

Abstract
The FeMoco of nitrogenase is an iron–sulfur cluster with exceptional bond-reducing abilities. ENDOR studies have suggested that E4, the state that binds and reduces N2, contains bridging hydrides as part of the active-site iron-sulfide cluster. However, there are no examples of any isolable iron-sulfide cluster with a hydride, which would test the feasibility of such a species. Here, we describe a diiron sulfide hydride complex that is prepared using a mild method involving C–S cleavage of added thiolate. Its reactions with nitrogenase substrates show that the hydride can act as a base or nucleophile and that reduction can cause the iron atoms to bind N2. These results add experimental support to hydride-based pathways for nitrogenase.
In natural nitrogen fixation, nitrogenase enzymes catalyze the conversion of N2 into ammonia (NH3). The most active nitrogenases utilize an active site cofactor (FeMoco) that is a cluster of iron, molybdenum, carbon, and sulfur atoms.1 The mechanism by which FeMoco converts N2 into NH3 is a topic of intense study. Though chemists have speculated about hydrides in nitrogenase for decades,2 it is only recently that spectroscopic evidence for Fe–H bonds in FeMoco has emerged.3 ENDOR spectra of the E4 state in an α-70Ile variant are consistent with two hydrides bridging iron centers,3 which is significant because the Thorneley–Lowe kinetic scheme indicates that the E4 state is the N2 binding state.4 These ideas recently gained further support through the H2 and N2 dependence of the E4 ENDOR signal in wild-type nitrogenase.5 The accumulated data strongly implicate a FeMoco-hydride as the key N2-binding species.
The postulation of an intermediate with hydrides is surprising because there are no previous examples in the literature of any synthetic iron–sulfide that has a hydride ligand.6 Herein, we dispel doubts about the feasibility of an iron–hydride coexisting with sulfide through the synthesis and full characterization of the first iron hydride sulfide complex.
The precursor to the new complex is the known dimeric iron hydride complex [LMeFeH]2 (1 in Scheme 1) where LMe is a bulky β-diketiminate ligand.7 Stirring a benzene solution of 1 with 1 equiv of sodium dodecanethiolate (NaSC12H25) at 60 °C for 16 h yields a maroon solution. Mössbauer analysis of the crude product mixture shows two distinct high-spin iron(II) products with 77% and 23% relative area (Figure S-11). Concentrated solutions of the product mixture in diethyl ether afford dark red crystals of the major product, 2. X-ray diffraction analysis of 2 shows an anionic iron hydride sulfide complex, with the sodium coordinated to the sulfide and to an arene ring (Figure S-23). The Fe–S bond distances of 2.2394(6) Å and 2.2553(7) Å differ by less than 0.02 Å (Table 1). The bridging hydride may be refined with an individual displacement parameter, revealing equivalent Fe–H distances of 1.66(3) Å and 1.68(3) Å and S–Fe–H angles of 86(1)° and 85.4(9)°. The second product has not been characterized by X-ray crystallography, but high performance electrospray mass spectrometry (7T ICR) shows a signal at m/z = 875.6 (Figure S-22), indicating that the side product is the iron(II) bis(thiolate) species [LMeFe(SC12H25)2]−.
Scheme 1. Synthesis of Diiron(II) Sulfide Hydride Complexes.

Table 1. Mössbauer and Metrical Parameters.
| compound | δ (mm/s) | |ΔEQ| (mm/s) | Fe–S (Å)a | Fe–S–Fe (deg) |
|---|---|---|---|---|
| 2 | 0.72 | 1.17 | 2.2474(9) | 76.20(2) |
| 3 | 0.67 | 0.73 | 2.218(2) | 76.34(4) |
| 4 (outer) | 0.73 | 2.11 | 2.264(4) | 82.4(1) |
| 4 (inner) | 0.83 | 1.63 | 2.264(4) | 82.4(1) |
| 5 | 0.87 | 1.91 | 2.298(2) | 100.51(9) |
Crystallographically inequivalent Fe–S distances are averaged.
