

Abstract
Objective:
To delineate molecular and clinical characteristics of 3 families with PRNP P105L mutation, a variant of Gerstmann-Sträussler-Scheinker syndrome whose main motor symptoms were parkinsonism and/or involuntary movements.
Methods:
The causative mutation was first determined in the affected patients of family 1 using whole-exome sequencing, and then mutational analysis was extended to families 2 and 3. The clinical features of the patients of these 3 families were summarized. Haplotype analysis was performed using high-density single nucleotide polymorphism array.
Results:
The whole-exome sequencing revealed that the heterozygous mutation c.314C>T (p.P105L) in PRNP was the only known pathogenic mutation shared by the 3 patients of the family with autosomal dominant parkinsonism. We further identified the same mutation in patients of the other 2 families with autosomal dominant parkinsonism and/or involuntary movements. The clinical features of our patients with PRNP P105L mutation included various motor symptoms such as parkinsonism and involuntary movements in addition to progressive dementia. The clinical features in part overlapped with those of other forms of inherited prion diseases, such as fatal familial insomnia and Huntington disease-like type 1. The patients with PRNP P105L mutation shared a haplotype spanning 7.1 Mb around PRNP, raising the possibility that the mutations in the patients originated from a common founder.
Conclusion:
Most of the patients presented with parkinsonism in addition to progressive dementia. Although spastic paraparesis has been emphasized as the main clinical feature, the clinical spectrum of patients with PRNP P105L is broader than expected.
Inherited prion diseases are caused by mutations of the prion protein gene (PRNP). At least 30 single nucleotide substitutions of PRNP have been reported.1 The clinical features of patients with PRNP mutations are characterized by varying combinations of neurologic symptoms and signs, including weakness, spasticity, ataxia, involuntary movements, autonomic disturbance, dementia, psychiatric symptoms, and insomnia. Various names have been assigned to some of the inherited prion diseases in the literature. The existence of genotype-phenotype correlations has been suggested for these diseases.
Seven families with the missense substitution of leucine for proline at codon 105 of PRNP have been reported since 1993.2–13 They include 6 Japanese families and 1 British family with de novo mutation. The prominent clinical features associated with this mutation are protracted clinical duration and spastic paraparesis in addition to progressive dementia. Substantial clinical variations have also been described,2,5 including ataxia, extrapyramidal signs, and involuntary movements.
Parkinsonism has not attracted much attention as a cardinal motor symptom in prion diseases. Only a few reports have described parkinsonism as the main symptom.2,6,14 We recently found that the PRNP P105L mutation is responsible for atypical parkinsonism with progressive dementia in a Japanese family and have identified 2 additional Japanese families carrying the same mutation. We herein report the clinical and molecular genetic analyses of the 3 families with the PRNP P105L mutation.
METHODS
Participants.
In this study, we conducted clinical and molecular genetic analyses of 9 patients and 2 unaffected members of 3 families. The pedigree charts of the families are presented in figure 1, and the clinical features of all the affected participants are summarized in table 1. The detailed clinical features of the affected individuals of the families are described briefly in appendix e-1 at Neurology.org/ng. Genomic DNA was prepared from peripheral blood leukocytes of the participants in accordance with the standard procedure.
Figure 1. Pedigree charts of 3 families with PRNP P105L.

Affected individuals are indicated by filled symbols. Individuals whose DNA was available are indicated by stars. “P” indicates proband.
