

Abstract
T-cell depletion therapy is used to prevent acute allograft rejection, treat autoimmunity and create space for bone marrow or hematopoietic cell transplantation. The evolved response to T-cell loss is a transient increase in IL-7 that drives compensatory homeostatic proliferation (HP) of mature T cells. Paradoxically, the exaggerated form of this process that occurs following lymphodepletion expands effector T-cells, often causing loss of immunological tolerance that results in rapid graft rejection, autoimmunity, and exacerbated graft-versus-host disease (GVHD). While standard immune suppression is unable to treat these pathologies, growing evidence suggests that manipulating the incipient process of HP increases allograft survival, prevents autoimmunity, and markedly reduces GVHD. Multipotent adult progenitor cells (MAPC) are a clinical grade immunomodulatory cell therapy known to alter γ-chain cytokine responses in T-cells. Herein, we demonstrate that MAPC regulate HP of human T-cells, prevent the expansion of Th1, Th17, and Th22 effectors, and block the development of pathogenic allograft responses. This occurs via IL-1β-primed secretion of PGE2 and activates T-cell intrinsic regulatory mechanisms (SOCS2, GADD45A). These data provide proof-of-principle that HP of human T-cells can be targeted by cellular and molecular therapies and lays a basis for the development of novel strategies to prevent immunopathology in lymphodepleted patients.
Introduction
Patients receiving T-cell depletion therapy often experience tissue destructive immunopathology due to iatrogenic dysregulation of T-cell homeostasis.1,2,3,4,5,6,7 Growing evidence suggests that modulation of incipient T-cell repopulation can target immune dysregulation to prevent allograft loss, GVHD, or autoimmunity.8,9,10,11 This study examines the potential of a clinical grade cell therapy to block the development of disease-causing pathogenic effector T-cells in a relevant human model system.
T-cell homeostasis requires a balance in programs underlying activation and apoptosis with those controlling quiescence and survival. In healthy, T-cell replete animals naive CD4 T-cells are sustained by self-peptide and IL-7 signals.12,13,14 However, upon T-cell depletion, loss of IL-7R-bearing T-cells leads to an increase in IL-7 concentration that facilitates stimulatory, rather than tonic responses to self-peptide, driving homeostatic proliferation (HP) of mature T-cells.12,13,15,16 The HP of naive CD4 T-cells proceeds with slow kinetics and results in the formation of effector memory-like progeny; a process that primes/expands predisposing T-cell clones to exacerbate GVHD, accelerate allograft rejection and cause autoimmunity.17,18,19,20,21,22 Similar mechanisms govern HP of memory cells.14
In the clinic, lymphodepletion is increasingly used to successfully prevent acute allograft rejection, remedy autoimmune pathology, and create space for bone marrow transplantation (BMT). While immune reconstitution is an immediate concern for lymphopenic patients, growing evidence suggests that lymphopenia-induced immune dysregulation significantly contributes to patient mortality and morbidity.1,4,5,23,24,25 During immune reconstitution, acutely lymphopenic patients experience an increase in circulating IL-7 levels, and have a T-cell compartment characterized by low naive cell frequency and a predominance of rapidly dividing, proinflammatory (Th1) memory T-cells, displaying STAT5 phosphorylation and decreased activation threshold.23,26,27 TCR specificity may also become highly restricted and auto- or allo-reactive T-cells appear to undergo preferential expansion.2,3,28 Consequently, clinical CD4 T-cell lymphopenia (and/or IL-7 levels) associates with increased mortality, exacerbated GVHD, enhanced allograft rejection, and causes autoimmunity.1,4,5,6,7,29,30,31 These pathologies present a challenge since systemic immunosuppressive drugs fail to target the IL-7 pathway, and HP-induced effector memory cells are resistant to steroids or costimulatory blockade.3,7,32 Furthermore, recovery of normal T-cell homeostasis takes 2–20 years and is dependent upon the contribution of an often involuted or damaged thymus.24,25 Taken collectively, these data indicate that lymphodepletion causes acute and chronic immunopathology via IL-7-mediated HP and enhanced inflammatory potential of pre-existing auto- or alloreactive T-cells. Targeting the IL-7 axis or HP has recently shown marked therapeutic potential in rodent models of autoimmunity, GVHD, and solid organ transplant.8,9,11,33 However, despite an urgent clinical need and clear rationale, the mechanisms regulating HP are poorly understood and there are no therapies to treat effector T-cell driven immunopathology in lympodepleted patients.
Multipotent adult progenitor cells (MAPC) are a class of in vitro expanded, adherent, bone marrow-derived precursors.34 Both MAPC and prototypic mesenchymal stromal cells (MSC) have demonstrable immunomodulatory potential in vivo, suppress human T-cell activation, and have a record of safety in clinical trials.35,36,37,38,39 The vast majority of ongoing trials evaluating MSC/MAPC are conducted within T-cell depleted cohorts, and anecdotal clinical and preclinical data suggest efficacy may relate to an impact on HP.40,41 However, a formal mechanistic study of how these cells impact the process of HP has yet to be completed. We and others have recently shown that MSC and MAPC control T-cell responses to γ-chain cytokines by an unknown mechanism.35,42 In this study, we use a robust human model to test the hypothesis that MAPC (and MSC) can block IL-7-driven expansion of pathogenic effector T-cells via defined molecular mechanisms and aim to identify novel pathways, drug targets or biomarkers to assist in the development of therapies that could prevent immunopathology in T-cell depleted patients.
Results
MAPC suppress HP in a monocyte CD4 T-cell coculture system
To study HP of human CD4 T-cells, we developed a reductionist coculture system based on a previously described model.43 Cell trace violet (hereafter CTV)-labeled CD4 T-cells were combined with autologous CD14+ monocytes in the presence of exogenous IL-7 and dye-dilution measured at 6 days. Neither IL-7 or autologous monocytes alone were sufficient to induce HP of CD4 T-cells, which occurred without exception when IL-7 and monocytes were used in combination (Figure 1a). This was largely dependent upon T-cell contact with APC (Figure 1b), proceeded with kinetics that were consistent with the documented pace of slow HP (1–4 divisions in 6 days), and exhibited dye-dilution patterns distinct to antigen-specific or polyclonal activation (Figure 1a; Supplementary Figure S1a). Thus, the monocyte-co-culture system faithfully emulates the characteristic features of HP that have been observed in vivo.
Figure 1.

