e-2). The patient was compound heterozygous for a missense mutation c.1202G>A encoding the p.Cys401Tyr substitution (mutation nomenclature based on ADAM22 variant 1, RefSeq ID NM_021723) and a 1-bp frameshift deletion c.2396delG (p.Ser799IlefsTer96). The missense mutation is present in the heterozygous state in 2 individuals of European ancestry in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), yielding an allele frequency of 1.658 × 10−5. The deletion mutation is novel. The missense substitution, p.Cys401Tyr, occurs in a highly conserved cysteine residue that forms a disulfide bond with Cys394 in the metalloproteinase domain25 (figure 2, B and C and figure e-3). The p.Cys401Tyr substitution is predicted to be deleterious by all 4 in silico methods used. The frameshift deletion is predicted to alter the last ∼100 residues of ADAM22, thus abolishing the PDZ-binding motif (figure 2B). The deletion occurs in exon 27 of the longest isoform of ADAM22 (RefSeq ID NM_021723), which encodes residues in the cytoplasmic tail and is subjected to alternative splicing26,27 (figures e-4 and e-5, see also Discussion).
Figure 2. Compound heterozygous mutations in ADAM22.

(A) Segregation of the mutations in the family. Arrow indicates the exome-sequenced patient. +, wild-type. (B) Domain structure of ADAM22 and location of the mutations. Exon numbering and mutation positions are based on the longest isoform of ADAM22 (variant 1, NM_021723). (C) ClustalX (http://www.clustal.org/clustal2/) comparison of amino acid sequences surrounding Cys401 shows full conservation of Cys401 (arrow) across different vertebrate species. Fully conserved, strongly similar, and weakly similar amino acid residues are labeled with asterisks, colon, and periods, respectively.
Eight variants passed the filtering under the “de novo” hypothesis, where we looked for novel heterozygous variants (figure e-1, table e-1). Because the parents were not exome- sequenced, we analyzed segregation of these variants by capillary sequencing. All 8 variants were inherited from an unaffected parent and thus were not considered pathogenic. Variants in mitochondrial DNA were known polymorphisms (data not shown).
Analysis of CNVs in the proband-parent trio did not yield likely pathogenic variants (data not shown).
ADAM22 mutants show aberrant binding to LGI1.
Using a cell surface binding assay, we first examined whether LGI1 interacts with the ADAM22 mutants. When LGI1 was cotransfected with wild-type ADAM22, LGI1 efficiently bound to ADAM22 on the cell surface as previously reported1 (figure 3A). In contrast, LGI1 did not bind to the Cys401Tyr and Ser799IlefsTer96 mutants on the cell surface. Although the localization of the Cys401Tyr mutant was similar to that of the wild type, most of the Ser799IlefsTer96 mutant protein was localized in the endoplasmic reticulum (ER), labeled by anti-KDEL antibody (figure 3, A and B).
Figure 3. ADAM22 mutant proteins do not bind to LGI1.

(A) Fluorescent confocal microscope images from the cell surface binding assay, in which indicated ADAM22 complementary DNAs were cotransfected with wild-type FLAG-tagged LGI1 into COS7 cells. Surface-bound FLAG-tagged LGI1 was labeled before cell permeabilization (red) and then ADAM22 was stained (green). Both ADAM22 mutants fail to bind to LGI1. Bar: 20 μm. (B) Ser799IlefsTer96 mutant (green) is localized in the endoplasmic reticulum labeled by the anti-KDEL antibody (red). Regions outlined with white squares are magnified in the upper right of the images. Bar: 20 μm. (C, D) Immunoprecipitation of ADAM22 mutants with FLAG-tagged LGI1 (C) or PSD-95 (D) in HEK293T cells. Neither ADAM22 mutant binds to LGI1 (left panel in C). The Cys401Tyr mutant binds to PSD-95, but the Ser799IlefsTer96 mutant does not (left panel in D). Arrows and arrowheads indicate the positions of immature and mature ADAM22, respectively. Immature ADAM22 is often observed in overexpressed cells and seems to nonspecifically bind to LGI1 under these conditions, whereas in the brain the immature band is not observed.