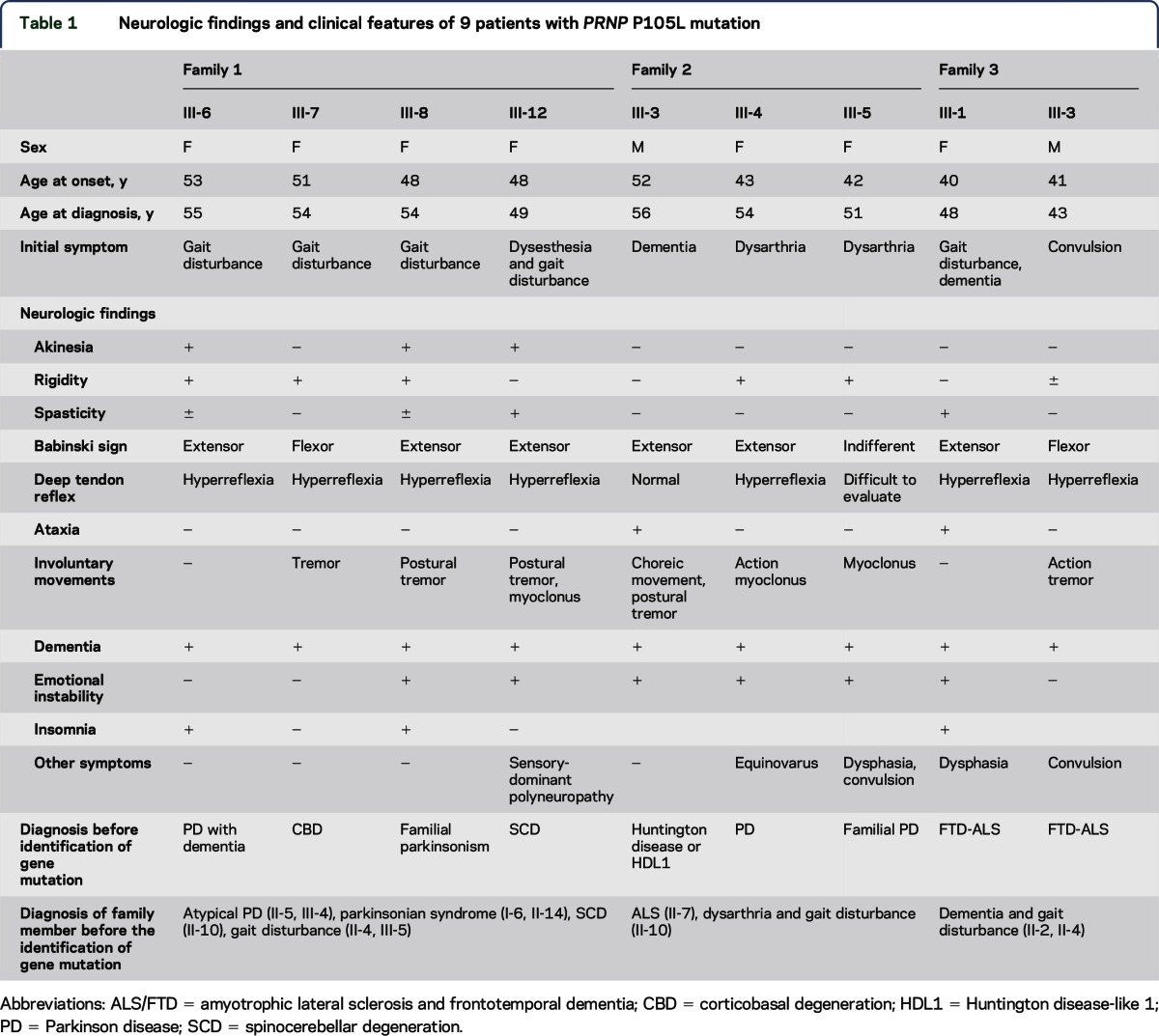
Table 1.
Neurologic findings and clinical features of 9 patients with PRNP P105L mutation
Family 1.
Family history revealed that there were 11 affected members in 3 successive generations (figure 1). Four affected members, III-6, III-7, III-8, and III-12, independently consulted different hospitals with which the authors were affiliated. Two of them, III-6 and III-8, were admitted to The University of Tokyo Hospital. We tentatively diagnosed them as having autosomal dominant familial parkinsonism with dementia.
The clinical features of III-8 are described below, and those of the other affected members are presented in the supplemental data. III-8 was a woman. At the age of 48 years, she started to walk slowly and stumble easily. She consulted with a neurologist at the age of 51, and neurologic examinations revealed lead-pipe rigidity of the extremities, anteflexion of the neck, postural tremor in the upper extremities, mild cognitive impairment, and emotional instability. Several antiparkinson drugs, including levodopa, zonisamide, ropinirole, and entacapone, were prescribed, but no improvement was noted. At the age of 53, her speech became weaker and faster. One year later, she began to report visual hallucinations and insomnia. On neurologic examination at the age of 54, bilateral hyperreflexia and extensor Babinski signs were noted in addition to parkinsonism and dementia. MRI showed mild diffuse atrophy of the cerebral cortex (figure e-1) with no abnormality on diffusion-weighted imaging. EEG showed background activities consisting of a low amount of slow alpha waves at 8–9 Hz intermixed with diffuse theta waves.
Family 2.
