

Abstract
In the United States, estimates indicate there are between 250,000 and 400,000 individuals with Down syndrome (DS), and nearly all will develop Alzheimer's disease (AD) pathology starting in their 30s. With the current lifespan being 55 to 60 years, approximately 70% will develop dementia, and if their life expectancy continues to increase, the number of individuals developing AD will concomitantly increase. Pathogenic and mechanistic links between DS and Alzheimer's prompted the Alzheimer's Association to partner with the Linda Crnic Institute for Down Syndrome and the Global Down Syndrome Foundation at a workshop of AD and DS experts to discuss similarities and differences, challenges, and future directions for this field. The workshop articulated a set of research priorities: (1) target identification and drug development, (2) clinical and pathological staging, (3) cognitive assessment and clinical trials, and (4) partnerships and collaborations with the ultimate goal to deliver effective disease-modifying treatments.
Keywords: Down syndrome, Alzheimer's disease, Dementia, Animal models, Ts65Dn, Drug discovery, Neuroinflammation, Cognitive assessment, Amyloid precursor protein, Beta-amyloid, Tau, Clinical trials, Biomarkers, Trisomy 21, ADNI, DS-Connect, Neuroimaging, Workshop
1. Introduction
Down syndrome (DS) is the most common genetic condition in the United States, currently affecting approximately one in 700 live births. In addition to intellectual disability and characteristic physical traits such as small stature and facial dysmorphism, people with DS are at increased risk for cardiac defects and certain blood diseases, such as leukemia during childhood, autoimmune disorders and recurrent infections, and are at a markedly increased risk of Alzheimer's disease (AD). Because life expectancy for people with DS has more than doubled over the past 30 years, their increased risk for AD has become an important concern. Virtually all adults with DS develop neuropathology consistent with AD by their 40s and by age 55–60 years, their current lifespan, at least 70% will develop dementia [1–4]. Unfortunately, as they live longer their risk for developing Alzheimer's will increase for there are no treatments to slow, stop, or prevent AD. Thus, with prevalence varying between 250,000 and 400,000 in the United States and more than 5 million worldwide, DS represents the largest group of individuals with early onset AD (EOAD) (i.e. individual under the age of 65 years).
The high incidence of AD among adults with DS, combined with the ability to identify these individuals at or before birth, suggests that it may be possible to target this population for early intervention or prevention. With this in mind, the Alzheimer's Association, in partnership with the Linda Crnic Institute for Down Syndrome and the Global Down Syndrome Foundation, convened a workshop of AD and DS experts to explore the pathogenic and mechanistic links between the two disorders, discuss whether individuals with DS comprise an appropriate population for AD clinical trials, and strategize about how to advance collaborative research toward the parallel goals of providing effective treatment for individuals with DS and AD and expediting AD drug development. The workshop articulated a set of research priorities to move the field forward, including further exploration of shared pathophysiologic mechanisms, exploration of the course of pathogenesis long before the onset of dementia, identification of additional and possibly unique biomarkers that herald the onset of pathogenesis and/or that differentiate AD + DS from the other types of early or late onset AD, establishment of a national repository of AD + DS brains and other tissues, discovery and validation of novel therapeutic targets, and implementation of clinical trials designed to anticipate and prevent pathogenesis. Since this meeting, this group has gone on to establish a Professional Interest Area for DS-AD under the auspices of the International Society To Advance Alzheimer's Research and Treatments of the Alzheimer's Association (AA). Furthermore, the AA, Linda Crnic Institute for Down Syndrome, and the Global Down Syndrome Foundation have sponsored a grant program to support these efforts.
2. Pathologic and mechanistic links between DS and AD
Post-mortem studies of DS brains reveal both pathological similarities to and differences from AD brains. Amyloid-β (Aβ) protein is deposited into extracellular plaques and blood vessel walls in brain in both AD and DS, however, Aβ deposition occurs decades earlier in DS compared with non-DS individuals with the most common form of AD, late-onset Alzheimer's disease (LOAD) (i.e. individuals over the age of 65) [3–5]. In a study of 29 DS subjects between the ages of 3 and 73, Aβ deposition was seen as early as age 12 in individuals with DS and was universal by age 31 [6], beginning with diffuse Aβ42 deposits and progressing to compacted, fibrillar plaques often containing Aβ40, that was associated with neurodegeneration. Similar to AD, Aβ42 is always more abundant than Aβ40 in DS individuals, and N-terminally truncated Aβ is observed in both conditions [7]. Imaging studies, using the Pittsburgh compound B (PiB) [8], suggest that early accumulation of amyloid follows a fronto-striatal pattern similar to PS-1 mutation carriers, while a study from using [(18)F]FDDNP (2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl)amino]-2-naphthyl}-ethylidene)malononitrile) [9] reported early accumulation of AD pathology in the frontal cortex rather than in the temporal cortex, raising the possibility that there may be regional differences in the pathological progression between AD and DS + AD.
Neurofibrillary tangles (NFTs) accumulate later, with the hippocampus, entorhinal cortex, and neocortex among the most affected regions [10]. The distribution of plaques and tangles in DS brains is very similar to AD, although the density is greater in DS [11]. There may also be less robust progression of neuronal loss in DS compared with AD; however, the DS brain starts out with fewer neurons [12], suggesting a lower brain “reserve”, especially in the frontal and temporal cortices.
Other similarities between AD and DS neuropathology include increased build-up of plaque-associated proteins, microglial activation, astrogliosis, inflammation, and oxidative stress [1,13–17]. Although the pathogenesis proceeds over decades in DS, the staging of AD neuropathology is not as clearly established in DS as it is in AD.
The partial overlap in neuropathology of DS and AD suggests that some common pathogenic mechanisms may exist. More than 50 years ago, DS was shown to result from trisomy of chromosome 21, which contains hundreds of genes, including the gene for amyloid precursor protein (APP). Numerous lines of evidence point to the importance of APP in AD; increased copies of the APP gene is sufficient to cause EOAD with cerebral amyloid angiopathy (CAA) [18]. APP promoter polymorphisms that increase protein expression are also associated with EOAD [19] and may increase susceptibility to sporadic AD [20]. In one 78-year-old individual with DS but only partial trisomy 21 (missing APP and several other genes), there was no dementia or AD pathology at autopsy [21], supporting that APP plays a role in DS + AD.
