

Cerebral folate deficiency is a genetically heterogeneous condition.1 Mutations in FOLR1 are responsible for a rare but treatable form of cerebral folate deficiency (OMIM #613068).1 The gene codes for folate receptor alpha (FRα), a specific CNS folate transporter. Individuals with FOLR1-related folate deficiency present with ataxia, dyskinesia, spasticity, seizures, and regression in cognitive abilities and motor skills during early childhood.2 Seizures commonly observed include generalized tonic-clonic, atonic, and myoclonic.3 To date, there have been 18 individuals with FOLR1-related cerebral folate deficiency diagnosed in childhood and reported in the literature.3–5 Early diagnosis is crucial, as high-dose folinic acid (2–5 mg/kg/day) has been reported to be an effective treatment that can ameliorate or even prevent further neurodegeneration, although no long-term treatment studies have been performed.1,3,5,6 We present the late diagnosis of adult siblings with cerebral folate deficiency due to FOLR1 mutations and their subsequent treatment.
The eldest sibling (sibling 1), aged 33 years, met her early developmental milestones until 22 months of age, when she developed myoclonic seizures, ataxia, and developmental regression. At 15 years of age, she had learned 75–100 words, but this decreased to only 12 words by age 30 years. By 7 years of age, she was wheelchair-dependent. There was a period of remission of her seizures from 11 to 18 years of age, but the generalized tonic-clonic seizures recurred in her 20s, particularly during sleep. As an adult, she had an average of 1 seizure per day and was treated with gabapentin.
Her younger sister (sibling 2), aged 28 years, is more severely affected. She also had a period of normal development until 18 months of age, when she developed progressive ataxia and developmental regression. This was followed by myoclonic seizures that evolved to generalized tonic-clonic seizures. She had feeding difficulties and required gastrostomy tube placement. She was also wheelchair-dependent by 5 years of age. She developed only a few words of speech, although she has some receptive language. Her condition stabilized in her teens, but she made no developmental progress. During her 20s, her motor function gradually declined and she had an average of 1 seizure daily, particularly during sleep, and she is treated with carbamazepine and levetiracetam. An MRI of sibling 2 (at age 25 years) showed extensive periventricular and deep cerebral white matter changes with frontal lobe and cerebellar atrophy (figure).
Figure. MRI of sibling 2.
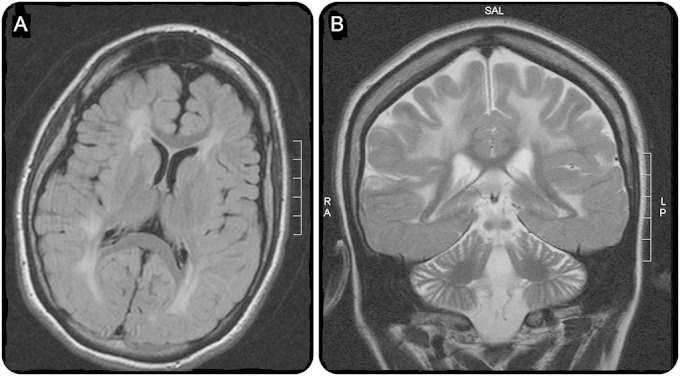
(A) Axial fluid-attenuated inversion recovery image showing extensive white matter disease and sparing of U-fibers. (B) Atrophy of frontal hemispheres and cerebellum with sparing of temporal lobes.
There was no family history of intellectual disability or epilepsy. There are 3 unaffected sisters, and the parents are first cousins of Italian ethnic origin.
The siblings were recruited into the national Care4Rare research project to identify the genetic etiology of their disease using whole-exome sequencing, and informed consent was obtained from the family. A novel homozygous mutation in FOLR1, NM_016724, c.128A>G. p.H43R was identified in both siblings. The parents were confirmed to be heterozygous carriers, and neither of the unaffected sisters was homozygous for the variant. The variant was not present in control databases, and in silico prediction programs (SIFT, PolyPhen-2) predicted it to be pathogenic. CSF studies in sibling 2 demonstrated diminished 5-methyltetrahydrofolate at <10 nmol/L (reference: 40–120 nmol/L) and reduced homovanillic acid at 26 nmol/L (reference: 145–324 nmol/L), consistent with cerebral folate deficiency. Oral folinic acid replacement was promptly initiated at 2 mg/kg/day. This resulted in a reduction in seizure frequency by approximately half. In sibling 2, seizures reduced from an average of 42 (range: 18–73) per month to 20 (range: 12–26) per month (t testone-sided, p = 0.000006), and in sibling 1 seizures reduced from 20 (range: 7–37) per month to 13 (range: 2–36) per month (t testone-sided, p = 0.012). Sibling 1 showed an increase in her vocalizations and use of words and improved fine motor skills, and sibling 2 appeared more alert. The dose of antiepileptic medication has been reduced for both sisters. The late molecular diagnosis of a treatable cause of seizures and intellectual disability in these adult siblings demonstrates the potential impact of next-generation sequencing for early diagnosis to inform disease-altering therapy. Although initiated later in life than has previously been reported, treatment with folinic acid showed a marked reduction in the frequency of seizures for both siblings, improving their quality of life and permitting a reduction in doses of antiepileptic medication. The molecular diagnosis also enabled accurate recurrence risk counseling for family members seeking information for family planning. This case highlights the value of a molecular diagnosis for adults with epilepsy and the finding that even late initiation of treatment for FOLR1 deficiency provides important benefits.7
Supplementary Material
Acknowledgments
Acknowledgment: The authors thank the family for their generosity in participating in this work.
Footnotes
Supplemental data at Neurology.org/ng
Author contributions: Dr. Patrick Ferreira contributed to drafting/revising the manuscript for content, study design, and interpretation of data as well as acquisition of data. Stephanie M. Luco was involved in drafting/revising the manuscript for content, study design, and interpretation as well as statistical analysis. Dr. Sarah L. Sawyer contributed drafting/revising the manuscript for content as well as acquisition of data. Dr. Jorge Davila contributed to analysis of data and drafting/revising the manuscript for content. Dr. Kym M. Boycott contributed to drafting/revising the manuscript for content, study design, and analysis of data. Dr. David A. Dyment contributed to drafting/revising the manuscript for content, study design, and analysis of data.
Study funding: This study was performed as part of the Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec, and Children's Hospital of Eastern Ontario Foundation. D.A.D. is the recipient of a CIHR clinical investigators award.
Disclosure: Dr. Patrick Ferreira, Stephanie M. Luco, Dr. Sarah L. Sawyer, Dr. Jorge Davila, Dr. Kym M. Boycott, and Dr. David A. Dyment report no disclosures relevant to the manuscript. Go to Neurology.org/ng for full disclosure forms. The Article Processing Charge was paid by the authors.
Contributor Information
Collaborators: Care4Rare Canada Consortium, Kym Boycott, Alex MacKenzie, Jacek Majewski, Michael Brudno, Dennis Bulman, and David Dyment
References
- 1.Steinfeld R, Grapp M, Kraetzner R, et al. Folate receptor alpha defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am J Hum Genet 2009;85:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramaekers VT, Häusler M, Opladen T, Heimann G, Blau N. Psychomotor retardation, spastic paraplegia, cerebellar ataxia and dyskinesia associated with low 5-methyltetrahydrofolate in cerebrospinal fluid: a novel neurometabolic condition responding to folinic acid substitution. Neuropediatrics 2002;33:301–308. [DOI] [PubMed] [Google Scholar]
- 3.Grapp M, Just IA, Linnankivi T, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain 2012;135:2022–2031. [DOI] [PubMed] [Google Scholar]
- 4.Ohba C, Osaka H, Iai M, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics 2013;14:225–232. [DOI] [PubMed] [Google Scholar]
- 5.Al-Baradie RS, Chaudhary MW. Diagnosis and management of cerebral folate deficiency. A form of folinic acid-responsive seizures. Neurosciences (Riyadh) 2014;19:312316. [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen FJ, Blau N. Cerebral folate deficiency: life-changing supplementation with folinic acid. Mol Genet Metab 2005;84:371–373. [DOI] [PubMed] [Google Scholar]
- 7.van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab 2014;111:428–438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.