Family history revealed that there were 5 affected members in 2 successive generations (figure 1). Three affected members, III-3, III-4, and III-5, independently consulted different hospitals with which the authors were affiliated. III-5 was admitted to The University of Tokyo Hospital.
III-3 was initially suspected of having Huntington disease. III-4 and III-5 were suspected of having atypical Parkinson disease. The clinical features of these 5 affected members are described in detail in the supplementary data. We could obtain CSF data from only 1 patient (III-5). The CSF contained 2 white blood cells/μL and 46 mg/dL protein. 14-3-3 protein level was not elevated. Total tau protein measured by ELISA and PrPSc measured by real-time quaking-induced conversion were negative. Genetic diagnosis excluded the possibilities of Huntington disease, dentatorubral-pallidoluysian atrophy, and spinocerebellar ataxia type 17. We considered it necessary to perform a differential diagnosis of Huntington disease-like diseases.
Family 3.
Family history revealed that there were 4 affected members in 2 successive generations (figure 1). The proband of this family, III-1, consulted The University of Tokyo Hospital, followed by III-3. III-1 and III-3 were admitted to The University of Tokyo Hospital.
III-1 and III-3 were suspected of having amyotrophic lateral sclerosis/frontotemporal dementia. They later presented with parkinsonism. The clinical features of the 4 affected members are described in detail in the supplemental data.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all the participants. The present study was approved by the institutional review board of the University of Tokyo.
Whole-exome sequencing analysis.
Exonic sequences were enriched using SureSelect Human All Exon 50 Mb Kit (Agilent Technology, Santa Clara, CA), followed by massively parallel sequence analysis (2 × 100 bp paired-end reads) using Genome Analyzer IIx and Hiseq 2000 (Illumina, San Diego, CA). The sequences were aligned to the human reference genome (hg19) using Burrows–Wheeler Aligner.15 SAMtools16 was used for extracting single nucleotide variants (SNVs) and small insertions and deletions.
To classify the extracted SNVs with regard to novelty and pathogenicity, we checked our in-house exome database of 373 Japanese controls and the Human Gene Mutation Database (http://www.hgmd.org/).
Mutational analysis.
PCR was conducted using LA taq (Takara Bio, Shiga, Japan) with specific primers for PRNP (table e-1). Direct nucleotide sequence analyses were conducted using an ABI 3100 sequencer and BigDye Terminator v3.1 (Life Technologies, Carlsbad, CA).
Determination of the phase of the c.314C>T (p.P105L) mutation and the c.385A>G (p.M129V) polymorphism of PRNP.
Genomic segments containing the c.314C>T (p.P105L) and c.385C>G (p.M129V) loci of PRNP were amplified by PCR followed by subcloning into the pTA2 vector (Toyobo, Osaka, Japan). The primer sequences are shown in table e-2. Plasmid DNA purified from 8 colonies using a QIAprep Spin Miniprep kit (Qiagen, Venlo, the Netherlands) was subjected to direct nucleotide sequence analysis to determine the phase of the c.314C>T (p.P105L) mutation and the c.385A>G (p.M129V) polymorphism of PRNP.
Haplotype analysis.
Genome-wide high-density single nucleotide polymorphism (SNP) typing was conducted on 9 patients and 2 married-in members (II-8 of family 2 and II-1 of family 3) using the Genome-Wide Human SNP array 6.0 (Affymetrix, Santa Clara, CA) following the manufacturer's instructions. Haplotypes were reconstructed by homozygosity haplotyping17 followed by visual inspection.
RESULTS
Whole-exome sequencing analyses.
Initially, family 1 was tentatively diagnosed as having a form of familial parkinsonism with autosomal dominant inheritance. Whole-exome sequencing analyses revealed 181,547; 182,254; and 220,616 SNVs and 16,126; 16,098; and 18,963 small insertions and deletions in III-6, III-7, and III-8 of family 1, respectively (table e-3). Of these SNVs, 10 nonsynonymous variants, which were shared by the 3 patients and were absent in 373 controls in the in-house exome database, were identified as the candidate causative mutations (table e-4). Through a search of the Human Gene Mutation Database, the heterozygous nonsynonymous substitution mutation c.314C>T (p.P105L) in PRNP was found to be the only known pathogenic mutation. We confirmed by direct nucleotide sequence analyses that all 4 affected patients of family 1 carried the PRNP P105L mutation in a heterozygous state (figure 2A).
Figure 2. Identification of PRNP P105 and haplotype analysis of 3 families.