APP overexpression is also linked to endocytic changes that are seen in both AD and DS. Endocytosis is critical for transmission and transport of neurotrophic factors in both the retrograde and anterograde direction. APP is processed by β- and γ-secretases in the endosomes, which are markedly enlarged in the AD brain early in the disease process. In DS, significant endosome enlargement is seen in the fetus at 28 weeks gestation, decades before the development of AD pathology, suggesting a failure of endosomal trafficking and/or recycling [22]. Endosomal dysfunction results in the disruption of many cellular processes essential for neuronal functioning, including protein turnover at synapses, local signaling, and subsequent effects on the cytoskeleton, protein synthesis, and retrograde signaling. In mouse models of AD and DS, APP overexpression resulted in endosomal dysfunction, increased nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF) signaling, especially at gamma-aminobutyric acid (GABA) synapses, failed retrograde transport of NGF and BDNF, and neurodegeneration of basal forebrain cholinergic neurons (BFCNs) [23].
BFCNs are important for memory function, and are shown to degenerate progressively in humans with DS, in idiopathic AD, and in the Ts65Dn mouse model of DS [24]. Ts65Dn mice are born with a normal density of BFCNs. However, like patients with DS, older Ts65Dn mice show a progressive decline in density of BFCNs in parallel with progressive loss of spatial reference and working memory [25]. Furthermore, reduced staining for the high-affinity NGF receptor, trkA, is observed in BFCNs not only from Ts65Dn mice, but also from individuals with DS and idiopathic AD, suggesting a biological mechanism for the cell loss involving this growth factor. Recently, Iulita et al [26] demonstrated parallel changes in neurotrophin maturation and cleavage mechanisms in the temporal and frontal cortices of both humans with DS + AD, and in Ts65Dn mice, providing potential mechanisms for BFCN degeneration and a target for development of pharmacotherapy for this population. Endosomal dysfunction is related to increased TrkB signaling, especially in inhibitory synapses, which may underlie the progression of AD [27]. Also notable, endosome enlargement and associated abnormal acceleration of endocytosis are mediated specifically by the β-cleaved carboxyl-terminal fragment (β-CTF) of APP, independently of Aβ [28].
Endocytosis is also critical for apoE function. The ApoE ε4 allele is the strongest genetic risk factor for LOAD, and in individuals with DS a doubling of amyloid burden is observed [10], and a significantly higher risk for AD and an earlier age of onset [29,30]. In patients with sporadic AD, the presence of the ApoE ε4 allele leads to significantly larger endosomes [22]. Interestingly, there is some evidence that the ApoE ε2 allele is protective against the development of dementia in individuals with DS [31].
Of course, APP is not the only gene triplicated in DS, for there are thought to be 635 genes according to the European Bioinformatics Institute listing; certainly other genes play important roles in producing the AD phenotype. One of the other genes triplicated in DS is the gene for dual-specificity tyrosine phosphorylated and regulated kinase 1a (DYRK1A), a protein that is overexpressed in both the DS and AD brain [32], with somewhat lower levels of overexpression in the AD brain. DYRK1A affects alternative splicing of tau by phosphorylating several splicing factors [33], priming tau for abnormal hyperphosphorylation by GSK-3 [34]. Overexpression of DYRK1A may also indirectly contribute to the early onset of neurofibrillary degeneration through phosphorylation of alternative splicing factors, leading to an imbalance between 3R-tau and 4R-tau. This imbalance results in a several-fold increase in the number of DYRK1A-positive and 3R-tau-positive NFTs in DS subjects in comparison to subjects with sporadic AD [35]. Moreover, the enhanced phosphorylation of APP at Thr 688 by overexpressed DYRK1A [36] facilitates amyloidogenic APP cleavage, which elevates Aβ40 and 42 levels resulting in possibly higher level of toxic Aβ oligomers that contributes to the earlier onset of Aβ pathology [37]. Hence, the inhibition of DYRK1A activity can be explored as a potential therapeutic approach to treat the developmental defects and the early onset Alzheimer-type pathology in DS.
Another gene encoded on human chromosome 21 (HSA21) is synaptojanin 1 (SYNJ1), a lipid phosphatase that plays an important role in synaptic transmission. SYNJ1 protein negatively regulates the levels of phosphatidylinositol-4,5-biphosphate [PtdIns(4,5)P(2)], a signaling phospholipid involved in many cellular processes. Studies in DS transgenic mice suggest that overexpression of SYNJ1 and the resulting perturbation of PtdIns(4,5) P(2) metabolism is associated with cognitive impairment [38]. Furthermore, trisomy for SYNJ1 in DS is functionally linked to the enlargement of early endosomes [39]. Conversely, SYNJ1 haploinsufficiency appears to be protective against the synaptotoxic action of Aβ [40], indicating that SYNJ1 protein levels may be relevant both in DS and AD.
Neuroinflammation has also been linked to cognitive deficits, and many inflammatory genes found on chromosome 21 may contribute to neurodegeneration in DS [16]. For example, the astrocyte-derived growth factor S100β has been implicated in the formation of neuritic plaques in AD. In DS, elevated numbers of S100β positive astrocytes are present throughout life and are associated with neuritic dystrophy [41]. Other inflammatory molecules, including ApoE and alpha1-antichymotrypsin, have also been shown to be upregulated in astrocytes and microglia in both AD and DS and are found in amyloid plaques [42]. Interestingly, a recent study demonstrated alterations in the transcriptome between a pair of monozygotic twins, where only one twin had DS [43]. The investigators found widely occurring gene expression dysregulation domains (GEDDs), suggesting that nuclear components of trisomic cells undergo modifications of the chromatin environment that can influence the overall transcriptome and give rise to specific DS-related phenotypes. The investigators also discovered that similar GEDDs were observed along the mouse chromosomes in Ts65Dn mice that were syntenic to HSA21, suggesting that this mouse model may, indeed, be an appropriate mouse model for DS at least in terms of genome wide alterations. Genetic alterations observed in the trisomic twin were related to an inflammatory response and cytokine-cytokine receptor interactions, confirming that neuroinflammation is an important pathological trait in DS brains. Further support is provided by DS mouse models exhibiting age-related and progressive activation of both microglial cells and astrocytes in the hippocampus and other limbic areas, and BFCN loss, and memory loss that can be prevented by treatment with the tetracycline derivative minocycline [44]. Finally, complement proteins, associated with innate immunity, have also been observed in association with amyloid plaques, dystrophic neuritis, and NFTs in AD and DS [45], supporting an important role of neuroinflammation genes in DS and AD pathophysiology.
Trisomy of chromosome 21 in DS also leads to other cellular changes. Cells may respond to changes in gene dose by altering DNA methylation of genes not only on chromosome 21, but on other genes throughout the genome. Methylation can alternatively silence or activate genes, and may be manipulated by drugs and nutrients. Studies of T-lymphocytes from adults with DS showed a characteristic pattern of methylation abnormalities in genes involved in the immune response [46]. In further studies of other tissues, particularly brain tissue from individuals with DS, researchers have begun to identify changes in methylation in neurons and glia that appear early in development. More research will be needed to determine the consequences of these changes and whether they can be modified therapeutically.
There is some evidence that trisomy may contribute to other forms of sporadic and familial AD. Trisomy 21 mosaicism has been found in fibroblasts from people with all forms of AD, both sporadic and familial [47]. In addition, hyperploidy has been demonstrated in neurons of AD brains at mild stages, before neuronal loss, and these neurons appear to be targeted for cell death as the disease progresses [48]. Studies in human cells and Xenopus egg extracts suggest that excess Aβ in hyperploid cells disrupts mitotic spindle formation by inhibiting kinesins (mitotic motor proteins), including Eg5, thus affecting neurogenesis and neuroplasticity [49]. In contrast, enhancement of neurogenesis and neuronal plasticity by treatment with a ciliary neurotrophic factor peptide was found to rescue cognitive impairment in Ts65Dn mice [50]. This suggests a mechanistic link between trisomy 21 and neurodegenerative disease [49], but the specifics of how small proportions of trisomic cells might contribute to brain-wide AD neuropathophysiology remain unclear.
3. DS animal models to understand AD
Mouse models are useful for studying disease processes and identifying potential interventions in genetically well-defined populations that can be manipulated in various ways. Of the several DS mouse models that have been generated, the most widely used is the Ts65Dn model for studying DS, which is trisomic for 88 of 161 of the ortholog protein coding genes on human chromosome 21 [51]. Ts65Dn mice are characterized by a progressive loss of working memory, cholinergic deficit in the basal forebrain, and neuroinflammation; changes observed in AD. Additionally, there is dysregulation of trophic factors and an altered plasmin pathway for neurotrophin cleavage [52,53]. Importantly, there is a striking correspondence between the developmental impact of trisomy in this mouse and humans with DS, resulting in similar phenotypes such as short stature, flat facies, flat nasal bridge, and a small cerebellum [54].
However, two facts regarding the Ts65Dn genetics must be kept in mind in evaluating it as a model for AD in DS. First, the Ts65Dn lacks trisomy of a number of HSA21 genes with functional features that are of compelling relevance to development of AD. These include S100 B (discussed previously), the Small Ubiquitin-like Modifier, SUMO3 and the serine protease inhibitor, CSTB, and other genes [55]. Second, the Ts65Dn is also trisomic for a small centromeric segment of mouse chromosome 17 that is not orthologous to HSA21. It has recently been shown that this segment is more gene rich than previously assumed, and indeed includes 50 protein coding genes [56], among them paralogs of some HSA21 genes, including SYNJ2 (see previously for paralog SYNJ1) and TIAM2, plus several Dynein light chain genes whose increased dosage could influence endosomal transport. Thus, the genetic basis for the phenotype of the Ts65Dn is more complex and may not be identical to DS.
The functions of orthologs of a few HSA21 genes have been studied individually in model organisms including mouse, fruit fly, Caenorhabditis elegans, and yeast. Typically, such studies suggest these orthologs regulate similar developmental pathways [57]. In mouse models overexpressing single HSA21 genes, hippocampal function is often affected, resulting in specific behavioral abnormalities and sometimes in excessive inhibitory input to the hippocampus, resulting in reduced long-term potentiation. In these studies, treatment with drugs that antagonize GABAergic transmission normalized these physiologic and behavioral outcomes [52]. In particular, studies focusing on the hippocampus indicate that down-regulation of GABA-A interneurons holds promise as a therapeutic agent. However, to date many other therapeutic interventions have also proved effective in the Ts65Dn, suggesting that other important mechanisms need to be explored [58].
In addition to the genetic concerns for replicating human trisomy in mice is the concern for the lack of plaques and tangles formation in DS mouse models to replicate the hallmarks of AD. However this may be due to the three amino acid difference in the mouse vs. the human β-amyloid sequence that increases the ability of the protein to aggregate in humans. Additionally, evidence has suggested that β-amyloid aggregation may be a trigger for tau aggregation, limiting the ability to develop tau abnormalities in mice. Therefore the DS models have interesting potential for studying AD by introducing human β-amyloid sequences into DS mouse models, allowing different trisomy models and their corresponding gene expression patterns to reflect on how they affect the pathophysiology of plaque and tangle development. Conversely, the lack of Aβ aggregation may allow the evaluation of other processes contributing to cognitive impairment and brain neurodegeneration independent of Aβ production or aggregation.
A problem with the current AD mouse models is that they do not reproduce the neuronal cell loss observed in humans; in fact, there is little to no neuronal loss. Of interest is the neurodegeneration in the Ts65Dn mouse, which may be due to increased APP production or one of its nonamyloid cleavage products, impaired NGF transport and/or cleavage to its mature form, gliosis in the hippocampus, impaired mitochondrial function, oxidative stress, neuroinflammation, calcium dysregulation or some combination of these changes [55]. The Ts65Dn model or other DS mouse models could therefore be useful to test whether intervening in these pathways, independent of plaques and tangles, is therapeutic to generally address neuronal cell loss. Although these animals do not produce plaques and tangles, these same cell death mechanisms may play an important role in cell loss downstream from these pathological changes. These similarities and differences in the DS and AD animal models have yet to be explored and may point to important aspects of underlying biochemical and structural changes that may drive the development of AD.
DS mouse models have provided a powerful tool for translational research in DS, and these models may also prove similarly useful in evaluating AD treatments, although the differences between mouse models and humans, as discussed previously, raise some concerns about relevance. Nonetheless, these models are enormous untapped areas of research with implications for drug development in both the DS and AD fields, overlapping in areas such as modifying APP levels and its cleavage products, modifiers of the endosomal/lysosomal system, and the interaction between basic metabolic pathways, such as those involving insulin, mTOR, immune signaling, amyloid and growth factor pathways. Along these lines are the several novel mouse models for DS that have been developed (for review, see Kleschevnikov et al. [59], Rueda et al. [60], Figure 1). It will be important in future studies to correlate phenotypes in the novel models with those already described for Ts65Dn mice, to confirm that a higher inclusion of syntenic genes leads to a more accurate behavioral and pathological model for the human condition. A recently developed mouse model [61] carries duplications of many of the gene regions corresponding to human chromosome 21. Initial studies suggest that this model exhibits additional memory impairment and pathological brain development, suggesting that it may serve as a more appropriate model; however, no aging studies or intervention studies have been carried out to date. The workshop participants identified a need to study the aging process in all DS mouse models, with particular concern for different background strains, to determine the most appropriate models to reflect AD progression in humans.
Fig. 1.
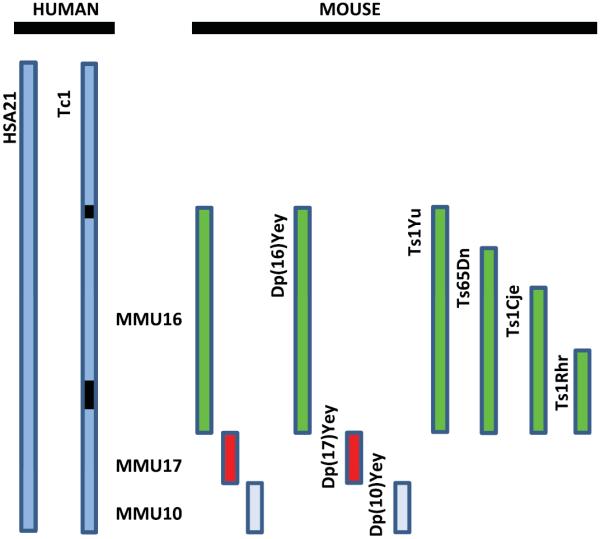
Mouse models of Down syndrome (DS). The diagram shows a representation of human chromosome 21, HSA21, and separate mouse chromosomes (10, 16, and 17) that are orthologous to HSA21that have been used to develop mouse models of DS [59,60]. The mouse models diagrammed here represent the triplication of different regions of the various murine chromosomes, except the Tc1 mouse model, that carries a transchromosome with the human HAS21 genes (black boxes are two internal deletions). The most commonly used model is the Ts65DN which represents the triplication of the mouse chromosome (MMU) 16 [79]. Ts1Cje, Ts1Yu and Ts1Rhr are segmental duplications or translocations of different lengths of MMU16 [60].
4. Diagnosing AD in individuals with Down syndrome
AD and DS follow a similar trajectory of decline; however, diagnosis of dementia in individuals with DS presents challenges, the most obvious related to the baseline functioning of individuals with substantial pre-existing intellectual disabilities that vary in type and severity within this population. Universal norms used in assessing the activities of daily living (ADLs) in AD are difficult when assessing individuals with DS, for some tasks may never have been performed previously, such as handling their own finances or driving a car. Thus, changes in ADLs function in DS need to be compared with the individual's baseline abilities as opposed to current standardized tests. Cognitive assessments must also take into account variability in baseline performance across cognitive domains, because some aspects of cognition are more affected than others. For example, a DS cognitive phenotype is characterized by relative weaknesses in morpho-syntax, visual short-term memory, and explicit long-term memory compared with visual-spatial short term memory, associative learning, and implicit long-term memory [62].
The Arizona Cognitive Test Battery (ACTB) was developed to provide a focused instrument for assessing possible improvements in cognition in clinical trials [63]. While this battery was originally designed to tap into aspects of cognition affected by intellectual disability and other cognitive impairments, it contains assessments of cognitive domains expected to be affected in AD. However, the ACTB is not widely used at this point. The challenge of assessing cognition in individuals with DS is complex, especially as we try to assess individuals very early in the disease process. Multiple groups are developing empirically validated methods for assessing AD in adults with DS, which should be done in conjunction with the current initiatives to develop more sensitive tests to address the new diagnostic guidelines for prodromal or earlier stages of AD (http://www.alz.org/research/diagnostic_criteria/). Universal adoption of cognitive assessments would be helpful in developing national or international DS/AD programs, similar to the successful Alzheimer's Disease Neuroimaging Initiative (ADNI) that is providing valuable insights into the development of AD in the general population. ADNI and its global partners, Worldwide ADNI (http://www.alz.org/research/funding/partnerships/WW-ADNI_overview.asp), have together gathered and shared substantial biomarker data aimed at delineating the pathological progression of AD.
Biomarkers have the potential to aid not only in diagnosis of AD in individuals with DS, but more importantly, biomarkers track the progression of the disease in its earliest stages and the effectiveness of different treatments. However, biomarker data are sparse in the DS + AD field, but there is data from amyloid PET imaging suggesting the deposition of amyloid earlier in individuals with DS than late-onset AD [8,64,65], and with tau PET imaging on the horizon this will help in the identification of individuals at risk. Also, linking cognitive studies and functional imaging may allow prediction of cognitive impairment and provide a platform for early intervention [66]. Other techniques may be of value, such as high resolution electroencephalography to determine epileptic activity, because there appears to be a subset of individuals with DS and/or AD that have seizures [66]. Additionally, structural and functional magnetic resonance imaging in DS population, as in the general population, may reflect early AD neuropathophysiology [67,68].
Cerebrospinal fluid (CSF) biomarkers are thought by many investigators to be necessary to detect brain pathology; however they may be difficult to evaluate in DS individuals due to difficulties in performing lumbar punctures for research with “no prospect of direct benefit”, but with new treatments this could possibly change. Alternatively, the possible availability of plasma biomarkers, including telo-mere length as a marker [69], plasma Aβ [70], or new platforms under development such as the SomaLogic technology [71], may greatly increase acceptance and open the doors to dramatically increased study participation and numbers. A combination of these diagnostic tools may be extremely valuable in early assessment for individuals at risk for developing AD, both in individuals with DS and the general population.
A public-private partnership was recently launched by the Alzheimer's Disease Cooperative Study and Michael Rafii, study director, as a means to establish biomarkers of dementia in DS that would support clinical trials of AD preventive therapies [72]. The clinical study, named the Down Syndrome Biomarker Initiative (DSBI), will mirror the efforts of the ADNI, which was established in 2003 to validate imaging and blood/CSF biomarkers for AD treatment trials as a means of speeding drug development [73]. Like ADNI, DSBI is intended to establish an infrastructure that could eventually be used in the development of clinical trials for secondary prevention studies in individuals with DS and AD. Programs and databases to identify individuals and ways to share information is growing for both communities, as seen in the newly created DS-Connect (https://dsconnect. nih.gov) and the Global Alzheimer's Association Interactive Network (GAAIN) (www.gaain.org).
5. AD clinical trials in individuals with DS
The extremely high prevalence of AD in people with DS could provide a unique opportunity to study how to slow, stop or prevent Alzheimer's in these individuals. Given the complexity of AD in the setting of DS, a single drug may not be completely effective, but novel therapeutics under development for AD and other neurodegenerative diseases may be of interest in the AD 1 DS population, particularly when they target mechanisms that appear to be important in DS. For example, almost all individuals with DS and AD, and about one-third of cognitively normal elderly subjects, have Cerebral amyloid angiopathy (CAA). CAA is an independent risk factor for dementia and may need to be factored in regarding treatment options or for stratifying populations in clinical trials. Certainly, this has been a newly emerging aspect of AD where autopsied brains are showing mixed pathologies.
An added complication is the prevalence of Attention Deficit Hyperactivity Disorder (ADHD) in DS individuals, which is as high as 43% [74]. ADHD may, in part, be due to altered norepinephrine (NE) levels because DS individuals exhibit an early and progressive loss of neurons supplying NE, locus coeruleus neurons, also seen in AD. Thus, a novel treatment approach may be using NE enhancing drugs, such as Atomoxetine or Droxidopa [75]. Because NE signaling has been shown to regulate growth factor expression, inflammatory pathways, and amyloid processing, treatment with NE enhancing drugs may be neuroprotective and prevent or slow further neuropathology progression [76]. Other novel treatment options considered included stimulating innate immune system with a Toll-like receptor 9 ligand [77], inhibiting the interaction of apoE with Aβ, which may prevent or slow oligomerization and polymerization of Aβ, thus halting or reducing the pathogenic cascade [42]. It is unclear at this point whether treatment options for DS individuals will be the same as the general population or possibly require additional treatments in combination.
Currently, there are approximately 12 DS trials related to AD in development, recruitment or active/completed. These include anti-amyloid antibodies, myoinositol, epigallocatechin gallate, nicotine, memantine, and vitamin E. The DS efforts have been recently advanced by the NIH Down Syndrome Registry called DS-Connect™ which links individuals with DS to resources and research studies. Clearly the DS and the Alzheimer's communities will benefit from the crosstalk in these clinical trials and the advanced trials being conducted in the AD field, including the Dominantly Inherited Alzheimer's Network Trial Unit (DIAN-TU), the Anti-Amyloid Asymptomatic Alzheimer's Trial (A4), the TOMMORROW trial, and the two trials under Alzheimer's Prevention Initiative [78].
A final important note with regard to clinical trials in individuals with DS: Institutional Review Board (IRB) approval and enrollment can be a barrier, given that adults with DS typically lack the capacity to balance potential benefits against potential risks. Therefore, IRBs may require increased evidence of safety and tolerability. One strategy for gaining IRB approval is to contend that people with DS constitute a health disparity population at exceptionally high risk, and these individuals should have access to the latest interventions and clinical trials. Programs such as these could greatly benefit from a national IRB protocol as proven to be successful in cancer trials.
6. Future directions
As care for individuals with DS has significantly improved, including medical treatments, educational policies, societal attitudes, better support, and advanced services, so has their lifespan which has unfortunately put them at greater risk for developing AD. Even though this risk has been recognized, awareness is still low, and only a relatively small portion of the DS + AD models have been explored and adequately tested. Expanded prospective studies in the DS population are needed to understand the biological processes that drive the disease. Moreover, a strong, unified effort to deconstruct the problems and identify solutions would be of tremendous benefit, not only to the AD field as a whole, but also to the many individuals and families caring for these individuals afflicted with the disease. Because of the high incidence of AD in individuals with DS, and because DS + AD represents a fairly large group of individuals globally, there is a great need to incorporate individuals with DS in the Alzheimer's biobanks. Few brain banks or Alzheimer Centers have included these individuals in their study, and the workshop participants recognized this as an important area of improvement. Clinical trials of Alzheimer's treatments in individuals with DS will be extremely insightful. As an outcome of this DS/AD workshop, meeting participants recommended a number of research priorities for moving the field forward:
Target Identification and Drug Development
Develop mouse models that use the combination of the DS models with the AD models to investigate mechanism(s) of neuronal loss.
Develop DS mouse models to express the human amino acid sequence of Aβ.
Discover and validate novel therapeutic targets and promote clinical trials that may be mutually beneficial for both the DS and AD affected population.
Study the role of inflammation and innate immunity in DS as it relates to the development of AD.
Explore the links between pathogenesis of DS and AD, including the role of endosomal enlargement and dysfunction.
Test DYRK1A inhibition as a factor contributing both to developmental abnormalities and early onset Alzheimer-type pathology in a trisomy 21 background.
Explore further the links between NGF and brain-derived neurotrophic factor signaling and neurodegeneration of basal forebrain cholinergic neurons.
Investigate the role of the synaptojanin 1 (SYNJ1) protein and lipid phosphatase signaling in causing abnormal synaptic transmission and endosome enlargement.
Clinical and Pathological Staging
Develop better pathological staging in asymptomatic and prodromal individuals with DS.
Define early biomarkers in the brain and blood that herald the onset of pathogenesis, e.g. Aβ42-specific ligands, fluorodeoxyglucose-positron emission tomography (FGD-PET), serum growth factors, inflammatory/oxidative stress markers, CSF biomarkers, or novel biomarkers such as telomere length and lens pathology.
Characterize the preclinical course of AD in DS adults in longitudinal natural history studies (i.e.,DSBI) using biomarkers that would be sensitive to gender differences, traumatic brain injury, ApoE status, and seizure status.
Compare the trajectory of disease between individuals with DS and individuals with autosomal dominant mutations that predispose to AD (i.e., the Dominantly Inherited Alzheimer's Network observational or trial unit (DIAN or DIAN-TU)).
Examine whether genetic variants that increase the risk of AD influence the onset of dementia in DS.
Cognitive Assessment and Clinical Trials
Standardize a core battery of cognitive and functional tests that are applicable to individuals across the broadest possible spectrum of impairment and intellectual disabilities.
Standardize biomarker and neuroimaging protocols for DS trials and where possible to align with ongoing AD trials through the Alzheimer's Disease Centers and/or programs like Alzheimer's Disease Neuroimaging Initiative (ADNI).
Analyze how prevention trials in AD may be adapted to individuals with DS or how prevention trials in DS might apply to a non-DS population at risk for developing AD.
Examine the role of life style factors, including diet, and exercise, in the progression of dementia and AD pathology in DS.
Establish a national repository for DS-AD brains and other tissues and cells, and a database for all specimens that have been biobanked in different locations.
Partnership and Collaborations
Partner with colleagues around the world to acquire and analyze CSF samples from people with DS.
Facilitate communication and collaboration in the field by developing a database of all ongoing research, similar to National Alzheimer's Coordinating Center (NACC), ADNI or World Wide-Alzheimer's Disease Neuroimaging Initiative in the Alzheimer's field.
Form a roundtable comprised of representatives of academia, industry, governmental and regulatory agencies, and funding and advocacy organizations to explore, brainstorm, and develop a plan for moving forward aggressively. Encourage the participation of a broader array of industry representatives from not only the pharmaceutical industry but health care and food/nutrition companies.
RESEARCH IN CONTEXT.
Systematic review: A workshop of world experts on Alzheimer's disease (AD) and Down Syndrome (DS) was convened to discuss their collective knowledge and expert opinions concerning similarities and differences between idiopathic AD and that observed in individuals with DS developing AD. This discussion was put in context of the current literature.
Interpretation: The workshop articulated specific challenges and promising areas of research that included: (i) Pathologic and mechanistic links between DS and AD, (ii) DS animal models for understanding AD, (iii) Diagnosing AD in individuals with Down syndrome, and (iv) Conducting AD clinical trials in individuals with DS.
Future directions: The workshop articulated a set of research priorities: (i) Target identification and drug development, (ii) Clinical and pathological staging, (iii) Cognitive assessment and clinical trials, and (iv) Partnerships and collaborations with the ultimate goal of delivering effective disease-modifying treatments.
Acknowledgments
We thank Lisa Bain for helping to prepare this manuscript.
References
- [1].Wilcock DM, Griffin WS. Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lai F, Williams RS. A prospective study of Alzheimer's disease in Down syndrome. Arch Neurol. 1989;46:849–53. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- [3].Wisniewski K, Wisniewski H, Wen G. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol. 1985;17:278–82. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- [4].Mann DM. The pathological association between Down syndrome and Alzheimer disease. Mech Ageing Dev. 1988;43:99–136. doi: 10.1016/0047-6374(88)90041-3. [DOI] [PubMed] [Google Scholar]
- [5].Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, et al. Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease. N Engl J Med. 1989;320:1446–52. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- [6].Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- [7].Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, et al. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol. 2013;183:369–81. doi: 10.1016/j.ajpath.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, et al. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nelson LD, Siddarth P, Kepe V, Scheibel KE, Huang SC, Barrio JR, et al. Positron emission tomography of brain beta-amyloid and tau levels in adults with Down syndrome. Arch Neurol. 2011;68:768–74. doi: 10.1001/archneurol.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hyman BT, West HL, Rebeck GW, Lai F, Mann DM. Neuropathological changes in Down's syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol. 1995;52:373–8. doi: 10.1001/archneur.1995.00540280059019. [DOI] [PubMed] [Google Scholar]
- [11].Hof PR, Bouras C, Perl DP, Sparks L, Mehta N, Morrison JH. Age-related distribution of neuropathological changes in the cerebral cortex of patients with Down's syndrome. Arch Neurol. 1995;52:379–91. doi: 10.1001/archneur.1995.00540280065020. [DOI] [PubMed] [Google Scholar]
- [12].Mann DMA, Yates PO, Marcyniuk B, Ravindra CR. Loss of neurones from cortical and subcortical areas in Down's syndrome patients at middle age. J Neurol Sci. 1987;80:79–89. doi: 10.1016/0022-510x(87)90223-1. [DOI] [PubMed] [Google Scholar]
- [13].Lott IT, Head E. Down syndrome and Alzheimer's disease: a link between development and aging. Ment Retard Dev Disabil Res Rev. 2001;7:172–8. doi: 10.1002/mrdd.1025. [DOI] [PubMed] [Google Scholar]
- [14].Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13:237–46. doi: 10.1002/mrdd.20163. [DOI] [PubMed] [Google Scholar]
- [15].Zigman WB, Devenny DA, Krinsky-McHale SJ, Jenkins EC, Urv TK, Wegiel J, et al. Alzheimer's disease in adults with down syndrome. Int Rev Res Ment Retard. 2008;36:103–45. doi: 10.1016/S0074-7750(08)00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wilcock DM. Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer's disease. Curr Gerontol Geriatr Res. 2012;2012:170276. doi: 10.1155/2012/170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Head E, Silverman W, Patterson D, Lott IT. Aging and Down syndrome. Curr Gerontol Geriatr Res. 2012;2012:412536. doi: 10.1155/2012/412536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- [19].Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, et al. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78:936–46. doi: 10.1086/504044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lv H, Jia L, Jia J. Promoter polymorphisms which modulate APP expression may increase susceptibility to Alzheimer's disease. Neurobiol Aging. 2008;29:194–202. doi: 10.1016/j.neurobiolaging.2006.10.001. [DOI] [PubMed] [Google Scholar]
- [21].Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, et al. Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann Neurol. 1998;43:380–3. doi: 10.1002/ana.410430316. [DOI] [PubMed] [Google Scholar]
- [22].Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–86. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- [24].Lockrow JP, Fortress AM, Granholm AC. Age-related neurodegeneration and memory loss in Down syndrome. Curr Gerontol Geriatr Res. 2012;2012:463909. doi: 10.1155/2012/463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Granholm AC, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down's syndrome. Exp Neurol. 2000;161:647–63. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- [26].Iulita MF, Do Carmo S, Ower AK, Fortress AM, Aguilar LF, Hanna M, et al. Nerve growth factor metabolic dysfunction in Down's syndrome brains. Brain. 2014;137:860–72. doi: 10.1093/brain/awt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010;68:885–93. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, et al. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–5. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Prasher VP, Sajith SG, Rees SD, Patel A, Tewari S, Schupf N, et al. Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer's disease and mortality in persons with Down syndrome. Int J Geriatr Psychiatry. 2008;23:1134–40. doi: 10.1002/gps.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schupf N, Kapell D, Lee JH, Zigman W, Canto B, Tycko B, et al. Onset of dementia is associated with apolipoprotein E epsilon4 in Down's syndrome. Ann Neurol. 1996;40:799–801. doi: 10.1002/ana.410400518. [DOI] [PubMed] [Google Scholar]
- [31].Royston MC, Mann D, Pickering-Brown S, Owen F, Perry R, Raghavan R, et al. Apolipoprotein E epsilon 2 allele promotes longevity and protects patients with Down's syndrome from dementia. Neuroreport. 1994;5:2583–5. doi: 10.1097/00001756-199412000-00044. [DOI] [PubMed] [Google Scholar]
- [32].Dowjat WK, Adayev T, Kuchna I, Nowicki K, Palminiello S, Hwang YW, et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Qian W, Liang H, Shi J, Jin N, Grundke-Iqbal I, Iqbal K, et al. Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011;39:6161–71. doi: 10.1093/nar/gkr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–33. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wegiel J, Kaczmarski W, Barua M, Kuchna I, Nowicki K, Wang KC, et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J Neuropathol Exp Neurol. 2011;70:36–50. doi: 10.1097/NEN.0b013e318202bfa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. J Neurochem. 2008;104:1333–44. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- [37].Becker W, Soppa U, Tejedor FJ. DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS Neurol Disord Drug Targets. 2014;13:26–33. doi: 10.2174/18715273113126660186. [DOI] [PubMed] [Google Scholar]
- [38].Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, et al. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc Natl Acad Sci U S A. 2008;105:9415–20. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, et al. Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum Mol Genet. 2012;21:3156–72. doi: 10.1093/hmg/dds142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Berman DE, Dall'Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, et al. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci. 2008;11:547–54. doi: 10.1038/nn.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Griffin WS, Sheng JG, McKenzie JE, Royston MC, Gentleman SM, Brumback RA, et al. Life-long overexpression of S100beta in Down's syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998;19:401–5. doi: 10.1016/s0197-4580(98)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Potter H, Wisniewski T. Apolipoprotein e: essential catalyst of the Alzheimer amyloid cascade. Int J Alzheimers Dis. 2012;2012:489428. doi: 10.1155/2012/489428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, et al. Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature. 2014;508:345–50. doi: 10.1038/nature13200. [DOI] [PubMed] [Google Scholar]
- [44].Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann Neurol. 2004;56:675–88. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- [45].Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, et al. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–99. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kerkel K, Schupf N, Hatta K, Pang D, Salas M, Kratz A, et al. Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet. 2010;6:e1001212. doi: 10.1371/journal.pgen.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis. 1999;6:167–79. doi: 10.1006/nbdi.1999.0236. [DOI] [PubMed] [Google Scholar]
- [48].Arendt T, Bruckner MK, Mosch B, Losche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010;177:15–20. doi: 10.2353/ajpath.2010.090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Borysov SI, Granic A, Padmanabhan J, Walczak CE, Potter H. Alzheimer Abeta disrupts the mitotic spindle and directly inhibits mitotic microtubule motors. Cell Cycle. 2011;10:1397–410. doi: 10.4161/cc.10.9.15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Blanchard J, Bolognin S, Chohan MO, Rabe A, Iqbal K, Grundke-Iqbal I. Rescue of synaptic failure and alleviation of learning and memory impairments in a trisomic mouse model of down syndrome. J Neuropathol Exp Neurol. 2011;70:1070–9. doi: 10.1097/NEN.0b013e318236e9ad. [DOI] [PubMed] [Google Scholar]
- [51].Sturgeon X, Gardiner KJ. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mamm Genome. 2011;22:261–71. doi: 10.1007/s00335-011-9321-y. [DOI] [PubMed] [Google Scholar]
- [52].Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504:329–45. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- [53].Lockrow J, Boger H, Gerhardt G, Aston-Jones G, Bachman D, Granholm AC. A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. J Alzheimers Dis. 2011;23:471–89. doi: 10.3233/JAD-2010-101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial mal-development in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217:137–45. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- [55].Sturgeon X, Le T, Ahmed MM, Gardiner KJ. Pathways to cognitive deficits in Down syndrome. Prog Brain Res. 2012;197:73–100. doi: 10.1016/B978-0-444-54299-1.00005-4. [DOI] [PubMed] [Google Scholar]
- [56].Ahmed MM, Dhanasekaran AR, Tong S, Wiseman FK, Fisher EM, Tybulewicz VL, et al. Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum Mol Genet. 2013;22:1709–24. doi: 10.1093/hmg/ddt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gardiner K, Costa AC. The proteins of human chromosome 21. Am J Med Genet C Semin Med Genet. 2006;142c:196–205. doi: 10.1002/ajmg.c.30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gardiner KJ. Molecular basis of pharmacotherapies for cognition in Down syndrome. Trends Pharmacol Sci. 2010;31:66–73. doi: 10.1016/j.tips.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kleschevnikov AM, Belichenko PV, Salehi A, Wu C. Discoveries in Down syndrome: moving basic science to clinical care. Prog Brain Res. 2012;197:199–221. doi: 10.1016/B978-0-444-54299-1.00010-8. [DOI] [PubMed] [Google Scholar]
- [60].Rueda N, Florez J, Martinez-Cue C. Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012;2012:584071. doi: 10.1155/2012/584071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19:2780–91. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol. 2010;9:623–33. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- [63].Edgin JO, Mason GM, Allman MJ, Capone GT, Deleon I, Maslen C, et al. Development and validation of the Arizona Cognitive Test Battery for Down syndrome. J Neurodev Disord. 2010;2:149–64. doi: 10.1007/s11689-010-9054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Landt J, D'Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, et al. Using positron emission tomography and Carbon 11-labeled Pittsburgh Compound B to image Brain Fibrillar beta-amyloid in adults with down syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68:890–6. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- [65].Sabbagh MN, Fleisher A, Chen K, Rogers J, Berk C, Reiman E, et al. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-beta in a patient with Down syndrome and Alzheimer disease. Arch Neurol. 2011;68:1461–6. doi: 10.1001/archneurol.2011.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–21. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Teipel SJ, Hampel H. Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer's disease. Behav Genet. 2006;36:405–15. doi: 10.1007/s10519-006-9047-x. [DOI] [PubMed] [Google Scholar]
- [68].Mullins D, Daly E, Simmons A, Beacher F, Foy CM, Lovestone S, et al. Dementia in Down's syndrome: an MRI comparison with Alzheimer's disease in the general population. J Neurodev Disord. 2013;5:19. doi: 10.1186/1866-1955-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jenkins EC, Ye L, Velinov M, Krinsky-McHale SJ, Zigman WB, Schupf N, et al. Mild cognitive impairment identified in older individuals with Down syndrome by reduced telomere signal numbers and shorter telomeres measured in microns. Am J Med Genet B Neuropsychiatr Genet. 2012;159b:598–604. doi: 10.1002/ajmg.b.32066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schupf N, Zigman WB, Tang MX, Pang D, Mayeux R, Mehta P, et al. Change in plasma Ass peptides and onset of dementia in adults with Down syndrome. Neurology. 2010;75:1639–44. doi: 10.1212/WNL.0b013e3181fb448b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gold L, Janjic N, Jarvis T, Schneider D, Walker JJ, Wilcox SK, et al. Aptamers and the RNAworld, past and present. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ness S, Rafii M, Aisen P, Krams M, Silverman W, Manji H. Down's syndrome and Alzheimer's disease: towards secondary prevention. Nat Rev Drug Discov. 2012;11:655–6. doi: 10.1038/nrd3822. [DOI] [PubMed] [Google Scholar]
- [73].Carrillo MC, Bain LJ, Frisoni GB, Weiner MW. Worldwide Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2012;8:337–42. doi: 10.1016/j.jalz.2012.04.007. [DOI] [PubMed] [Google Scholar]
- [74].Ekstein S, Glick B, Weill M, Kay B, Berger I. Down syndrome and attention-deficit/hyperactivity disorder (ADHD) J Child Neurol. 2011;26:1290–5. doi: 10.1177/0883073811405201. [DOI] [PubMed] [Google Scholar]
- [75].Bari A, Robbins TW. Inhibition and impulsivity: behavioral and neural basis of response control. Prog Neurobiol. 2013;108:44–79. doi: 10.1016/j.pneurobio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- [76].Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, et al. Ascending monoaminergic systems alterations in Alzheimer's disease. Translating basic science into clinical care. Neurosci Biobehav Rev. 2013;37:1363–79. doi: 10.1016/j.neubiorev.2013.05.008. [DOI] [PubMed] [Google Scholar]
- [77].Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer's disease-related pathology. J Neurosci. 2009;29:1846–54. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Carrillo MC, Brashear HR, Logovinsky V, Ryan JM, Feldman HH, Siemers ER, et al. Can we prevent Alzheimer's disease? Secondary “prevention” trials in Alzheimer's disease. Alzheimers Dement. 2013;9:123–1311. doi: 10.1016/j.jalz.2012.12.004. [DOI] [PubMed] [Google Scholar]
- [79].Reinholdt LG, Ding Y, Gilbert GJ, Czechanski A, Solzak JP, Roper RJ, et al. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome. 2011;22:685–91. doi: 10.1007/s00335-011-9357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]