Development of a monocyte-CD4 T-cell coculture system to study multipotent adult progenitor cells (MAPC) suppression of IL-7-driven HP. Untouched, violet labeled CD4+ T-cells and CD14+ autologous monocytes were sorted from peripheral blood mononuclear cells and cocultured (10:1) in the presence and absence of IL-7 (50 ng/ml) and MAPC (1:2) for 6 days. (a) Representative fluorescence-activated cell sorting plots displaying proliferation of CD4 T-cells under the conditions indicated. (b) Proliferation of responding CD4 T-cells in the presence of IL-7 and monocytes either in contact (IL-7+Mo) or with T-cells and monocytes separated via a 1.0 μM pore transwell insert (IL-7+Mo, TW). (c) Proliferation of responding T-cells in the presence of IL-7 upon coculture with autologous monocytes in the presence or absence of third party MAPC and MSC (T-cell: MAPC ratio of 1:2) that were derived from the same bone marrow aspirate. (d) A dose titration of MAPC was added to monocytes and CD4 T-cells stimulated with IL-7 as above. (e) IL-7 was added to monocytes and CD4 T-cells from six different donors as above and proliferation measured in the presence and absence of MAPC (ratio of 1:2). Error bars in b–d represent the mean ± SEM of three donors. *P < 0.05; **P < 0.01; ***P < 0.001. Data are representative of three independent experiments. CTV, cell trace violet; MSC, mesenchymal stromal cell; NS, not significant.
As previously demonstrated, allogeneic MAPC (Figure 1a) and MSC (not shown) were nonimmunogenic when cultured alone with CD4 T-cells.35 However, addition of MSC or MAPC potently suppressed IL-7+Monocyte-mediated HP in a dose-dependent manner (Figure 1c,d). We observed no statistical difference in the level of suppression mediated by MSC versus MAPC derived from the same donor (Figure 1c). This effect was reproducible in experiments replicated with T-cell:monocytes from independent blood draws of the same donor (JR, unpublished data, April 2013) and T-cell:monocyte cultures from several different individuals (Figure 1e). Suppression of HP also was observed with multiple independent batches of unrelated MAPC in combination with different responder donors (Supplementary Figure S1b), irrespective of HLA type (Supplementary Table S1). MAPC and MSC also inhibited IL-7- and IL-15-driven effector expansion of CD4 and CD8 T-cells in whole peripheral blood mononuclear cell (PBMC) cultures (Supplementary Figure S1c–j).
MAPC prevent IL-7-mediated enhancement of cytokine production in CD4 T-cells
Th1 (IFN-γ and TNFα producing), Th17 (IL-17 producing), and Th22 (IL-22 producing) cells are known to mediate pathology in allogeneic transplant and autoimmune settings via the action of signature proinflammatory cytokines. IL-7-driven HP has been shown to enhance Th1 cytokine production, yet the effect of HP on the production of other cytokines in human T-cells is not entirely clear.22 As expected, IL-7-driven HP caused significantly elevated frequencies of Th1 cytokines IFN-γ and TNFα (Figure 2a–c), but also increased the frequency of IL-17- and IL-22-producing T-cells (Figure 2d–f). This effect was predominantly localized to divided cells, indicating that, like antigen-driven differentiation, effector potential increases upon cell division (Supplementary Figure S2a,b). MAPC coculture resulted in a dose-dependent suppression of all four effector cytokines, demonstrating that the presence of MAPC can impede both IL-7-driven cellular replication and the interdependent induction of effector memory potential (Figure 2a–f). We next examined whether suppression of cytokine induction was dependent upon inhibition of T-cell proliferation by establishing cocultures containing low MAPC:T-cell ratios, under which conditions proliferation was minimally affected. T-cells that had proliferated despite the presence of MAPC continued to exhibit diminished levels of IFN-γ, IL-22, and TNFα production (Supplementary Figure S2c,d), indicating that suppression of cytokine synthesis does not solely rely upon inhibition of proliferation. To test this in a transplantation-relevant system, we next isolated T-cells that had undergone HP in the presence and absence of a low ratio of MAPC then restimulated cells with allo-DC in secondary cultures. Low ratios of MAPC lead to a significant diminution of inflammatory allo-responses, despite only moderately inhibiting proliferation (Figure 2g,h). Thus MAPC modulation of HP blocks pathogenic allo-responses, and does so in a manner that is not dependent upon the ability to inhibit proliferation. This suggests that, at low ratios MAPC could prevent inflammatory effector cell responses while facilitating immune reconstitution. Finally, we tested whether MAPC suppression of was specific to naive or memory T cells. Both naive and memory cells exhibited significant responses to IL-7 and, although the magnitude was greater in memory cells, MAPC suppressed proliferation and expression of all cytokines tested in both subsets (Supplementary Figure S2e,f).
Figure 2.

Multipotent adult progenitor cells (MAPC) prevent HP-induced enhancement of effector T-cell cytokine responses. Untouched CD4+ T-cells were cocultured at a ratio of 1:5 with autologous monocytes in the presence and absence of IL-7 (50 ng/ml) with or without MAPC for 6 days. Cultures were then restimulated for 4 hours with 50 ng/ml PMA and 1 μg/ml Io. (a,d) Representative fluorescence-activated cell sorting (FACS) plots (MAPC at 1:2) and (b–f) bar graphs (ratios indicated) of data from ICS analysis of cocultures; gates were set on CD3+ viable cells. (g) Representative FACS plots and schematic of experimental design for sorted T-cell restimulation assays. CTV-labeled CD4 T-cells were cocultured with monocytes in the presence or absence of IL-7 as above with or without MAPC for 6 days at a low ratio of 1 MAPC: 40 CD4 T-cells. Under which conditions suppression of proliferation was minimal in the donors used. Untouched CD4 T-cells from all three conditions were FACS sorted and subsequently cocultured with allogeneic DC (1:8) for 72 hours. Harvested S/N were used for cytokine bead array analysis and the levels of (h) IFN-γ measured, *P < 0.05; NA, not activated; ns, not significant. Error bars represent the mean ± SEM of five donors. Data are representative of three independent experiments. CTV, cell trace violet; MSC, mesenchymal stromal cell.
MAPC suppress HP via a soluble factor following monocyte-dependent priming
We next sought to determine the mechanism by which MAPC suppressed HP. Suppression was observed through a transwell when MAPC and monocytes were cultured together in the upper chamber, but not when MAPC were cultured alone, thereby indicating that suppression occurred by a soluble factor that was only synthesized when MAPC and monocytes were in close proximity (Figure 3a–c). In agreement with this, cell-free conditioned media from MAPC-monocyte cocultures, but not MAPC alone suppressed HP (Supplementary Figure S3a,b). To determine if the soluble mediator of HP suppression was generated by MAPC following monocyte priming, or from MAPC-conditioned monocytes, we next compared the suppressive activity of several cell-free supernatants generated via serial media exchange (Figure 3d). Suppression of HP was exclusively observed using conditioned media (CM) from MAPC pretreated with monocyte supernatant (Figure 3e), demonstrating that, MAPC suppress HP via a soluble factor posterior to priming mediated by high local concentrations of a secreted, monocyte-derived molecule.
Figure 3.

