

Ataxia with oculomotor apraxia type 4 (AOA4) is an autosomal recessive (AR) disorder recently delineated in a Portuguese cohort and caused by mutations in the PNKP (polynucleotide kinase 3′-phosphatase) gene.1 AOA4 is a progressive, complex movement disorder that includes hyperkinetic features, eye movement abnormalities, polyneuropathy, varying degrees of cognitive impairment, and obesity. PNKP mutations were initially discovered to be the cause of the severe nonprogressive syndrome microcephaly, early-onset intractable seizures, and developmental delay (MCSZ).2 Here we describe a patient with compound heterozygous PNKP mutations presenting with an AOA4 phenotype. New features that we report include both mutations, presence of chorea, absence of oculomotor apraxia (OMA), and slow disease progression.
Case description.
The patient, a 28-year-old Swedish woman, was affected by insidious dystonia and chorea since the age of 5 (video at Neurology.org/ng). These hyperkinesias receded spontaneously over time. Learning disabilities were noticed during primary school and an assessment at age 12 concluded that the patient met the criteria for pervasive developmental disorder, which motivated attendance at a special school. From this point, progressive ataxia was documented. At age 15, progressive weakness and areflexia were found. Electroneurography 2 years later revealed widespread sensorimotor polyneuropathy, which was more severe in her legs. This predominant feature led to finger contractures, distal muscle wasting, and bilateral foot drop necessitating the use of foot orthosis. Gait has become severely impaired over time; the patient has depended on a wheelchair for mobility since the age of 25 and is able to walk only with support. A recent examination yielded a Scale for the Assessment and Rating of Ataxia score of 22.5 and an Inventory of Non-Ataxia Symptoms score of 5. Leg weakness and complete loss of proprioception and vibration in the feet are evident upon examination. Eye examination revealed broken smooth pursuit and nystagmus but absence of OMA; video-oculography found increased saccade latency. The patient went through a gastric binding procedure 4 years ago because of obesity. Her body mass index then was 39.4 and decreased to 30.1 postsurgery. Hypoalbuminemia and hypercholesterolemia were present before this procedure; 2 years ago a mild elevation of α-fetoprotein (AFP) was found but her immunoglobulin levels were normal (table e-1). Treatment with simvastatin was started and the patient was recommended a protein-enriched diet. Brain MRIs at ages 17 and 28 displayed progressive cerebellar atrophy (figure 1). Polyglutamine spinocerebellar ataxias and Friedreich ataxia were ruled out first. Colony-stimulating assay, Western blot of the ataxia-telangiectasia mutated (ATM) protein, sequencing, and multiplex ligation-dependent probe amplification analysis of the APTX and SETX genes were normal, ruling out ataxia telangiectasia (AT), AOA1, and AOA2. Whole-exome sequencing of the patient and her parents revealed 2 new compound heterozygous variants, c.1196T>C (p.Leu399Pro) and c.1385G>C (p.Arg462Pro), in the kinase domain of the PNKP gene (supplemental data, figure e-1). Both variants were confirmed by Sanger sequencing and predicted to be pathogenic by an in silico analysis. Reduced cellular levels of PNKP protein were found in Epstein-Barr virus–transformed lymphocytes derived from the patient (figure e-2).
Figure 1. Midsagittal T1-weighted brain MRI from a 28-year-old woman affected by ataxia with oculomotor apraxia type 4.
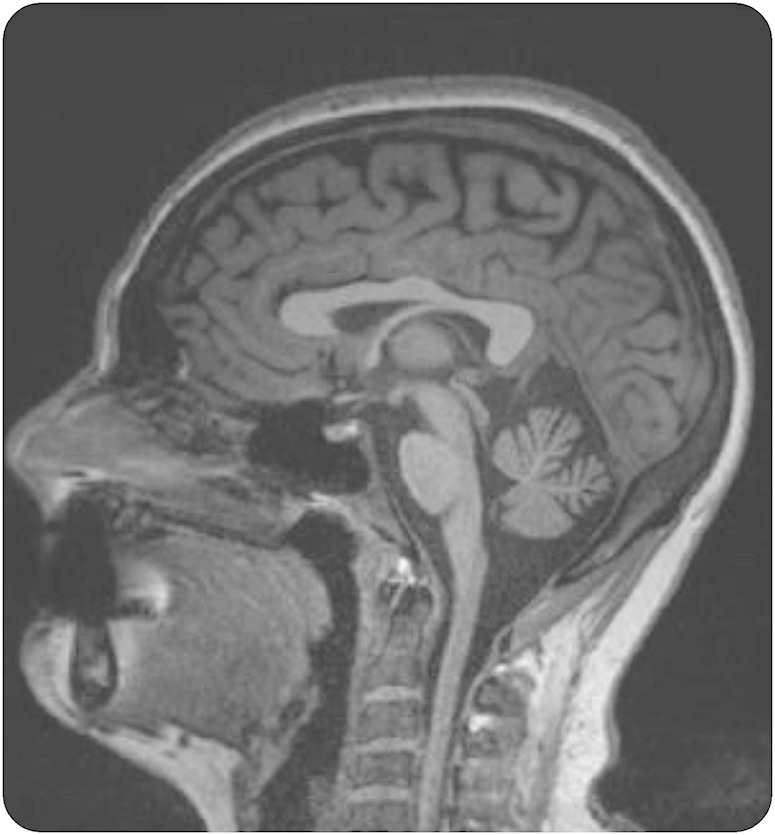
This section displays moderate cerebellar atrophy but no evidence of brainstem atrophy.
Discussion.
Phenotype heterogeneity in AOA4 is similar to that in other diseases in the AOA disease spectrum.3 All the patients described in the Portuguese cohort displayed ataxia, predominant polyneuropathy, OMA, and cerebellar atrophy.1 Some also had brainstem atrophy. However, there are some unique features in AOA4. For instance, dystonia that receded completely over time was the first symptom in the Portuguese patients and in our patient. Extrapyramidal hyperkinesias are usually persistent in the AOA disease spectrum. In our patient, the presence of chorea and the absence of OMA are described for the first time in AOA4. Also of note in our patient is the slower disease progression rate: time to wheelchair was 20 years compared to the average of 13 years in the Portuguese cohort.1 Some patients with AOA4 had laboratory abnormalities found in both AOA1 and AOA2, such as hypercholesterolemia, hypoalbuminemia, and elevated AFP levels. What also makes AOA4 different from other AOA diseases is the striking presence of obesity in some, as also seen in our patient (tables e-2 and e-3). AOA4 is the second most common AR ataxia disorder in Portugal; its prevalence in other countries is still unknown.1 Like other genes in the AOA disease spectrum, the PNKP gene encodes a DNA-repairing enzyme.2,4,5 Despite this and in contrast to AT, there is no evidence of increased risk for malignancies or immunodeficiency in those with PNKP mutations. In AT there is a correlation between ATM activity and phenotype severity.6 Whether this is also the case in PNKP mutations remains to be studied. Similar to MCSZ, we found reduced levels of PNKP protein in AOA4.2 All mutations associated with AOA4 are located in the kinase domain of PNKP. Only 2 cases with the same homozygous mutation (c.1250_1266dup) in this region have been described: in 1 case it was associated with MCSZ and in a second it was associated with combined features of MCSZ and AOA4.2,7 The remaining mutations associated with MCSZ are located in the forkhead and phosphatase domains.
Supplementary Material
Acknowledgments
Acknowledgment: The authors are grateful to the patient for consenting to this report and to her father for providing valuable early video recordings. The authors also thank Prof. Keith Caldecott, University of Sussex, for providing cells from the patient with MCSZ.
Footnotes
Supplemental data at Neurology.org/ng
Author contributions: Dr. M. Paucar, Dr. H. Malmgren, Prof. M. Taylor, and Dr. R. Press: study concept, data collection, and writing of the manuscript. Prof. M. Taylor: performing the CSA and Western blots for ATM, aprataxin, and senataxin. Dr. J. Reynolds: performing the Western blots for PNKP and editing the revised manuscript. Prof. P. Svenningsson, Prof. M. Taylor, and Prof. A Nordgren: study concept and editing of the manuscript.
Study funding: Stockholm County Council.
Disclosure: Dr. Paucar has received research support from the Stockholm County Council. Dr. Malmgren reports no disclosures. Dr. Taylor has served on the scientific advisory board of Ataxia Telangiectasia Society of UK and has received research support from Cancer Research UK. Dr. Reynolds has received research support from the Medical Research Council. Dr. Svenningsson has served on the scientific advisory board of CBD Solutions, has served on the editorial board of PLoS ONE, and has received research support from the Swedish Research Council. Dr. Press and Dr. Nordgren report no disclosures. Go to Neurology.org/ng for full disclosure forms. The Article Processing Charge was paid by the authors.
References
- 1.Bras J, Alonso I, Barbot C, et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet 2015;96:474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen J, Gilmore EC, Marshall CA, et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 2010;42:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med 2012;366:636–646. [DOI] [PubMed] [Google Scholar]
- 4.Jilani A, Ramotar D, Slack C, et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3'-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem 1999;274:24176–24186. [DOI] [PubMed] [Google Scholar]
- 5.Karimi-Busheri F, Daly G, Robins P, et al. Molecular characterization of a human DNA kinase. J Biol Chem 1999;274:24187–24194. [DOI] [PubMed] [Google Scholar]
- 6.Verhagen MM, Last JI, Hogervorst FB, et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: a genotype-phenotype study. Hum Mutat 2012;33:561–571. [DOI] [PubMed] [Google Scholar]
- 7.Poulton C, Oegema R, Heijsman D, et al. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics 2013;14:43–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.