(A) Identification of the causative mutation in PRNP. The upper panel shows the aligned short reads shown using Integrative Genomics Viewer.20 The lower panel shows the sequence data of direct nucleotide sequence analysis. (B) Haplotypes of 3 families. Haplotypes around a PRNP locus are shown. Single nucleotide polymorphisms (SNPs) in the blue-shaded area are the haplotypes containing the PRNP P105L mutation.
Mutational analysis of PRNP.
Given the PRNP P105L mutation in family 1, we performed mutational analysis of PRNP in 5 patients of 2 additional families (families 2 and 3). All 5 patients had the heterozygous nonsynonymous substitution mutation c.314C>T (p.P105L) in PRNP.
Intriguingly, all 9 patients had the heterozygous M129V (c.385A>G) polymorphism of PRNP. There were no other variants in the coding region of PRNP in the 9 patients.
The phases of c.314C>T (p.P105L) and c.385A>G (p.M129V) in PRNP were determined for all the patients, as described in the Methods section. Both the mutant sequence (c.314T) and the c.385G polymorphism replacing the methionine at codon 129 with a valine were located on the same allele in all the patients (figure e-2).
Haplotype analysis.
We reconstructed the haplotypes containing the PRNP P105L mutation using dense SNP markers and compared the haplotypes among all 3 families. Patients in families 2 and 3 shared the common haplotype spanning 18.3 Mb around PRNP with telomeric and centromeric boundaries defined by rs6106883 and rs2144765, respectively. Families 1, 2, and 3 shared the common haplotype spanning 7.1 Mb around PRNP with telomeric and centromeric boundaries defined by rs6051365 and rs221020, respectively (figure 2B).
DISCUSSION
We determined the PRNP c.314C>T (p.P105L) mutation in 9 patients from3 families who presented with parkinsonism or chorea with progressive dementia with autosomal dominant inheritance. The mean age at onset was 47.2 ± 4.5 (SD) years. On average, from the time of onset, they became wheelchair bound in 5 years and bedridden in 10 years. The patients presented various motor symptoms, dementia that was attributable to a diffuse cerebral cortical dysfunction, emotional instability, and insomnia. Intriguingly, 8 patients showed parkinsonism. The patients tended to show postural tremor rather than resting tremor. Levodopa was ineffective in all patients who were prescribed it. Their disease progression was relatively rapid, and dementia developed in the early stage (table 1). These features are in striking contrast to the clinical features of idiopathic Parkinson disease.
The age at onset and disease duration of the patients were similar to those of the previously reported cases of the PRNP P105L mutation. However, the clinical features of previously reported cases were spastic paraparesis and progressive dementia. Only 3 reports described parkinsonism as the main symptom.2,6,14 Figure 3 shows a summary of neurologic findings of our 9 patients and previously reported cases. The present study revealed that the main symptoms of P105L prion disease were not only spastic paraparesis but also parkinsonism.
Figure 3. Prevalence of neurologic findings in 20 patients with PRNP P105.

Neurologic findings of previously reported patients (10), our reported patients (9), and their family member (1). We summarized only members from whom we were able to obtain detailed neurologic findings. Black bars indicate previously reported patients. Gray bars indicate newly diagnosed patients. Seven patients had tremor, 4 of whom had postural tremor.
This study further broadened the spectrum of the clinical presentations of patients with the PRNP P105L mutation. Because they showed a long disease course and did not show myoclonus or periodic synchronous discharges on EEG, elevated tau and 14-3-3 levels, or high-intensity signal on diffusion-weighted MRI, it took a long time for our patients to be diagnosed with P105L prion disease. Because they presented mainly with parkinsonism, an inherited prion disease with the PRNP P105L mutation should be considered as a differential diagnosis of atypical familial parkinsonism with dementia. The famous Indiana kindred of Gerstmann-Sträussler-Scheinker syndrome with the PRNP F198S mutation presented with progressive ataxia, dementia, and parkinsonian features. Patients in this kindred tend to present with dementia and cerebellar ataxia early in the disease and rigidity and bradykinesia later in the disease.18 Rare variants in the octarepeat region of the PRNP gene have been found in patients with parkinsonism,19 but there were no variants in the octarepeat region of the PRNP gene in our patients. We have not performed the functional brain imaging tests of dopamine transporters in all our patients. Autopsies were performed for some patients with the PRNP P105L mutation.4,6,9,11 PrP plaques and neuronal loss were observed in the basal ganglia in the terminal phase regardless of the presence of extrapyramidal symptoms. These pathologic findings in the basal ganglia might be related to parkinsonian features of the patients with the PRNP P105L mutation.
The 3 families shared a long common haplotype around PRNP, which spanned at least 1.8 Mb, suggesting that they were descendants from a common ancestor. All our patients had the mutation sequence (c.314T, P105L) and the polymorphism (c.385G, p.M129V) on the same haplotype. All the other previously reported cases had the Val 129 polymorphism, although the phase of the P105L mutation and the Val 129 polymorphism of PRNP on the same allele was confirmed in only 1 study.8 The allele frequency of Val 129 in the Japanese population is 0.04 according to the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/). Consequently, the existence of a very strong linkage disequilibrium between the Leu 105 mutation allele and the rare Val 129 polymorphic allele in previously reported cases suggests that all Japanese patients might have a common founder.
There were phenotypic variations of patients in families with the PRNP P105L mutation. The variants of genes other than PRNP, for example, genes related to the degradation and elimination pathways of prion protein, might influence the phenotype of patients with the PRNP P105L mutation as genetic modifying factors. We need further studies to elucidate the phenotypic variations of patients with inherited prion disease.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Ms. Mio Takeyama, Ms. Keiko Hirayama, and Ms. Zhenghong Wu for their support in the laboratory experiments; Dr. Takamichi Hattori and Dr. Nobuyuki Araki for providing clinical information on patients; and Dr. Katsuya Satoh for measuring 14-3-3 protein, total tau protein, and PrPSc of CSF.
GLOSSARY
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Drs. Tsuji, Goto, Mano, Matsukawa, Mitsui, Ishiura, Tokushige, Takahashi, Sato, Nakamoto, Ichikawa, Nagashima, Terao, Shimizu, Hamada, Uesaka, Oyama, and Ogawa: drafting/revising the manuscript for content, including medical writing for content. Drs. Tsuji, Goto, Mano, Matsukawa, Mitsui, Ishiura, and Takahashi: study concept and design. Drs. Tsuji, Goto, Mano, Matsukawa, Mitsui, Ishiura, Yoshimura, Doi, and Morishita: analysis or interpretation of data. Drs. Mano, Matsukawa, Mitsui, Ishiura, Tokushige, Takahashi, Sato, Nakamoto, Ichikawa, Nagashima, Terao, Shimizu, Hamada, Uesaka, Oyama, and Ogawa: acquisition of data. Drs. Tsuji and Goto: study supervision or coordination. Dr. Tsuji: obtaining funding.
STUDY FUNDING
This work was supported in part by KAKENHI (Grant-in-Aid for Scientific Research on Innovative Areas) from the Ministry of Education, Culture, Sports, Science and Technology in Japan, a Grant-in-Aid from the Ministry of Health, Labour and Welfare.
DISCLOSURE
Dr. Mano reports no disclosures. Dr. Matsukawa received funds from a Grant-in-Aid for JSPS Fellows (25·3206) and GlaxoSmithKline. Dr. Mitsui is funded by JSPS KAKENHI Grant Number 25860700 and 15K09334 and has received research support from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Dr. Ishiura received a Grant-in-Aid for Young Scientist (Startup, 24890044); has received research support from The Cell Science Research Foundation, the Ministry of Health Labour and Welfare, the Japan Agency for Medical Research and Development, the Cell Science Research Foundation, and the Ministry of Education, Culture, Sports, Science and Technology of Japan; and has received a speaker honorarium from Sanofi and Sumimoto Dainoppon Pharma. Dr. Tokushige reports no disclosures. Dr. Takahashi received funds from Takeda Science Foundation, The Ichiro Kanehara Foundation for the Promotion of Medical Science & Medical Care, ALS Foundation, Japan ALS Association, and a Grant-in-Aid for Scientific Research (C, 25471270). Dr. Sato and Dr. Nakamoto report no disclosures. Dr. Ichikawa is funded by JSPS KAKENHI (Grant Number 23591233) and has received a speaker honorarium from Japan Blood Products Organization. Dr. Nagashima received funds from a Grant-in-Aid for JSPS Fellows (PD, 25-6016), a Grant-in-Aid for Young Scientists (B, 14441789), and a Grant-in-Aid (H26 Ninchisho Kenkyu Kaihatsu Jigyo 26340401) from the Ministry of Health, Labour and Welfare, Japan. Dr. Terao was supported by a Research Project Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant Number 22590594) and has received research support from the Japan Society for the Promotion of Science. Dr. Shimizu is funded by Grant-in-Aid for Scientific Research (C, 24591289 and 15K09347) and has received research support from the Ministry of Health, Labour and Welfare of Japan. Dr. Hamada is funded by JSPS KAKENHI (Grant Number 15H01658, 15K19476), Magnetic Health Science Foundation, and the Japan Foundation for Neuroscience and Mental Health; has served as a medical advisor for Pfizer Japan Inc.; has served on the editorial board of Brain Stimulation; and has received research support from the Japan Society for the Promotion of Science Postdoctoral Fellowships for Research Abroad. Dr. Uesaka is funded by Ministry of Health, Labour and Welfare of Japan as a member of the research for “wake-up stroke” and “subacute myelo-optico-neuropathy” and has received lecturer's fees from Bayer Yakuhin, Ltd., Pfizer Seiyaku K.K., Boehringer Ingelheim Japan, Inc., Otsuka Pharmaceutical Co. Ltd., Sanofi K.K., Bristol-Myers Squibb Co., Mitsubishi Tanabe Pharma Co., and Glaxo SmithKline K.K. Dr. Oyama is funded by KAKEN grants # 25860724 and # 24591304 and the Uehara Memorial Foundation and has received speaker honoraria from Glaxo Smith Kline, Medtronic, Novartis Pharma, MSD, Nihon Medi Phisics, and Otsuka Pharmaceutical. Dr. Ogawa has served on the scientific advisory boards of MediHealth and BioTechnic Corp.; has received a gift (laptop) from Advent Pharmaceuticals; has received travel funding from Jenson Pharmaceuticals and GeneBio Corp.; has served on the editorial Boards of Genetic Study and Immunologic Response; holds a patent for Subthalamic Transcranial Magnetic device; receives publishing royalties for Gene Therapy (Hochstein & Thorpe Ltd, 2007) and Microbiology Fourth Edition (Dayton Books, 2013); has been an employee of Jensen Labs; has been a consultant for Birex Labs and the University of New Dalton; has served on the speakers' bureau for Birex Labs; has had involvement with Myasthenia Gravis Section of Georgian Hospital, MRI, 45%; has received research support from GeneBio Corp, NIH/National Institute of Neurological Disorders and Stroke, Parkinson Association, and Tardive Dyskinesia Foundation; holds stock/stock options/Board of Directors compensation with GeneBio Corp. and Birex Labs; receives license fee payments from OptiVisor and Birex Labs; and gave expert testimony in a case involving GeneBio Corp. Dr. Yoshimura and Dr. Doi report no disclosures. Dr. Morishita has served on the editorial board of the Journal of BMC Genomics and the Journal of Bioinformatics and Computational Biology and is funded by Grant-in-Aid for Scientific Research on Innovative Areas (22129008, 221S0002) (in part) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT). Dr. Tsuji is funded by KAKENHI [Grant-in-Aid for Scientific Research on Innovative Areas (22129001 and 22129002)] from the Ministry of Education, Culture, Sports, Science and Technology in Japan, and a Grant-in-Aid [H26-Itaku(Nan)-Ippan-006] from the Ministry of Health, Labour and Welfare; has received speaker honoraria from Kowa Pharmaceutical Co., Ltd., Japan Blood Products Organization, Mitsubishi Tanabe Pharma Co., Bayer Japan, Janssen Pharmaceutical K.K., Genzyme Japan K.K., Eisai Co., Ltd., FP-Pharmaceutical Co., Dainippon Sumitomo Pharma Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Actelion Pharmaceuticals Japan Ltd., Sony Corporation, Nikkei Business Publications Inc., and Leave a Nest Co., Ltd.; and has received research support from Sanofi K.K., Japan Blood Products Organization, Mitsubishi Tanabe Pharma Co., Pfizer Japan Inc., Ono Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Kowa Pharmaceutical, Co., Ltd., GlaxoSmithKline K.K., and the Japan Agency for Medical Research and Development. Dr. Goto is funded by JSPS KAKENHI (Grant Number 26461265) and has been a consultant for Mochida Pharmaceutical Co., Ltd. Go to Neurology.org/ng for full disclosures.
REFERENCES
- 1.Jeong BH, Kim YS. Genetic studies in human prion diseases. J Korean Med Sci 2014;29:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwasaki Y, Kizawa M, Hori N, Kitamoto T, Sobue G. A case of Gerstmann-Sträussler-Scheinker syndrome with the P105L prion protein gene mutation presenting with ataxia and extrapyramidal signs without spastic paraparesis. Clin Neurol Neurosurg 2009;111:606–609. [DOI] [PubMed] [Google Scholar]
- 3.Yamada M, Itoh Y, Fujigasaki H, et al. A missense mutation at codon 105 with codon 129 polymorphism of the prion protein gene in a new variant of Gerstmann-Sträussler-Scheinker disease. Neurology 1993;43:2723–2724. [DOI] [PubMed] [Google Scholar]
- 4.Kitamoto T, Amano N, Terao Y, et al. A new inherited prion disease (PrP-P105L mutation) showing spastic paraparesis. Ann Neurol 1993;34:808–813. [DOI] [PubMed] [Google Scholar]
- 5.Shiraishi A, Mizusawa H, Yamada M. Early and persistent sensory-psychiatric symptoms in an inherited prion disease with a PrP P105L mutation. J Neurol 2002;249:1740–1741. [DOI] [PubMed] [Google Scholar]
- 6.Yamazaki M, Oyanagi K, Mori O, et al. Variant Gerstmann-Sträussler syndrome with the P105L prion gene mutation: an unusual case with nigral degeneration and widespread neurofibrillary tangles. Acta Neuropathol 1999;98:506–511. [DOI] [PubMed] [Google Scholar]
- 7.Itoh Y, Yamada M, Hayakawa M, et al. A variant of Gerstmann-Sträussler-Scheinker disease carrying codon 105 mutation with codon 129 polymorphism of the prion protein gene: a clinicopathological study. J Neurol Sci 1994;127:77–86. [DOI] [PubMed] [Google Scholar]
- 8.Yamada M, Itoh Y, Inaba A, et al. An inherited prion disease with a PrP P105L mutation: clinicopathologic and PrP heterogeneity. Neurology 1999;53:181–188. [DOI] [PubMed] [Google Scholar]
- 9.Amano N, Yagishita S, Yokoi S, et al. Gerstmann-Sträussler syndrome—a variant type: amyloid plaques and Alzheimer's neurofibrillary tangles in cerebral cortex. Acta Neuropathol 1992;84:15–23. [DOI] [PubMed] [Google Scholar]
- 10.Kubo M, Nishimura T, Shikata E, Kokubun Y, Takasu T. A case of variant Gerstmann-Sträussler-Scheinker disease with the mutation of codon P105L [in Japanese]. Rinsho Shinkeigaku 1995;35:873–877. [PubMed] [Google Scholar]
- 11.Nakazato Y, Ohno R, Negishi T, Hamaguchi K, Arai E. An autopsy case of Gerstmann-Sträussler-Scheinker's disease with spastic paraplegia as its principal feature [in Japanese]. Rinsho Shinkeigaku 1991;31:987–992. [PubMed] [Google Scholar]
- 12.Terao Y, Hitoshi S, Shimizu J, Sakuta M, Kitamoto T. Gerstmann-Sträussler-Scheinker disease with heterozygous codon change at prion protein codon 129 [in Japanese]. Rinsho Shinkeigaku 1992;32:880–883. [PubMed] [Google Scholar]
- 13.Beck JA, Poulter M, Campbell TA, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 2010;31:E1551–E1563. [DOI] [PubMed] [Google Scholar]
- 14.Tunnell E, Wollman R, Mallik S, Cortes CJ, DeArmond SJ, Mastrianni JA. A novel PRNP-P105S mutation associated with atypical prion disease and a rare PrPSc conformation. Neurology 2008;71:1431–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyazawa H, Kato M, Awata T, et al. Homozygosity haplotype allows a genomewide search for the autosomal segments shared among patients. Am J Hum Genet 2007;80:1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farlow MR, Yee RD, Dlouhy SR, Conneally PM, Azzarelli B, Ghetti B. Gerstmann-Sträussler-Scheinker disease. I. Extending the clinical spectrum. Neurology 1989;39:1446–1452. [DOI] [PubMed] [Google Scholar]
- 19.Wang V, Chuang TC, Soong BW, Shan DE, Kao MC. Octarepeat changes of prion protein in Parkinson's disease. Parkinsonism Relat Disord 2009;15:53–58. [DOI] [PubMed] [Google Scholar]
- 20.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.