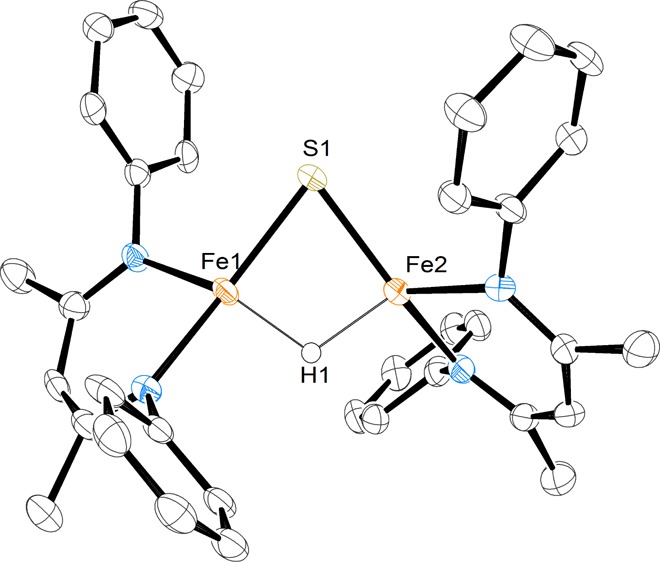
Thorough purification of 2 is difficult due to its high solubility in nonpolar solvents. To alleviate this issue, a chelating agent was used to create a discrete cation–anion pair. Complex 2 was dissolved in pentane and added dropwise to a pentane solution of a 2.2.2-cryptand, generating a purple precipitate that was purified through crystallization from toluene/pentane to give analytically pure 3 in an overall yield of 55% (from 1). X-ray structural analysis (Figure 1) reveals an iron hydride sulfide anion in which the sodium ion is chelated by the 2.2.2-cryptand (3 in Scheme 1). The Fe–S bond distances are 2.215(1) Å and 2.220(1) Å, the Fe–H distances are equivalent at 1.68(3) Å and 1.66(4) Å, and the S–Fe–H angles are both 86(1)°. These values closely resemble the analogous ones in 2. However, the N–Fe–S angles of 3 are 153.6(1)° and 106.4(1)° as compared to 150.07(5)° and 110.26(5)° in 2, indicating that the coordination of the sodium to the arene in 2 influences the geometry of the diketiminate ligand. Electrospray mass spectrometry further confirms the assigned composition of the anion in 3 (Figure S-21). Compound 3 has markedly lower thermal stability than 2, as approximately 50% decomposition was detected by 1H NMR spectroscopy in THF-d8 solution after 5 d at room temperature.
Figure 1.
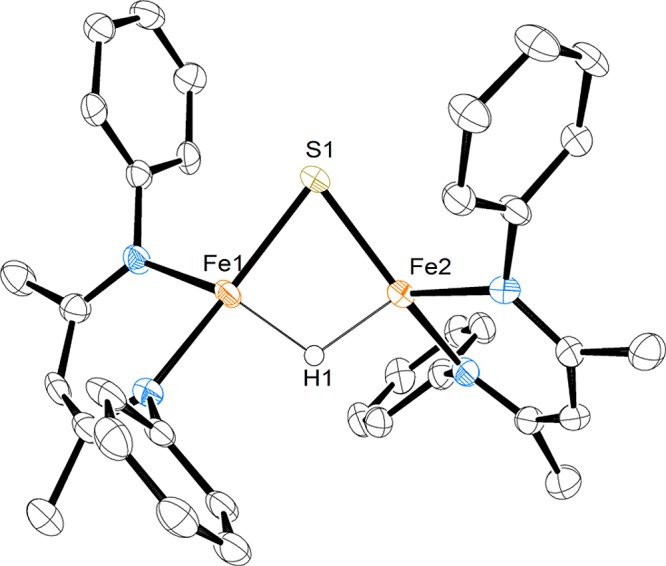
Molecular structure of the anion in 3, with thermal ellipsoids shown at 50% probability. The hydrogen atoms (except for the hydride), isopropyl groups, and sodium cryptand counterion have been omitted for clarity.
The use of thiolate C–S cleavage to generate 2 is crucial, because other potential routes (addition of hydride or acids to known iron–sulfide complexes;8 addition of other sulfur transfer reagents to 1; see Supporting Information) are ineffective. Thus, this C–S cleavage represents a mild way of introducing the sulfido group.9 GC/MS analysis of the crude reaction mixture from 1 and sodium dodecanethiolate indicate the presence of dodecane. In order to examine this reaction further, the deuterium analogue of 1 (1-D)7c was utilized in an analogous reaction to generate the deuteride sulfide complex (2-D). GC/MS analysis of the reaction mixture showed the presence of singly deuterated dodecane (C12H25D), supporting the idea that the S atom on the dodecyl group is exchanged with a hydride during the reaction.
The choice of alkali metals is surprisingly important. Using lithium dodecanethiolate in place of its sodium analogue gives a close analogue of 3 (3-Li), as shown by the formation of a product with a nearly identical 1H NMR spectrum (Figure S-3). However, analogous reactions with potassium dodecanethiolate do not yield any 3, as judged by 1H NMR spectroscopy. Adding the 2.2.2-cryptand to a solution of 1 prior to adding sodium dodecanethiolate does not form any detectable amount of 3. The need for an uncomplexed Li or Na suggests that the cation plays a crucial role in the formation of 2. Additional studies on the mechanism of this interesting C–S cleaving reaction will be reported in due course.
The 1H NMR spectrum of 3 in THF-d8 has broad resonances at room temperature. After the sample was cooled to −80 °C, these decoalesce into roughly 15 peaks with chemical shifts that range from 10.1 to −23.7 ppm, indicating that some fluxional process occurs in solution. The deuterium analogue of 3 (3-D) exhibits a paramagnetic isotope effect on chemical shift (PIECS) at −80 °C, shifting some peaks as much as 0.4 ppm relative to the nondeuterated isotopologue (Figure S-4). In general, PIECS is seen with bridging hydride complexes7c,10 and is likely to result from the slight difference in Fe–Fe distance when bridged by the smaller deuteron. Thus, the detection of PIECS suggests that the bridging hydride observed in the solid-state structure of 3 is also bridging in solution.
The Mössbauer spectrum of 3 consists of a quadrupole doublet with an isomer shift that is normal for high-spin iron(II) centers with soft ligands (Figure 2).11 The Mössbauer spectrum of 2 (Figure S-11) shows a major component with a nearly identical isomer shift and a noticeably different quadrupole splitting, consistent with the crystallographic observation that Na+ affects the geometry of the iron ligands (see above). Solid-state magnetometry measurements (Figure S-20) show that the high-spin iron ions in 3 are antiferromagnetically coupled, with an exchange coupling of J = −164 cm–1.
Figure 2.
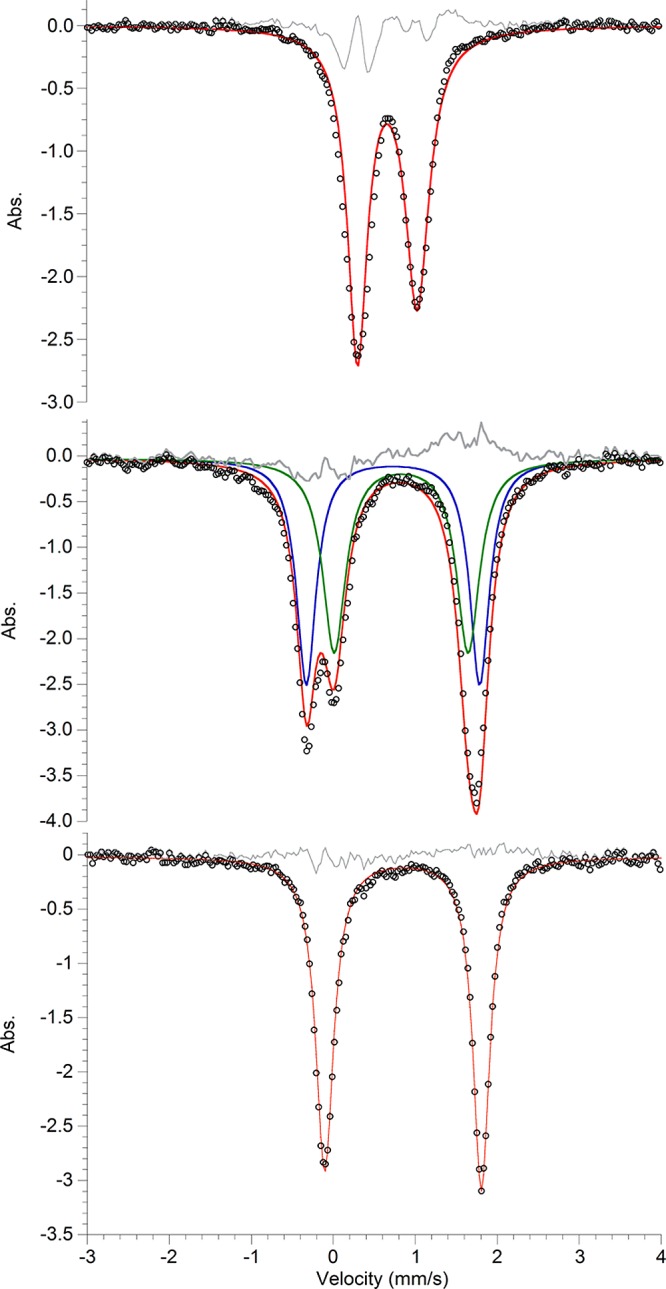
Mössbauer spectra of 3 (top), 4 (middle), and 5 (bottom) at 80 K. Residuals are shown in gray.
The isolation of a verified iron–sulfur cluster with a hydride enabled us to test the reactivity of hydride in an iron–sulfur environment. Nitrogenase has been reported to hydrogenate internal and terminal alkynes.1 To test the reactivity of 3 with alkynes, 1 equiv of 3-ethynyltoluene was added to a solution of 3 in THF. Over 1 h, the solution turned to crimson red, and it was possible to isolate dichroic red/yellow crystals from a concentrated THF solution in 54% yield. X-ray analysis of the crystals revealed a terminally bound alkynyl product [NaCrypt-222][LMeFe(CH3C6H4CC)(μ-S)FeLMe] (4; Scheme 2, Figure 3). Examination of the headspace via gas chromatography indicated an 87(7)% yield of H2 gas. Solid state Mössbauer spectra of 4 (Figure 2) show two quadrupole doublets of equal intensity, consistent with two inequivalent high-spin iron(II) sites as observed in the structure. The tendency of 3 to deprotonate rather than reduce a terminal alkyne suggests that the hydride is a better base than a nucleophile. By analogy, it is possible that FeMoco-bound hydrides may have limited capabilities for nucleophilic attack on alkyne substrates and may instead favor elimination of H2 as in a recently proposed hypothesis for N2 binding.12 However, the selectivity in the synthetic complex could be influenced by steric effects, and future studies will explore smaller supporting ligands to test this idea.
Scheme 2. Formation of the Alkynyl (4) and Formate Sulfide (5) Products.

Figure 3.
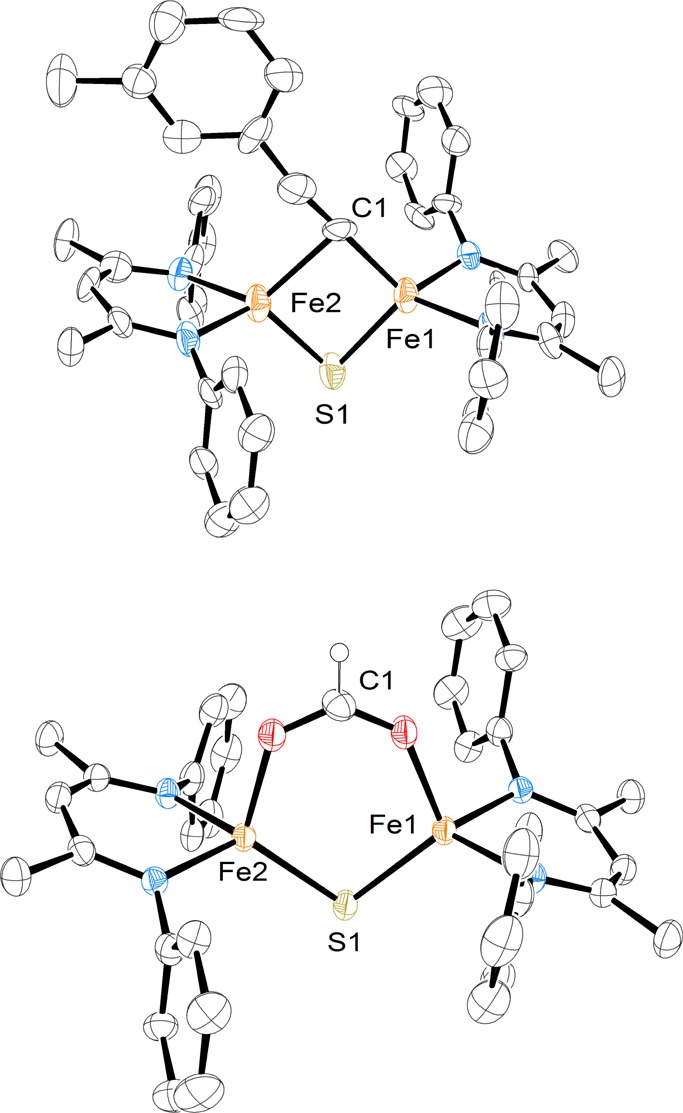
Molecular structures of 4 (top) and 5 (bottom) with thermal ellipsoids shown at 50% probability. The hydrogen atoms (except for the formyl H), isopropyl groups, and sodium cryptand counterion have been omitted for clarity.
Molybdenum-dependent nitrogenases have been shown to reduce CO2 to CO, formic acid, or methane.1,13 We anticipated that 3 could be used to predict the first elementary step to be expected in CO2 reduction if it were initiated by a Fe-hydride. Thus, a concentrated solution of 3 in THF was exposed to 2.5 equiv of CO2. After 3 h, red crystals formed in 75% yield. X-ray analysis of the crystals revealed a formate sulfide complex [NaCrypt-222][LMeFe(μ-CHOO)(μ-S)FeLMe] (5 in Scheme 2 and Figure 3). To our knowledge, this is the first example of an iron sulfide formate complex.14 Though the hydrogen atom of the formate was not clearly visible in the Fourier map, the bridging CO2 unit was assigned as formate based on the O–C–O bond angle of 129.8(9)°. Mössbauer spectroscopy of 5 revealed a single quadrupole doublet (Figure 2) for the equivalent high-spin iron(II) environments. The FTIR spectrum of 5 shows a C–O stretching band at 1599 cm–1 that shifts to 1556 cm–1 with 13CO2 (Figure S-14). This frequency is similar to the value of 1628 cm–1 for the bridging formate complex [LMeFe(μ-CHOO)]2.7a This hydride insertion suggests that the nitrogenase-catalyzed reduction of CO2 to methane13 could use a mechanism where the CO2 is initially hydrogenated by a metal hydride.
Reduction of 3 with 2 equiv of potassium/graphite (KC8) under an atmosphere of N2 led to K2[LMeFe(μ-S)FeLMe] and K2[LMeFe(μ-N2)FeLMe] in 10% and 24% spectroscopic yields, respectively (Figure S-7). Each of these compounds has been reported previously.8b,15 The most intriguing aspect with respect to nitrogenase is the formation of the formally diiron(0)–N2 complex when the amount of KC8 added was only sufficient to reduce each iron(II) to the formal iron(I) oxidation level. Formation of the diiron(0) N2 complex implies that another reduction event occurred. Because reductive elimination of H2 seemed possible, the reaction headspace was examined via gas chromatography, showing the presence of H2 in 27(2)% yield (relative to a theoretical yield of 0.5 mol of H2 per mol of 3). The reaction produced a substantial amount of unidentified byproducts, and so it is not possible to draw a firm conclusion regarding the source of electrons for the iron(0) species. However, it is significant that this iron–sulfide–hydride complex can bind N2 and form H2, as found in the key N2-binding step for nitrogenase.5
In conclusion, we have prepared the first synthetic example of an iron sulfide complex that also contains a hydride ligand. This hydride is bridging, like the ones in the E4 state of nitrogenase.5 The hydride can reduce a multiple bond in CO2, but does not reduce a terminal alkyne. Upon reduction it releases H2 with N2 binding, which gives an important experimental precedent for the idea that an iron–sulfide species can use hydrides to generate iron sites with N2 binding ability.
Acknowledgments
P.L.H. thanks the National Institutes of Health (GM065313) for support. M.A.J. thanks the Air Force Office of Scientific Research (FA9550-13-1-0007) for support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b06841.
The authors declare no competing financial interest.
Supplementary Material
References
- a Burgess B. K.; Lowe D. J. Chem. Rev. 1996, 96, 2983. 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- a Thorneley R. N. F.; Eady R. R.; Lowe D. J. Nature 1978, 272, 557. 10.1038/272557a0. [DOI] [Google Scholar]; b Crabtree R. H. Inorg. Chim. Acta 1986, 125, L7. 10.1016/S0020-1693(00)85472-9. [DOI] [Google Scholar]; c Leigh G. J.; McMahon C. N. J. Organomet. Chem. 1995, 500, 219. 10.1016/0022-328X(95)00516-S. [DOI] [Google Scholar]; d Henderson R. A. In Recent Advances in Hydride Chemistry; Peruzzini M., Poli R., Eds.; Elsevier: New York, 2001; pp 463–505. [Google Scholar]; e Dance I. Biochemistry 2006, 45, 6328. 10.1021/bi052217h. [DOI] [PubMed] [Google Scholar]
- a Igarashi R. Y.; Laryukhin M.; Dos Santos P. C.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2005, 127, 6231–6241. 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]; b Lukoyanov D.; Barney B. M.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1451–1455. 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lukoyanov D.; Yang Z.-Y.; Duval S.; Danyal K.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Inorg. Chem. 2014, 53, 3688. 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Lowe D. J. Met. Ions Biol. 1985, 7, 221. [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Khadka N.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2015, 137, 3610. 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A borohydride complex of an FeS cluster has been reported:Koutmos M.; Coucouvanis D. Inorg. Chem. 2004, 43, 6508. 10.1021/ic049275w. [DOI] [PubMed] [Google Scholar]
- a Yu Y.; Sadique A. R.; Smith J. M.; Dugan T. R.; Cowley R. E.; Brennessel W. W.; Flaschenriem C. J.; Bill E.; Cundari T. R.; Holland P. L. J. Am. Chem. Soc. 2008, 130, 6624. 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dugan T. R.; Holland P. L. J. Organomet. