

Ligands for NKG2D, the NK cell–activating receptor, have been explored as targets for chimeric antigen receptor T-cell (CART) therapy of cancer in preclinical mouse models for over a decade, and a phase I clinical trial is currently recruiting patients to test this platform. In this issue of Molecular Therapy, VanSeggelen and colleagues report on the observation of lethal toxicity in mice following administration of chimeric NKG2D (chNKG2D) CART therapy for targeting NKG2D ligands.1 CART-mediated toxicities were mouse strain-dependent, highlighting the influence of intraspecies differences that are often overlooked when using genetically inbred mice for preclinical studies. These findings warrant careful consideration as chNKG2D CART therapeutic regimens move into the clinic and raise broader questions regarding the variability in, and prognostic value of, syngeneic mouse models for preclinical validation of CART therapy.
CART therapy can redirect a patient's own T cells against tumor cells by linking an extracellular tumor antigen recognition domain (usually a scFv from a monoclonal antibody) to intracellular T-cell activation–signaling elements.2 CART therapy has shown significant clinical success in treating patients with CD19+ leukemia and lymphoma with impressive response rates, up to 90% in patients with B-cell acute lymphoblastic leukemia.3,4,5 In some patients, side effects can be quite severe and generally result from a cytokine storm or tumor lysis syndrome associated with activation of the transferred CAR T cells. Through careful monitoring and therapeutic intervention, these toxicities are generally transient and manageable. Another common side effect in CD19 CART–treated patients is long-term B-cell aplasia, resulting from on-target, off-tumor recognition of normal CD19+ B cells. Although serum immunoglobulin replacement therapy can offset this toxicity, off-tumor recognition of antigens on vital organs remains a major challenge in translating CAR T-cell therapy to other cancer types.
The numerous ligands for NKG2D (NKG2DLs) include MICA, MICB, ULBP1–4 in humans and Rae1α–ε, H60a–c, and MULT-1 in mice. NKG2DLs are expressed on many types of tumor cells6 but are also upregulated on healthy cells in response to stress stimuli, such as pathogen infection. Because expression in tissues is thought to be low under normal conditions, targeted therapy for NKG2DL+ cancers is an area of great interest in cancer research. The NKG2D receptor is naturally expressed on natural killer cells and CD8+ T cells, facilitating interactions between these immune subsets and cancer cells, but receptor activation requires coexpression of adapter molecules DAP10 in humans and DAP10 or DAP12 in mice.7
In 2005, Sentman and colleagues developed the first engineered T-cell platform targeting NKG2DLs by fusing full-length NKG2D with the CD3ζ intracellular T-cell activation signaling domain (chNKG2D), and established the feasibility of targeting NKG2DLs using both mouse and human engineered chNKG2D T cells.8,9 A large body of work targeting various NKG2DL+ tumors in C57BL/6 mice, including several studies using the ID8 ovarian carcinoma model, has since been reported. This work established the mechanistic requirements for chNKG2D CAR T-cell efficacy, including IFNγ signaling, perforin/granzyme-B activity, activation of host adaptive immunity, and targeting of NKG2DLs on other cells in the tumor microenvironment including suppressive CD4+Foxp3+ Tregs, myeloid-derived suppressor cells, and tumor vasculature.10,11,12 Over the many years of investigation in the C57BL/6 model, no severe adverse events have been reported.
It is well established that incorporation of costimulatory domains in CAR constructs augments CAR T-cell cytokine production, proliferation, and persistence in preclinical models and in patients. Essentially all CAR platforms in clinical translation now utilize at least one costimulatory signaling domain in addition to CD3ζ. Lehner et al.13 and Song et al.14 have explored the addition of costimulatory domains CD28 and 41BB, respectively, to chNKG2D platforms in human T cells; however, the impact of costimulation in mouse models has not been evaluated.
As DAP10 is necessary for NKG2D expression and activation, VanSeggelen and colleagues reasoned that inclusion of exogenous DAP10 (NKz10) may increase chNKG2D CAR (NKz) expression and/or functional capacity. A traditional second-generation CAR was also constructed utilizing just the extracellular domain of NKG2D coupled to a CD8α hinge, CD28 transmembrane, and intracellular signaling domain before CD3ζ (NK28z). In this configuration, the NKG2D extracellular domain is essentially utilized only to impart antigen specificity (similar to the platforms in human T cells13,14) and is not expected to signal through the downstream NKG2D/DAP10 axis. These CAR formats were then evaluated for functional activity in C57BL/6, as well as BALB/c T cells.
Inclusion of DAP10 (NKz10) greatly enhanced NKz CAR expression in both BALB/c and C57BL/6 CD8+ and CD4+ T cells, whereas NK28z CAR T cells displayed intermediate expression. BALB/c T cells showed a general trend for higher CAR expression as well as antigen-induced cytokine production compared to C57BL/6. In addition, BALB/c T cells clearly outperformed C57BL/6 in cytolytic assays with NKG2DL+ target cells.
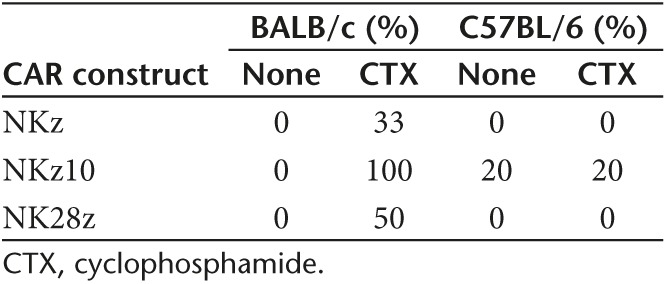
Transfer of CAR T cells resulted in acute signs of toxicity. The degree of toxicity was influenced by mouse strain (BALB/c > C57BL/6), lymphodepleting preconditioning with cyclophosphamide (CTX) (CTX > none), and CAR construct (NKz10 > NK28z > NKz), as summarized in Table 1. Whereas CTX greatly exacerbated toxicity in BALB/c mice, temperature changes and weight loss were only slightly increased in C57BL/6 mice treated with NKz and NKz10 cells. Nearly all C57BL/6 mice recovered, and no differences in survival were observed compared with mice without CTX preconditioning.
Table 1. Morbidity in mice treated with chNKG2D CAR T cells: the effects of strain, preconditioning, and construct.
The acute toxicity in BALB/c mice was mirrored by an acute cytokine storm 8 hours after T-cell transfer. CTX increased toxicity associated with NKz, but there were essentially no differences in cytokine levels between nonconditioned and CTX-pretreated animals. Conversely, with NK28z, many cytokine levels were significantly higher following CTX, suggesting preconditioning is particularly effective at increasing the activity of CD28–costimulated chNKG2D T cells.
Lung immunopathology was observed in CTX-treated NKz10-treated BALB/c mice. To address whether the observed lung pathology was a result of on-target off-tumor CAR T-cell activation, the authors measured mRNA levels of four known NKG2DLs before and after CTX. Rae1 transcript was moderately upregulated in BALB/c lung 24 and 32 hours after CTX treatment, suggesting a mechanistic link between upregulated target ligand expression and increased toxicity. It is possible that the absence of severe toxicity in CTX-treated C57BL/6 mice reflects an inherent strain difference in NKG2DL response to CTX, but NKG2DL expression data were not provided for C57BL/6 mice. Since immunopathology was only assessed in CTX-treated NKz10 T cell–treated BALB/c mice, it also remains unknown whether the degree of lung pathology would have corresponded to the differences in survival observed using different constructs or strains of mice. The authors have made an interesting observation, but there are still many questions about the mechanism driving these findings.
It is currently unclear whether the severe differences in toxicity observed using C57BL/6 and BALB/c T cells rely more on T-cell functional capacity than on variance in NKG2DL expression. CAR expression and in vitro functionality was certainly higher for BALB/c T cells at baseline. Interestingly, another group modeling CD19 CAR T cells in fully syngeneic mouse models similarly observed toxicity associated with second-generation CD28-containing constructs, compared to first-generation constructs.15 As in the current study, acute toxicities were only observed in BALB/c mice (not C57BL/6 or C3H), with CTX or total-body irradiation preconditioning, and at doses of >5 × 106 CAR T cells. Other groups utilizing CD19 CAR T cells in C57BL/6 also did not observe autotoxicity,16 consistent with the chNKG2D experiments in C57BL/6 vs. BALB/c mice. The observation that CD19-directed BALB/c T cells were also more toxic than C57BL/6 in an independent study suggests that strain differences in T-cell function probably play a role. However, variability in NKG2DL expression could also have a large impact on toxicity. In particular, C57BL/6 mice do not express H60a and have lower levels of Rae1, so differing levels of target antigen in normal tissues like the lung are also likely a contributing factor.
The current findings by VanSeggelen et al. are largely compatible with previous experiments using chNKG2D CAR T cells. The Sentman group only used first-generation chNKG2D (analogous to NKz) in C57BL/6 mice without CTX preconditioning. Under these conditions, VanSeggelen and colleagues observed moderate, recoverable weight loss and survival in all treated mice. In addition, the newer study utilized T cells at day 4 post activation, whereas protocols in the previous work waited until day 7–8. Although overall doses were similar (5 × 106–10 × 106 T cells per dose), as a result of the large degree of expansion between days 4 and 8 in vitro, the newer study likely provided a much higher effective dose, which may account for the transient symptoms observed here.
Previous studies have implicated CD4+-engineered T cells as mediators of toxicity in vivo.15,17 Therefore, the applied CD4+/CD8+ CAR T-cell ratio becomes an important issue. The Sentman group generally utilized >90% CD8+ T cells (or purified CD8+ T cells), and in their hands, chNKG2D expression was very low on CD4+ owing to the lack of endogenous DAP10 expression. Interestingly, the current study demonstrated moderate NKz CAR expression in CD4+ and CD8+ T cells. Although they do not report the ratio of CD4+ to CD8+ in the infused T cells, one might imagine that a greater frequency of CAR+CD4+ T cells, particularly likely for NKz10 and NK28z constructs, which would not rely on endogenous DAP10 expression, could account in part for the increased toxicity observed in the current study.
The extensive preclinical literature in C57BL/6 mice has led to the initiation of a phase I clinical trial to test the safety of chNKG2D CAR T cells in patients with myeloid malignancies (ClinicalTrials.gov Identifier: NCT02203825). The planned trial will evaluate the first-generation CAR (chNKG2D or NKz without costimulation), without lymphodepleting preconditioning, and starting at very low doses (1 × 106 T cells). Under analogous conditions, no major toxicities were observed in C57BL/6 or BALB/c mice. However, the findings by VanSeggelen et al. merit extra caution when implementing NKG2DL-targeting T cells in patients, as there are many complicating factors to consider when attempting to utilize mouse models of CART therapy to inform expected clinical outcomes. For example, one of the few reported patient deaths resulting from transfer of CAR T cells occurred using Her2-redirected CAR T cells and was presumably caused by CAR T-cell recognition of low levels of target antigen in the lung.18 This toxicity has not been reproducible in mouse models using transgenic mice with expression of human Her2 in normal tissues19,20 (except at T-cell doses exceeding 25 × 106, probably as a result of cytokine storm20). These findings highlight the difficulty in translating mouse models to clinical experience even when the same tumor antigen was evaluated.
Predicting the toxicity of NKG2D-based platforms becomes especially problematic because of the diversity of target ligands. Both efficacy and toxicity will probably be affected by the ligand tissue distribution, density, and affinity with which NKG2D binds the respective ligand. None of these parameters are very well characterized and likely vary greatly between mice and humans. NKG2DL genes differ significantly between mice and humans. MICA and MICB, the best-characterized human ligands, do not have orthologous genes in mice, and show a large degree of allelic variance within the human population. Therefore, targeting these ligands has not been evaluated using mouse models. The current findings highlight that even within the few common strains of research mice, responses to CART therapy can greatly vary, and that large variations between different strains of mice could predict even larger variability in genetically diverse human patients.
References
- VanSeggelen, H, Hammill, JA, Dvorkin-Gheva, A, Tantalo, DGM, Kwiecien, JM, Denisova, GF et al. (2015). T cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther 23: 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, G, Waks, T and Eshhar, Z (1989). Expression of immunoglobulin–T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 86: 10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp, SA, Kalos, M, Barrett, D, Aplenc, R, Porter, DL, Rheingold, SR et al. (2013). Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Davila, ML, Riviere, I, Park, J, Wang, X, Cowell, LG et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5: 177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, DW, Kochenderfer, JN, Stetler-Stevenson, M, Cui, YK, Delbrook, C, Feldman, SA et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nausch, N and Cerwenka, A (2008). NKG2D ligands in tumor immunity. Oncogene 27: 5944–5958. [DOI] [PubMed] [Google Scholar]
- Lanier, LL (2008). Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T, Lemoi, BA and Sentman, CL (2005). Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood 106: 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T, Barber, A and Sentman, CL (2006). Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res 66: 5927–5933. [DOI] [PubMed] [Google Scholar]
- Barber, A, Rynda, A and Sentman, CL (2009). Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol 183: 6939–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber, A and Sentman, CL (2009). Chimeric NKG2D T cells require both T cell- and host-derived cytokine secretion and perforin expression to increase tumor antigen presentation and systemic immunity. J Immunol 183: 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T and Sentman, CL (2013). Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D-modified T cells. J Immunol 190: 2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner, M, Gotz, G, Proff, J, Schaft, N, Dörrie, J, Full, F et al. (2012). Redirecting T cells to Ewing's sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One 7: e31210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, DG, Ye, Q, Santoro, S, Fang, C, Best, A and Powell, DJ Jr (2013). Chimeric NKG2D CAR-expressing T cell–mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther 24: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle, EJ, Sheard, V, Rothwell, DG, Bridgeman, JS, Ashton, G, Hanson, V et al. (2014). Differential role of Th1 and Th2 cytokines in autotoxicity driven by CD19-specific second-generation chimeric antigen receptor T cells in a mouse model. J Immunol 192: 3654–3665. [DOI] [PubMed] [Google Scholar]
- Kochenderfer, JN, Yu, Z, Frasheri, D, Restifo, NP and Rosenberg, SA (2010). Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116: 3875–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnasamy, D, Yu, Z, Theoret, MR, Zhao, Y, Shrimali, RK, Morgan, RA et al. (2010). Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngeneic tumors in mice. J Clin Invest 120: 3953–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Yang, JC, Kitano, M, Dudley, ME, Laurencot, CM and Rosenberg, SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, LX, Westwood, JA, Moeller, M, Duong, CP, Wei, WZ, Malaterre, J et al. (2010). Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res 70: 9591–9598. [DOI] [PubMed] [Google Scholar]
- Globerson-Levin, A, Waks, T and Eshhar, Z (2014). Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor–modified T cells. Mol Ther 22: 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]