Multipotent adult progenitor cells (MAPC)-suppression of homeostatic proliferation occurs via soluble factors following monocyte-dependent priming. (a) Schematic and (b) representative FACS plots from transwell experiments. CTV-labeled, sort purified, untouched CD4 T-cells were cocultured with autologous monocytes (5:1) and 50 ng/ml IL-7 and proliferation measured by dye-dilution at 6 days. MAPC (1:8) were added either in direct contact or in the top chamber of a transwell, either alone or in combination with additional monocytes (1:1). (c) Bar graph displaying proliferation of responder CD4 T-cells in the lower chamber of the transwell. (d) Schematic and (e) bar graph for culture media exchange assays. Monocytes and MAPC were cultured separately for 6 days (1 × 105/well) in 200 μl in 96-well plates. Alternatively, after 3 days, CM was removed from either cell type, wells were washed gently in media without detachment of adherent cells, then 200 μl cell free CM from MAPC added to monocytes and vice versa. All CM was collected at 6 days and added to a fresh monocyte-CD4 T-cell + IL-7 coculture at 50% volume and proliferation measured at 6 days. Media alone, or MAPC cells (1:8) were used as controls. Error bars represent the mean ± SEM for three donors. Results are indicative of at least three independent experiments. *P < 0.05, **P < 0.01. CM, conditioned media; CTV, cell trace violet; FACS, fluorescence-activated cell sorting; HP, homeostatic proliferation; NS, not significant.
MAPC suppress HP via an indoleamine 2,3-deoxygenase (IDO)-independent mechanism
We and others have previously demonstrated that IDO-mediated tryptophan catabolism is required for MAPC/MSC suppression of antigen-driven T-cell responses.35,37 Therefore, we examined whether MAPC suppression of HP occurred via IDO by conducting HP and anti-CD3/28-driven proliferation assays using the monocyte-T-cell coculture system in the presence and absence of the chemical inhibitor 1-methyl-L-tryptophan (1-MT) (Figure 4). As observed previously, T-cell proliferation in anti-CD3/28-stimulated cultures was potently suppressed by MAPC, consistent with IDO-mediated tryptophan catabolism and production of kynurenine (Figure 4a,c). Addition of 1-MT resulted in restoration of tryptophan levels and a significant recovery in T-cell proliferation (Figure 4a,c). In stark contrast, when MAPC were added to HP cultures neither tryptophan consumption nor kynurenine production was apparent, coincident with negligible effects on either molecule in the presence of 1-MT and a failure of the inhibitor to rescue T-cell proliferation (Figure 4b,d). Therefore, unlike anti-CD3/28 responses, MAPC inhibit HP via an IDO-independent mechanism, suggesting that MAPC suppress heterologous T-cell responses via context-dependent mechanisms. MAPC suppression of HP was subsequently found to be independent of IL-10, TGFβ and IL-6 in antibody blocking experiments (Supplementary Figure S3c).
Figure 4.

Multipotent adult progenitor cells (MAPC) suppress homeostatic proliferation via an IDO-independent mechanism. Monocyte-CD4 T-cell coculture experiments were set up as above with stimulation via 1:10 anti-CD3/28 (a,b) or 50 ng/ml IL-7 (c,d) in the presence and absence of 1:4 MAPC and 1 mmol/l 1-MT. Proliferation (b,d), tryptophan and kynurenine (a,c) were measured by dye dilution and mass spectrometry, respectively at 6 days. Error bars represent the SEM of two (a,c) and three (b,d) donors. *P < 0.05. IDO, indoleamine 2,3-deoxygenase.
MAPC suppress HP via prostaglandin E2 (PGE2) following monocyte priming
An established property of MAPC (and MSC) is the ability to produce the immunoregulatory molecule PGE2, which can suppress GVHD in vivo.41,44 To assess a role for any potential prostanoid in MAPC-mediated suppression of HP, we first measured concentrations of these molecules in the coculture supernatants from MAPC that were untreated or exposed to monocytes and/or T-cells. Levels of PGF2a, PGI2, PGD2, TXA2 were undetectable or close to background and where present were not induced by monocyte or monocyte-T-cell coculture, while PGE2 was consistently and potently produced by MAPC under the same conditions (Supplementary Figure S3d,e). Similarly MAPC produced PGE2 after treatment with monocyte CM (Figure 5a), coincident with induction of suppressive potential observed above (See Figure 3e). Monocytes induced MAPC to produce PGE2 at least partially via IL-1β, since treatment of MAPC with rhIL-1β but not IFN-γ- or TNFα-induced PGE2 (Figure 5b) and PGE2 secretion could be significantly reduced by the inclusion of anti-IL-1β blocking antibody in monocyte-MAPC cocultures (Figure 5c). Addition of PGE2 to HP cultures lead to dose-dependent suppression of proliferation, recapitulating the effect of PGE2-containing supernatant (Figure 5d). We next blocked PGE2 production using the COX-2 inhibitor NS-398 in anti-CD3/28 (Figure 5e,f) and IL-7 (Figure 5g,h) stimulated cultures, leading to significant depletion of PGE2 levels (Figure 5e,g). In line with a dominant role for IDO, MAPC suppression of CD3/28-driven proliferation was unaffected by PGE2 blockade (Figure 5f). However, NS-398 treatment completely ablated the suppression of HP by MAPC (Figure 5h). Importantly, the activity of NS-398 could be reversed by supplementation of MAPC+NS-398 treated cocultures with PGE2, confirming pathway specificity (Figure 5i). Therefore, PGE2 is induced at least partially by IL-1β-dependent monocyte priming and is accountable for MAPC suppression of IL-7-induced CD4 T-cell proliferation. Given that suppression of cytokine production by MAPC appeared to occur independently of effects on proliferation, we tested if PGE2 was also responsible for MAPC-mediated attenuation of IL-7-enhanced cytokine synthesis. Interestingly, whilst NS-398 ablated suppression of proliferation, COX-2 inhibition only partially recovered cytokine production; thereby indicating that suppression of cytokine induction may involve additional pathways (Figure 5j).
Figure 5.

Multipotent adult progenitor cells (MAPC) suppress HP via PGE2. (a) PGE2 levels were measured in supernatants from 6-day cultures containing monocytes, MAPC, monocytes treated with 3-day MAPC CM or MAPC treated with 3-day Monocyte CM (see legend of Figure 3d for more detail). (b) PGE2 levels were measured in 6-day supernatants from MAPC cultures treated with the cytokines indicated at 25 ng/ml or (c) cocultures containing allogeneic monocytes (1:2) in the presence or absence of 5 μg/ml IgG1 isotype control or anti-IL-1β antibodies. (d) Proliferation in IL-7-driven CD4 T-cell monocyte cocutlures was measured in the presence and absence of exogenous PGE2 at the concentrations indicated. (e–h) Monocyte: CD4 T-cell cocultures (5:1) were stimulated with 1:10 anti-CD3/28 (e,f) or 50 ng/ml IL-7 (g,h) and PGE2 levels (e,g) or proliferation (f,h) measured at 6 days in the presence and absence of 5 μmol/l NS-398. (i) IL-7 HP cocultures were treated with or without 1:8 MAPC in the presence and absence of 5 μmol/l NS-398 and/or 50 ng/ml PGE2. (j) % Suppression of proliferation and IFN-γ production were measured according to dye-dilution and ICS after PMA/Io, respectively for monocyte+IL-7 stimulated T-cells in the presence or absence of MAPC treated with or without NS-398 as above. Error bars represent the SEM for two (a) or three (b,c) experiments or three donors (e–j). *P < 0.05; **P < 0.01. Data are representative of three independent experiments. CM, conditioned media; HP, homeostatic proliferation; NA, not activated; NS, not significant.
Suppression of HP activates T-cell intrinsic regulatory mechanisms
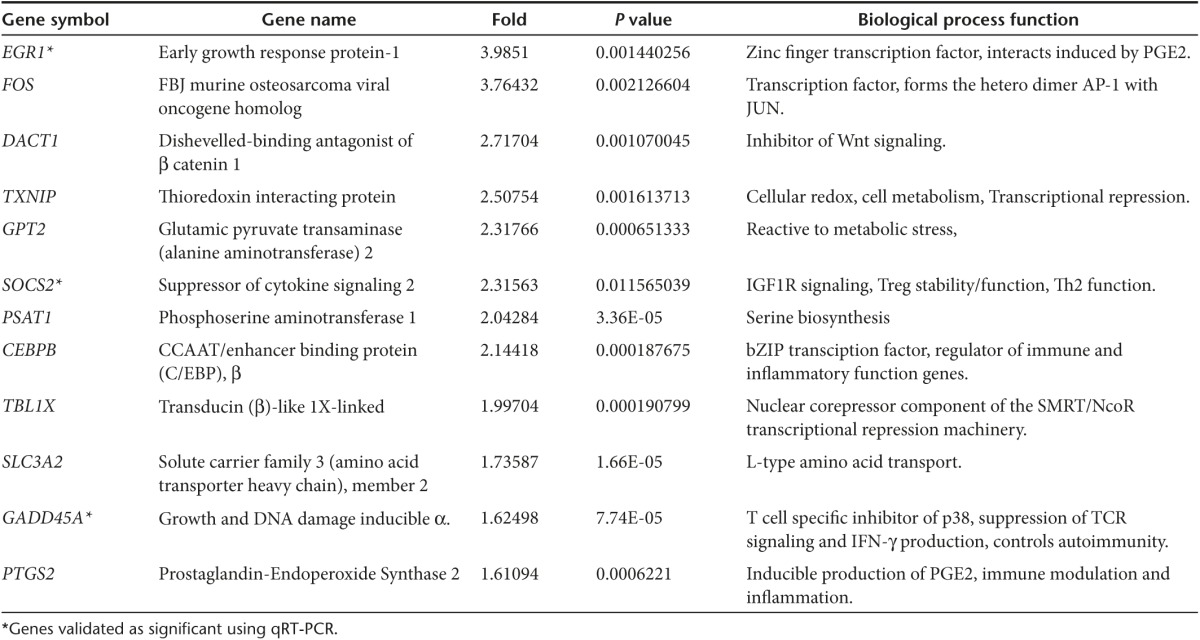
We next sought to determine the impact of HP suppression on T-cell molecular dynamics. Our experimental design was based upon the rationale that undivided T-cell populations within MAPC-containing cultures would comprise an admixture of resting T-cells, cells that had failed to respond to IL-7 stimulation, those that were engaged to proliferate and those that had undergone suppression. Therefore, to resolve a gene signature specific to suppressed cells we performed dye-dilution experiments and compared the gene expression profile of FACS-sorted resting, nonresponding and proliferating cells to that of MAPC-suppressed cells (Figure 6a). Highly reproducible levels of suppression (79% ± 3.1) were observed in all three experiments using independent donors to source RNA (Supplementary Figure S4a). Initially, unbiased principal component analysis independently grouped samples from each of the four conditions into discrete clusters, indicating treatment-specific differences in global gene expression (Figure 6b). Proliferated cells showed the highest degree of variance with respect to the other three groups, whilst suppressed cells exhibited a closer relationship to nonresponding, and resting cells (Figure 6b). Relative to resting cells, IL-7 treatment resulted in the significant differential upregulation of genes involved in cell-cycle regulation and DNA biosynthesis (10 most upregulated shown in Supplementary Table S2), confirming that the experimental system could be successfully applied to extract biologically relevant treatment-specific differences in gene regulation. We next compared global gene expression in MAPC-suppressed T-cells to that of all other groups. Fifty-nine genes were significantly (P < 0.05) upregulated in MAPC-treated, undivided T-cells (heat map in Figure 5c shows the 40 most markedly upregulated loci, raw data online at www.ebi.ac.uk accession number E-MTAB-3228). Twelve of these genes exhibited a reproducibly higher level of expression in MAPC-suppressed cells relative to all other treatment groups for all 3/3 donors (example of validation provided in Supplementary Figure 4b). These 12 loci (EGR1, FOS, DACT1, TXNIP, GPT2, SOCS2, PSAT1, CEBPB, TBL1X, SLC3A2, GADD45A, PTGS2, Table 1) could be functionally classified according to roles in transcriptional regulation, cellular metabolism/stress and PGE2 responsiveness (Figure 6d; Table 1). To confirm these data, replicate experiments were performed with PGE2-suppressed CD4 T-cells used as an additional control (Supplementary Figure S4c,d). qRT-PCR analysis confirmed that the average expression of 8/8 loci tested was higher in MAPC-suppressed T-cells (Figure 6e; Supplementary Figure S4e). Three of these genes, EGR1, SOCS2, and GADD45a were expressed at significantly higher levels in suppressed T-cells, relative to each of the three other treatment conditions for 3/3 donors, comprising a core index to identify CD4 T-cells suppressed by PGE2 or MAPC during HP (Figure 6e). In seven out of eight of the genes tested by qPCR, there was a trend for expression to be higher in MAPC-suppressed T-cells versus those inhibited by PGE2 (significant for GADD45a) including 3 genes (PSAT1, GPT2, SLC3A2) that were exclusively induced by MAPC (Figure 6e, Supplementary Figure S4e), suggesting that HP suppression by MAPC induces both PGE2-dependent and PGE2-independent gene induction. Thus, despite some discernable differences, suppression of HP by MAPC or PGE2 leads to a conserved pattern of molecular reprograming in CD4 T-cells.
Figure 6.

Suppression of HP activates T-cell intrinsic regulatory mechanisms. (a) CD4 T-cells were cocultured with autologous monocytes (5:1) in the absence of simulation (header NA, not activated), with 50 ng/ml IL-7 (header IL-7) or with 50 ng/ml IL-7 plus 1:4 MAPC for 6 days (header IL-7+MAPC. Cells from each condition were FACS sorted as four populations representing resting (Rest.), proliferated (Prolif.), nonresponding (NR.), or suppressed (Supp.) cells as indicated. Experiments were completed for three independent donors and RNA used for microarray analysis. (b) Unbiased clustering of samples from microarray data via principal component analysis. (c) Heat map showing comparative analysis of the 40 most differentially expressed genes significantly upregulated (P < 0.05) in MAPC (suppressed) samples versus the mean expression of all three other groups ordered by hierarchical clustering of samples (columns) and fold change (rows). Twelve most consistently upregulated genes for 3/3 donors for all Illumina probe sets are marked (*). (d) Venn diagram displaying functional classification of the 12 most consistently upregulated genes in MAPC-supp. cells; the number of genes within each functional category is shown, those common to multiple categories are indicated in the overlapping sections. (e) Confirmation of microarray data by qRT-PCR showing relative expression levels of three genes denoted in the y-axis of each bar graph. The fold expression value relative to resting cells (NA) for all other groups is shown. Bar graph in bottom right shows expression index of all three genes combined as a single value. Data were normalized to B2M (shown) and EIF4a expression with similar results. *P < 0.05, NS, nonsignificant. Error bars represent the mean ± SEM of three donors. NA, not activated (i.e., Rest.). CTV, cell trace violet; FACS, fluorescence-activated cell sorting; HP, homeostatic proliferation; MAPC, multipotent adult progenitor cells; NS, not significant.
Table 1. List of the 12 consistently upregulated genes in MAPC-suppressed CD4 T-cells versus all other conditions as determined by microarray analysis.
Discussion
T-cell depletion therapy is increasingly used in the clinic for a multitude of transplant and autoimmune indications. The consequent phenomenon of HP is responsible for both immune reconstitution and immunopathology. However, a human model of HP has not previously been reported, and there is a paucity of data regarding the regulation of this process, highlighting a scientific gap that underpins an urgent, unmet clinical need. In line with rodent studies, our data indicate a two-signal model in which both cytokine and self-peptide signals are required to drive human HP, suggesting that pharmacological inhibition of TCR and/or cytokine signals may successfully modulate cell division and effector potential.14 Furthermore, the polyfunctional inflammatory profile we discovered for HP-induced T-cells (IFNγ, IL-17, IL-22 induction) extends beyond the previously described Th1 bias, providing a potential pathological basis for the spectrum of lymphopenia-associated tissue destruction observed in the clinic.
This report is the first example of context-dependent suppression mechanisms being deployed by MSC/MAPC according to the nature of the ongoing T-cell response. Our results collectively illustrate that PGE2 and IDO are coordinately induced during antigen-driven responses, where IDO plays a predominant role. In contrast, only PGE2 is induced during IL-7-driven proliferation, where it is necessary and sufficient for regulation of HP with a subsequent impact on antigen-driven responses. It is not presently clear whether IDO is able to suppress HP, since this enzyme is not active in our HP model; however it is noteworthy that in vivo MAPC-derived PGE2 alone is able to block GVHD in the absence of IDO, potentially via effects on HP.41 Moreover, though it is most likely that the low levels of IFN-γ in HP cultures account for a lack of IDO induction, the proliferation rate/metabolic demand of dividing T-cells, or involvement of APC function may also be key factors determining system-dependent IDO versus PGE2 dominance.
The induction of PGE2 target genes in our microarray suggests a direct effect of the molecule on T-cell populations. However, PGE2 is known to alter APC function, including that of monocytes, thus the contribution of direct (MAPC>T cell) or indirect (MAPC>Mono>T-cell) suppression of HP on the arrest or molecular reprogramming of T-cells is not presently clear.37 GADD45a, SOCS2, and EGR-1 were all consistently induced by MAPC or PGE2 during suppression of proliferation. However, compared to PGE2, MAPC induced an increased breadth and magnitude of gene expression during T-cell regulation (e.g., induction of PSAT1, GPT2, SLC3A2). These loci may be involved in the suppression of cytokine synthesis, which was not a potent function of PGE2. It remains interesting that at lower ratios MAPC inhibited cytokine induction but not proliferation. Similarly, MAPC exerted selective effects on cytokines; suppressing IFNγ, TNFα, and IL-22 but not IL-17 in divided cells. These results may reflect preferential inhibition of IL-7 versus TCR signals or selective interference of transduction events within the IL-7 signaling cascade (e.g., JAK1/3, STAT 1/3/5 phosphorylation). To this end, GADD45a is a known inhibitor of T-cell function, operating in part via p38 kinase blockade.45 Given that p38 kinase is involved in the proliferative response to IL-7, it remains a strong possibility that MAPC (and PGE2) suppress HP via GADD45a-mediated p38 kinase attenuation.46 Similarly, SOCS2 maintains Treg stability and an anti-inflammatory phenotype by preventing the induction of proinflammatory cytokines and thus may directly contribute to MAPC or PGE2 T-cell inhibition.47 On aggregate, the coordinate expression of SOCS2 and GDD45a may represent a molecular basis for HP suppression, but also form part of a general T-cell tolerance signature, represent MAPC-specific biomarkers, or serve as therapeutic targets for future immunotherapy development.
T-cell depletion is currently an integral component of life-sparing transplantation regimens. However, this strategy introduces a wealth of acute and chronic immunological complications that are clinically challenging due to risks associated with both immune insufficiency (e.g., opportunistic infection and malignancy) and immunopathology (e.g., graft rejection and autoimmunity). For example, in allogeneic BMT/HSCT lymphopenic hosts require immune reconstitution to prevent reactivation of latent viruses and malignant disease relapse, yet the major cause of patient mortality, acute GVHD, necessitates potent prophylactic immune suppression. This complex patient need is not only unmet, but likely exacerbated by common pharmacologics that fail to prevent effector cell expansion and/or limit immune reconstitution or competency.3,32 Thus, a revised therapeutic paradigm is required to treat T-cell-depleted hosts, in which context IL-7-driven HP appears a suitable target. In the clinic, delayed administration of rhIL-7 (e.g., +100 days post BMT) can safely boost immune recovery in patients without historical or current GVHD, while day +7–14 post-BMT IL-7 levels associate with acute GVHD. In contrast, prophylactic blockade of IL-7 can prevent GVHD in vivo.9,29,48 Thus, manipulation of the IL-7 axis offers great therapeutic promise but must be undertaken in a patient, time, and indication-specific manner. We and others have demonstrated efficacy of cell therapies in rodent transplant or GVHD models resulting from modified cytokine or antigen-driven T-cell proliferation.33,49,50,51 Our results herein suggest clinical grade MAPC, a therapy active in clinical trials of T-cell-depleted cohorts, may represent an ideal candidate for human HP modulation. A key finding in support of this is that low ratios of MAPC can block IL-7 driven generation of pathogenic cytokine responses without affecting T-cell proliferation, thereby hitting a potential “sweet spot” for normalized immune reconstitution. Similarly, PGE2 has been shown to improve T-cell reconstitution, suggesting that therapies inducing this molecule (including MAPC or novel inhibitors) may simultaneously promote naive cell export and suppress effector cell expansion.1,52,53 However, implementing this clinically will require future investigation and considerable optimization of treatment regimens.
The efficacy of MAPC to suppress HP in vivo will likely depend on the interdependent variables of route/time of administration, homing, cytokine environment and cellular milieu. Our data implies that the presence of APC are crucial in licensing MAPC and potentially in modulating the T-cell response. Indeed MSC/MAPC efficacy has been linked to induction of regulatory cell populations (e.g., MDSC, M2 macrophages, tDCs, and Tregs) in vivo.37,54 Our system also models a proximal interaction of MAPC, monocytes, and T-cells, emulating an in vivo model in which MAPC-derived PGE2 suppression of GVHD was only observed upon delivery of cells to sites of allo-priming.41 Taken collectively, we propose that local delivery of MAPC to the spleen, lymph node or bone marrow early after depletion or alongside transplantation would maximize the chance for licensed production of PGE2 (and possibly IDO), induction of infectious tolerance via APCs, and inhibition of IL-7 (or IL-15) effector cell expansion. Indeed, our preliminary preclinical work suggests that MAPC can significantly reduce IL-7-mediated T-cell derived IFN-γ and ki67 in vivo in a manner that may be dependent upon specific routes of administration and patterns of biodistribution (F. Carty, J. Reading, K. English, unpublished data, June 2015).
The present study adds to a growing body of clinical and preclinical data, which highlights that targeting of cytokine, and not just antigen-driven T-cell responses can influence fundamental therapeutic processes relevant to a broad spectrum of clinical indications. The cellular and molecular features of human HP identified here may serve as novel targets for future immunotherapy development, while clinical strategies deploying MSC/MAPC, PGE2 or GADD45a or SOCS2 inducing agents could prevent effector T-cell mediated immunopathology, such as that seen downstream of IL-7 in T-cell depleted patients.
Materials and Methods
MSC and MAPC generation. MAPC used in this study were clinical grade MultiStem cells. MSC were generated in parallel. MAPC and MSC used throughout the majority of this study were manufactured by Athersys (Cleveland, OH) from a single-donor bone marrow aspirate, from a fully consented 21-year-old Caucasian male donor and processed according to the previously described methods.35 Three subsequent MAPC batches from unrelated donors were used to verify results (Supplementary Figure S1b).
Cell culture. PBMC were isolated from anonymous leucocyte cones of healthy blood donors that had given informed consent (National blood service, NHS, UK) using density-gradient centrifugation with Lymphoprep (Axis Shield, Oslo, Norway) then labeled with 1 μmol/l cell trace violet according to the manufacturer's instructions (Life Technologies, Grand Island, NY) and cryopreserved as previously described.35 For monocyte-CD4 T-cell coculture, see information in figure legends and Supplementary Methods. All cultured cells were incubated in X-vivo-15 media (Lonza, Basel, Switzerland) containing penicillin-streptomycin (100 μg/ml) and amphotericin-B (both from Life Technologies) at 37 °C, 5% CO2, using 96-well round bottom plates with or without 1.0 μM transwell chambers (Corning, Corning, NY, USA). For details on tissue HLA genotyping of MAPC and responder donors, see Supplementary Methods.
IDO, PGE2, and antibody blocking assays. IDO and PGE2 inhibition assays and PGE2 addition experiments were carried out as indicated in the figure legends. For reagent information, see Supplementary Methods.
PGE2 ELISA. Supernatants were analyzed with the Prostaglandin E2 Parameter Assay Kit (R&D Systems NE, Minneapolis, MN) according to the manufacturer's instructions. For more details on measurement of other prostanoids, see Supplementary Methods.
Tryptophan and kynurenine measurement. Tryptophan and kynurenine measurements were performed by LC/MS/MS, for more details see Supplementary Methods.
Cytometric bead array analysis. Levels of IFN-γ in supernatants were determined by Flow cytomix (eBioscience, San Diego, CA) cytokine bead array according to the manufacturer's instructions.
Microarray. Microarray using RNA from sorted T-cells was performed on Illumina HumanHT-12 v4 Expression BeadChip as contracted research by AROS Applied Biotechnology (Aarhus, Denmark, http://www.arosab.com). After removal of one outlier in the rest/NA group, the raw data were Log2 transformed and quantile normalized using Bioconductor Lumi package. Data analysis (principal component analysis and hierarchical clustering) was carried out using Qlucore Omics Explorer (Qlucore, Lund, Sweden).
qRT-PCR. RNA was isolated using the RNAeasy mini kit (Qiagen, Limburg, The Netherlands), and reverse transcribed using the cDNA synthesis kit (Life Technologies). qPCR was carried out using Taqman probes and Taqman master mix (Life Technologies) in an ABI 7900HT instrument (Applied Biosystems, Grand Island, NY). For more information, see Supplementary Methods.
Flow cytometry. Dead cells were excluded with 7-aminoactinomycin D (7-AAD) (Sigma Aldrich, St Louis, MO) or Fixable Live/dead stains (Molecular probes, Invitrogen, Life technologies). Fluorochrome-labeled antibodies used (Biolegend) are listed in the Supplementary Methods. Intracellular staining was performed using the intracellular staining kit (Biolegend, San Diego, CA) according to the manufacturers' instructions. Flow cytometry acquisition was performed on a BD FACS Canto II and cell sorting completed using the BD FACS ARIA (BD Biosciences, Franklin Lakes, NJ), both equipped with FACS Diva software v6.0 (BD Biosciences) and data analyzed using Flowjo X (Treestar, Ashland, OR).
Data analysis. Data were analyzed using Prism version 6 (Graphpad) using relevant statistical tests.
All human subjects provided written, informed consent in accordance with the declaration of Helsinki protocol and King's College London institutional code of ethics.
SUPPLEMENTARY MATERIAL Figure S1. Characteristics of monocyte + IL-7-driven homeostatic proliferation and the suppression of homeostatic proliferation by MSC and MAPC in PBMCs. Figure S2. T-cell effector cytokine suppression by MAPC in dividing CD4 T cells and naïve versus memory T-cells. Figure S3. MAPC-mediated suppression of HP requires monocyte co-culture and is independent of IL-10, TGFβ and IL-6 and coincident with expression of PGE2, but not other COX-2 derived prostanoids. Figure S4. Suppression, candidate gene QC and relative gene expression in Microarray and qRT-PCR experiments. Table S1. MAPC from four separate individuals/batches and PBMC from four unrelated donors used for proliferation assays were DNA sequenced by PCR with primers specific for all major HLA alleles. Table S2. The 10 most markedly up-regulated genes and the 3 down-regulated genes in monocyte + IL-7-stimulated CD4 T-cells (both proliferated and non proliferated samples were pooled for analysis) when compared to resting CD4 T-cells (monocyte alone) as determined by microarray analysis.
Acknowledgments
The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. This work was supported by grants from the UK Technology Strategy Board (TSB), and an unrestricted research agreement from Athersys Inc. J.L.R. is a post-doctoral research associate at King's College London, partially funded by a sponsored, unrestricted research agreement from Athersys Inc. B.V. is an Employee of Regenesys ltd. a wholly-owned subsidiary of Athersys Inc. N.S.W., A.D., N.A.L., J.M.T., J.P., R.D., and A.E.T. are employees of Athersys Inc. R.D. and A.E.T. are shareholders in Athersys Inc. F.C. is an Irish Research Council scholar partially funded through an enterprise partnership scheme by Regenesys ltd. T.I.M.T. is Senior lecturer and principal investigator at King's College London, in receipt of an unrestricted research agreement from Athersys Inc. C.H., S.S., T.H., and K.E. declare no conflict of interest.
Supplementary Material
References
- Saas, P, Bamoulid, J, Courivaud, C, Rebibou, J, Gaugler, B (2013). CD4 T lymphopenia, thymic function, homeostatic proliferation and late complications associated with kidney transplantation. Curr Issues Futur Dir Kidney Transplant (Chapter 18).
- Pearl, JP, Parris, J, Hale, DA, Hoffmann, SC, Bernstein, WB, McCoy, KL et al. (2005). Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. Am J Transplant 5: 465–474. [DOI] [PubMed] [Google Scholar]
- Monti, P, Scirpoli, M, Maffi, P, Ghidoli, N, De Taddeo, F, Bertuzzi, F et al. (2008). Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J Clin Invest 118: 1806–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, K, Kim, HT, McDonough, S, Bascug, G, Warshauer, B, Koreth, J et al. (2010). Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J Clin Invest 120: 1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neujahr, DC, Chen, C, Huang, X, Markmann, JF, Cobbold, S, Waldmann, H et al. (2006). Accelerated memory cell homeostasis during T cell depletion and approaches to overcome it. J Immunol 176: 4632–4639. [DOI] [PubMed] [Google Scholar]
- Jones, JL, Thompson, SA, Loh, P, Davies, JL, Tuohy, OC, Curry, AJ et al. (2013). Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci USA 110: 20200–20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z, Bensinger, SJ, Zhang, J, Chen, C, Yuan, X, Huang, X et al. (2004). Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med 10: 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai, HL, Boeffard, F, Longis, J, Danger, R, Martinet, B, Haspot, F et al. (2014). IL-7 receptor blockade following T cell depletion promotes long-term allograft survival. J Clin Invest 124: 1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, B, Dudl, EP, Min, D, Barsky, L, Smiley, N and Weinberg, KI (2007). Prevention of graft-versus-host disease by anti IL-7Ralpha antibody. Blood 110: 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, LF, Logronio, K, Tu, GH, Zhai, W, Ni, I, Mei, L et al. (2012). Anti-IL-7 receptor-α reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci USA 109: 12674–12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaranda, C, Kuswanto, W, Hofmann, J, Kenefeck, R, Narendran, P, Walker, LS et al. (2012). IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci USA 109: 12668–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, JT, Dudl, E, LeRoy, E, Murray, R, Sprent, J, Weinberg, KI et al. (2001). IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA 98: 8732–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst, B, Lee, DS, Chang, JM, Sprent, J and Surh, CD (1999). The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 11: 173–181. [DOI] [PubMed] [Google Scholar]
- Boyman, O, Létourneau, S, Krieg, C and Sprent, J (2009). Homeostatic proliferation and survival of naïve and memory T cells. Eur J Immunol 39: 2088–2094. [DOI] [PubMed] [Google Scholar]
- Martin, B, Bécourt, C, Bienvenu, B and Lucas, B (2006). Self-recognition is crucial for maintaining the peripheral CD4+ T-cell pool in a nonlymphopenic environment. Blood 108: 270–277. [DOI] [PubMed] [Google Scholar]
- Hennion-Tscheltzoff, O, Leboeuf, D, Gauthier, SD, Dupuis, M, Assouline, B, Grégoire, A et al. (2013). TCR triggering modulates the responsiveness and homeostatic proliferation of CD4+ thymic emigrants to IL-7 therapy. Blood 121: 4684–4693. [DOI] [PubMed] [Google Scholar]
- Cho, BK, Rao, VP, Ge, Q, Eisen, HN and Chen, J (2000). Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med 192: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna, K and Ahmed, R (2000). Cutting edge: naive T cells masquerading as memory cells. J Immunol 165: 1733–1737. [DOI] [PubMed] [Google Scholar]
- Anderson, BE, McNiff, JM, Matte, C, Athanasiadis, I, Shlomchik, WD and Shlomchik, MJ (2004). Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood 104: 1565–1573. [DOI] [PubMed] [Google Scholar]
- Baccala, R and Theofilopoulos, AN (2005). The new paradigm of T-cell homeostatic proliferation-induced autoimmunity. Trends Immunol 26: 5–8. [DOI] [PubMed] [Google Scholar]
- Sener, A, Tang, AL and Farber, DL (2009). Memory T-cell predominance following T-cell depletional therapy derives from homeostatic expansion of naive T cells. Am J Transplant 9: 2615–2623. [DOI] [PubMed] [Google Scholar]
- Moxham, VF, Karegli, J, Phillips, RE, Brown, KL, Tapmeier, TT, Hangartner, R et al. (2008). Homeostatic proliferation of lymphocytes results in augmented memory-like function and accelerated allograft rejection. J Immunol 180: 3910–3918. [DOI] [PubMed] [Google Scholar]
- Lundström, W, Fewkes, NM and Mackall, CL (2012). IL-7 in human health and disease. Semin Immunol 24: 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackall, CL, Fleisher, TA, Brown, MR, Andrich, MP, Chen, CC, Feuerstein, IM et al. (1995). Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med 332: 143–149. [DOI] [PubMed] [Google Scholar]
- Hakim, FT, Cepeda, R, Kaimei, S, Mackall, CL, McAtee, N, Zujewski, J et al. (1997). Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood 90: 3789–3798. [PubMed] [Google Scholar]
- Gurkan, S, Luan, Y, Dhillon, N, Allam, SR, Montague, T, Bromberg, JS et al. (2010). Immune reconstitution following rabbit antithymocyte globulin. Am J Transplant 10: 2132–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvy, AP, Kho, MM, Klepper, M, Litjens, NH, Betjes, MG, Weimar, W et al. (2013). Kinetics of homeostatic proliferation and thymopoiesis after rATG induction therapy in kidney transplant patients. Transplantation 96: 904–913. [DOI] [PubMed] [Google Scholar]
- Williams, KM, Hakim, FT and Gress, RE (2007). T cell immune reconstitution following lymphodepletion. Semin Immunol 19: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiant, S, Yakoub-Agha, I, Magro, L, Trauet, J, Coiteux, V, Jouet, JP et al. (2010). Plasma levels of IL-7 and IL-15 in the first month after myeloablative BMT are predictive biomarkers of both acute GVHD and relapse. Bone Marrow Transplant 45: 1546–1552. [DOI] [PubMed] [Google Scholar]
- Kielsen, K, Jordan, KK, Uhlving, HH, Pontoppidan, PL, Shamim, Z, Ifversen, M et al. (2015). T cell reconstitution in allogeneic haematopoietic stem cell transplantation: prognostic significance of plasma interleukin-7. Scand J Immunol 81: 72–80. [DOI] [PubMed] [Google Scholar]
- Hickman, SP and Turka, LA (2005). Homeostatic T cell proliferation as a barrier to T cell tolerance. Philos Trans R Soc Lond B Biol Sci 360: 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts, DR, Shankar, S, Chastain, EM, Martin, A, Getts, MT, Wood, K et al. (2011). Current landscape for T-cell targeting in autoimmunity and transplantation. Immunotherapy 3: 853–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, A, Zhang, Q, Jiang, J, Yuan, G, Feng, Y, Hao, J et al. (2006). Co-transplantation of bone marrow stromal cells transduced with IL-7 gene enhances immune reconstitution after allogeneic bone marrow transplantation in mice. Gene Ther 13: 1178–1187. [DOI] [PubMed] [Google Scholar]
- Breyer, A, Estharabadi, N, Oki, M, Ulloa, F, Nelson-Holte, M, Lien, L et al. (2006). Multipotent adult progenitor cell isolation and culture procedures. Exp Hematol 34: 1596–1601. [DOI] [PubMed] [Google Scholar]
- Reading, JL, Yang, JH, Sabbah, S, Skowera, A, Knight, RR, Pinxteren, J et al. (2013). Clinical-grade multipotent adult progenitor cells durably control pathogenic T cell responses in human models of transplantation and autoimmunity. J Immunol 190: 4542–4552. [DOI] [PubMed] [Google Scholar]
- Duffy, MM, Ritter, T, Ceredig, R and Griffin, MD (2011). Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther 2: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, K and Mahon, BP (2011). Allogeneic mesenchymal stem cells: agents of immune modulation. J Cell Biochem 112: 1963–1968. [DOI] [PubMed] [Google Scholar]
- Vaes, B, Van't Hof, W, Deans, R and Pinxteren, J (2012). Application of MultiStem(®) Allogeneic Cells for Immunomodulatory Therapy: Clinical Progress and Pre-Clinical Challenges in Prophylaxis for Graft Versus Host Disease. Front Immunol 3: 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem, HK and Thiemermann, C (2010). Mesenchymal stromal cells: current understanding and clinical status. Stem Cells 28: 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perico, N, Casiraghi, F, Introna, M, Gotti, E, Todeschini, M, Cavinato, RA et al. (2011). Autologous mesenchymal stromal cells and kidney transplantation: a pilot study of safety and clinical feasibility. Clin J Am Soc Nephrol 6: 412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill, SL, Kelly, RM, O'Shaughnessy, MJ, Zhou, Q, Xia, L, Panoskaltsis-Mortari, A et al. (2009). Multipotent adult progenitor cells can suppress graft-versus-host disease via prostaglandin E2 synthesis and only if localized to sites of allopriming. Blood 114: 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocelli-Tyndall, C, Bracci, L, Schaeren, S, Feder-Mengus, C, Barbero, A, Tyndall, A et al. (2009). Human bone marrow mesenchymal stem cells and chondrocytes promote and/or suppress the in vitro proliferation of lymphocytes stimulated by interleukins 2, 7 and 15. Ann Rheum Dis 68: 1352–1359. [DOI] [PubMed] [Google Scholar]
- Ge, Q, Palliser, D, Eisen, HN and Chen, J (2002). Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc Natl Acad Sci USA 99: 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, K, Barry, FP, Field-Corbett, CP and Mahon, BP (2007). IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett 110: 91–100. [DOI] [PubMed] [Google Scholar]
- Jirmanova, L, Giardino Torchia, ML, Sarma, ND, Mittelstadt, PR and Ashwell, JD (2011). Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood 118: 3280–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley, JB, Rawlinson, L, Lali, FV, Page, TH, Saklatvala, J and Foxwell, BM (1997). T cell proliferation in response to interleukins 2 and 7 requires p38MAP kinase activation. J Biol Chem 272: 15023–15027. [DOI] [PubMed] [Google Scholar]
- Knosp, CA, Schiering, C, Spence, S, Carroll, HP, Nel, HJ, Osbourn, M et al. (2013). Regulation of Foxp3+ inducible regulatory T cell stability by SOCS2. J Immunol 190: 3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales, MA, Goldberg, JD, Yuan, J, Koehne, G, Lechner, L, Papadopoulos, EB et al. (2012). Recombinant human interleukin-7 (CYT107) promotes T-cell recovery after allogeneic stem cell transplantation. Blood 120: 4882–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacsovics-Bankowski, M, Streeter, PR, Mauch, KA, Frey, MR, Raber, A, van't Hof, W et al. (2009). Clinical scale expanded adult pluripotent stem cells prevent graft-versus-host disease. Cell Immunol 255: 55–60. [DOI] [PubMed] [Google Scholar]
- Tobin, LM, Healy, ME, English, K and Mahon, BP (2013). Human mesenchymal stem cells suppress donor CD4(+) T cell proliferation and reduce pathology in a humanized mouse model of acute graft-versus-host disease. Clin Exp Immunol 172: 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, S, Ding, Y, Tadokoro, CE, Olivares-Villagómez, D, Camps-Ramírez, M, Curotto de Lafaille, MA et al. (2005). Control of homeostatic proliferation by regulatory T cells. J Clin Invest 115: 3517–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L, Kim, HT, Nellore, A, Patsoukis, N, Petkova, V, McDonough, S et al. (2014). Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/β-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J 4: e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y, Desai, A, Yang, SY, Bae, KB, Antczak, MI, Fink, SP et al. (2015). Tissue Regeneration. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348: aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, EF, Tobin, LM, Carty, F, Mahon, BP and English, K (2015). Jagged-1 is required for the expansion of CD4(+) CD25(+) FoxP3(+) regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.