We next analyzed the heterologous expression of ADAM22 mutants in HEK293T cells by Western blotting. The expression level of Cys401Tyr was comparable to that of the wild type, whereas Ser799IlefsTer96 was less expressed in HEK293T cells and remained as an immature form (propeptide not cleaved, see inputs in figure 3, C and D). When LGI1 was cotransfected with wild-type ADAM22, ADAM22 was efficiently coimmunoprecipitated with LGI1 (figure 3C). In contrast, neither the Cys401Tyr nor the Ser799IlefsTer96 mutant was coimmunoprecipitated with LGI1 (figure 3C, arrowheads). We also found that the wild-type ADAM22 and Cys401Tyr mutant, which have a PDZ-binding motif at their C-terminus, bound to the postsynaptic scaffolding protein PSD-95, as shown previously for wild-type ADAM22,1 but Ser799IlefsTer96 lacking the motif did not (figure 3D).
DISCUSSION
We identified compound heterozygous loss-of-function mutations in ADAM22 in a patient with rapidly progressing severe encephalopathy with intractable seizures and profound intellectual disability. After initially normal CT scans, a remarkable feature of her disease was the rapidly progressing cerebral atrophy (within only a few weeks) with subdural effusions that became apparent approximately 2 months after seizure onset. The brain imaging changes were very similar to those seen in Menkes syndrome,28,29 and the patient's symptoms also resemble those seen in Alpers syndrome.30 The disease course of the patient was also unusual. The progression was very rapid in infancy after the onset of intractable seizures at 3 months of age, following the apparently normal first months of life. Thereafter the condition stabilized, leaving the patient nonambulatory with intractable seizures, and she is still alive at the age of 26 years. A rapidly progressing disease course in infancy may be a characteristic feature for ADAM22-associated disease, related to the role of ADAM22 in postnatal brain development.
We searched for pathogenic recessive and de novo variants in the exome and CNV data. The only variants passing the filtering and having a compatible segregation pattern in the family were compound heterozygous mutations in ADAM22 (figure e-1). The ability to dramatically reduce the number of candidate variants when only the proband was exome-sequenced was possible by filtering against comprehensive variant databases including population-matched individuals, in our case Finns in the ExAC database.
Our functional assays indicated that both mutations cause a nonfunctional ADAM22 protein. The Cys401Tyr mutant did not bind to LGI1 in cell surface binding and immunoprecipitation assays. The p.Cys401Tyr substitution is predicted to break a disulfide bond in the metalloproteinase-like (M) domain. The crystal structure of the extracellular domain of ADAM22 has been recently determined25 (figure e-3). Based on this structure, the authors suggested a modular movement model of ligand binding to ADAM22, where the ligand, LGI1, competes with the M domain in binding with the cysteine-rich domain (C), making the M domain move away from the C domain.25 Because the p.Cys401Tyr substitution likely affects the tertiary structure of the M domain, it may hinder the conformational change that is necessary for ADAM22 in implementing its protein–protein adhesion function.
Because the C-terminal PDZ-binding motif is intact in the Cys401Tyr mutant, its binding to PSD-95 appeared normal in the immunoprecipitation assay. On the contrary, the frameshift mutation is predicted to alter the ∼100 amino acids in the cytoplasmic part of the protein, including residues binding to cytoplasmic PSD-95 (figure 2B) and 14-3-3 proteins.1,5,31 Our binding assays showed that the frameshift mutation leads to a nonfunctional protein that is localized mainly in the ER and does not bind to LGI1 or to PSD-95.
The highly conserved exon 27 containing the frameshift deletion is subjected to alternative splicing (figure e-4), as are several other exons encoding parts of the cytoplasmic domain of ADAM22. Exon 27 is expressed in several regions of the human and mouse brain13,26,27,32 and has a progressive temporal expression pattern that differs from that of ADAM22 exons that are not subjected to alternative splicing (figure e-5).33 Accounting for all exons, ADAM22 expression is highest in postnatal stages (reference 1 and figure e-5). In embryonic stages, exon 27 is expressed in lower levels than other exons; the proportion of splice variants expressing exon 27 rapidly increases toward the end of the embryonic development and reaches a plateau at the first postnatal months. Exon 27 does not contain known peptide motifs, but its temporal expression pattern suggests that it contains residues playing a role in the CNS function in postnatal stages in particular. Reflecting the importance of LGI1–ADAM22/23 complexes in postnatal brain development, the Lgi1−/−, Adam22−/−, and Adam23−/− mice start developing seizures around 10 postnatal days.2,11–14 The expression pattern of ADAM22 and data from knockout mice are in line with the development of symptoms in our patient, who was apparently asymptomatic at birth, with the first symptoms appearing at approximately 2 months of age and the disease progressing rapidly thereafter.
Because not all ADAM22 transcripts express exon 27, the frameshift mutation is likely not a total null mutation but rather a hypomorphic one, its effect increasing temporally. This may also explain why the compound heterozygous mutations in our patient were not lethal in early life, in contrast to Lgi1−/−, Adam22−/−, and Adam23−/− mice, which die at 2–3 postnatal weeks.2,11–14 Furthermore, exon 27 is skipped from ADAM22 in the PNS,8,13,32 which may explain why sural biopsy shows normally myelinated nerves in the proband, whereas the knockout mice present with marked hypomyelination of the peripheral nerves.13
One epileptogenic mechanism of LGI1–ADAM22 dysfunction could stem from the observations that loss of neuronal LGI1 reduces AMPAR-mediated synaptic transmission1,2 and that loss of ADAM22 reduces both AMPAR- and NMDAR-mediated currents in excitatory synapses.5 This role of the LGI1-ADAM22 complex as the regulator of functional maturation of excitatory synapses in postnatal stages is mediated by interaction between the PDZ-binding motif of ADAM22 and PSD-95.5 When the complex is dysfunctional, it leaves synapses immature5 and could promote seizure activity, as has been shown for a monoallelic LGI1 mutant.4 Reduction of synaptic AMPARs as a consequence of the disruption of LGI1-ADAM22 interaction has also been suggested34 to be the underlying epileptogenic pathomechanism in autoimmune limbic encephalitis, which manifests with amnesia and seizures and is caused by autoantibodies against LGI1.35,36 It remains unknown whether seizure activity contributes to the rapidly progressive degeneration of cortical regions in our patient or whether ADAM22 dysfunction has a direct negative effect on the development of neuronal connections, increasing seizure susceptibility.
Our data suggest that loss of ADAM22 function in the CNS underlies the severe rapidly progressing infantile-onset encephalopathy with epilepsy in our patient. Given that heterozygous LGI1 mutations underlie ADLTE, previous studies have attempted to identify dominantly inherited mutations in ADAM22 in ADLTE families.37,38 However, the fact that the parents of the proband—who are heterozygous carriers of loss-of-function mutations—do not have epilepsy indicates that heterozygous deleterious mutations are tolerated in ADAM22 and that ADAM22 is primarily a recessive disease gene. This hypothesis is supported by the presence of 9 truncating variants of the canonical ADAM22 transcript in the ExAC database, whereas there are none for LGI1. Lack of rare biallelic mutations in ADAM22 in other exome data sets (appendix e-1) indicates that ADAM22-associated encephalopathy is rare, which may partially be because complete knockout mutations of ADAM22 are not tolerated and only hypomorphic mutations cause a phenotype similar to that in our study. Future studies are needed to confirm this genetic association and define the phenotypic presentations of ADAM22 dysfunction.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the family for participation in the study, Dr. Toshitaka Kawarai (Tokushima University Graduate School) for complementary DNA of human ADAM22, Carol Scott and Jillian Durham (Wellcome Trust Sanger Institute) for exome sequence processing, Sinikka Lindh for management of patient samples, Dr. Kaisa Kettunen (Institute for Molecular Medicine Finland) for whole-genome genotyping data generation, and CSC–IT Center for Science, Ltd., for the allocation of computational resources. Exome Aggregation Consortium, EuroEPINOMICS RES consortium, and The Deciphering Developmental Disorders study are acknowledged for data sharing.
GLOSSARY
- ADLTE
autosomal dominant lateral temporal lobe epilepsy
- cDNA
complementary DNA
- CNV
copy number variant
- ER
endoplasmic reticulum
- ExAC
Exome Aggregation Consortium
- GFP
green fluorescent protein
- OFC
occipital frontal circumference
- PNS
peripheral nervous system
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
M. Muona: analysis and interpretation of genetic data, drafting and revising the manuscript. Y. Fukata: functional analysis and interpretation, drafting and revising the manuscript. A.-K. Anttonen: patient evaluation, study concept and design, drafting and revising the manuscript. A. Laari: analysis and interpretation of genetic data, revising the manuscript. A. Palotie: study concept and design, revising the manuscript. H. Pihko: patient evaluation, revising the manuscript. T. Lönnqvist: patient evaluation, revising the manuscript. L. Valanne: patient evaluation, revising the manuscript. M. Somer: patient evaluation, revising the manuscript. M. Fukata: functional analysis and interpretation, drafting and revising the manuscript. A.-E. Lehesjoki: study concept and design, interpretation of data, drafting and revising the manuscript.
STUDY FUNDING
This study was supported by the Folkhälsan Research Foundation (A.-E.L.), Wellcome Trust grants 089062 and 098051 (A.P.), the European Commission FP7 project no. 242167 SynSys (A.P.), Health-2010 project no. 261433 BioSHare (A.P.), the Academy of Finland (grants no. 251704 and 263401) (A.P.), Emil Aaltonen Foundation (M.M. and A.-K.A.), Epilepsy Research Foundation (M.M.), University of Helsinki Funds (M.M.), Doctoral Programme in Biomedicine (M.M.), Arvo and Lea Ylppö Foundation (M.M. and A.-K.A.), Finnish Brain Foundation (M.M.), Paulo Foundation (M.M.), Biomedicum Helsinki Foundation (M.M.), Helsinki University Central Hospital Research Fund (A.-K.A.), and the Ministry of Education, Culture, Sports, Science, and Technology (15H04279 and 15H01299) (Y.F.) and (15H01570) (M.F.).
DISCLOSURE
Mr. Muona has received research support from the Emil Aaltonen Foundation, the Epilepsy Research Foundation, University of Helsinki Funds, the Doctoral Programme in Biomedicine University of Helsinki, the Arvo and Lea Ylppö Foundation, the Finnish Brain Foundation, the Paolo Foundation, and the Biomedicum Helsinki Foundation. Dr. Y. Fukata has received research support from the Ministry of Education, Culture, Sports, Science, and Technology; the Astellas Foundation for Research on Metabolic Disorders; the Takeda Science Foundation; and the Mochida Memorial Foundation for Medical and Pharmaceutical Research. Dr. Anttonen has received research support from the Arvo and Lea Ylppö Foundation and the Helsinki University Central Hospital Research Fund. Ms. Laari reports no disclosures. Dr. Palotie is a member of the Pfizer Genetics Scientific Advisory Panel, has received travel expenses and/or honoraria for lectures or educational activities not funded by industry, and has received research support from the Finnish Academy, the European Union NIH, National Institute of Neurological Disorders and Stroke, the Juselius Foundation, and the Finnish Foundation for Cardiovascular Research. Dr. Pihko receives publishing royalties for Textbook of Child Neurology, Duodecim 2014. Dr. Lönnqvist, Dr. Valanne, and Dr. Somer report no disclosures. Dr. M. Fukata has served on the editorial board of the European Journal of Neuroscience and has received research support from The Ministry of Education, Culture, Sports, Science, and Technology. Dr. Lehesjoki has received research support from the Academy of Finland, the Sigrid Juselius Foundation, the Liv och Hälsa Foundation, and the Folkhälsan Research Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 2006;313:1792–1795. [DOI] [PubMed] [Google Scholar]
- 2.Fukata Y, Lovero KL, Iwanaga T, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A 2010;107:3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duffy M, Mullooly M, O'Donovan N, et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin Proteomics 2011;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou YD, Lee S, Jin Z, Wright M, Smith SEP, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 2009;15:1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lovero KL, Fukata Y, Granger AJ, Fukata M, Nicoll RA. The LGI1-ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc Natl Acad Sci U S A 2015;112:E4129–E4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benarroch EE. ADAM proteins, their ligands, and clinical implications. Neurology 2012;78:914–920. [DOI] [PubMed] [Google Scholar]
- 7.Ogawa Y, Oses-Prieto J, Kim MY, et al. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci 2010;30:1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Özkaynak E, Abello G, Jaegle M, et al. Adam22 is a major neuronal receptor for Lgi4-mediated Schwann cell signaling. J Neurosci 2010;30:3857–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokoi N, Fukata Y, Kase D, et al. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med 2015;21:19–26. [DOI] [PubMed] [Google Scholar]
- 10.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu YE, Wen L, Silva J, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet 2010;19:1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chabrol E, Navarro V, Provenzano G, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain 2010;133:2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagane K, Hayakawa K, Kai J, et al. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci 2005;6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci 2009;42:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koskinen LL, Seppala EH, Belanger JM, et al. Identification of a common risk haplotype for canine idiopathic epilepsy in the ADAM23 gene. BMC Genomics 2015;16:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 2013;14:681–691. [DOI] [PubMed] [Google Scholar]
- 18.EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform 2013;11:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007;17:1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron 2004;44:987–996. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Shim AHR, He X. Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase: insights on ADAM function. J Biol Chem 2009;284:29077–29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada T, Nishie A, Torigoe K, et al. The specific expression of three novel splice variant forms of human metalloprotease-like disintegrin-like cysteine-rich protein 2 gene in brain tissues and gliomas. Jpn J Cancer Res 2000;91:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gödde NJ, D'Abaco GM, Paradiso L, Novak U. Differential coding potential of ADAM22 mRNAs. Gene 2007;403:80–88. [DOI] [PubMed] [Google Scholar]
- 28.Barkovich AJ, Good WV, Koch TK, Berg BO. Mitochondrial disorders: analysis of their clinical and imaging characteristics. AJNR Am J Neuroradiol 1993;14:1119–1137. [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobs DS, Smith AS, Finelli DA, Lanzieri CF, Wiznitzer M. Menkes kinky hair disease: characteristic MR angiographic findings. AJNR Am J Neuroradiol 1993;14:1160–1163. [PMC free article] [PubMed] [Google Scholar]
- 30.Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): a personal review. J Child Neurol 1990;5:273–287. [DOI] [PubMed] [Google Scholar]
- 31.Zhu P, Sang Y, Xu H, et al. ADAM22 plays an important role in cell adhesion and spreading with the assistance of 14-3-3. Biochem Biophys Res Commun 2005;331:938–946. [DOI] [PubMed] [Google Scholar]
- 32.Lonsdale J, Thomas J, Salvatore M, et al. ; GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature 2011;478:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 2013;33:18161–18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai M, Huijbers MGM, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabrol E, Gourfinkel-An I, Scheffer IE, et al. Absence of mutations in the LGI1 receptor ADAM22 gene in autosomal dominant lateral temporal epilepsy. Epilepsy Res 2007;76:41–48. [DOI] [PubMed] [Google Scholar]
- 38.Diani E, Di Bonaventura C, Mecarelli O, et al. Autosomal dominant lateral temporal epilepsy: absence of mutations in ADAM22 and Kv1 channel genes encoding LGI1-associated proteins. Epilepsy Res 2008;80:1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.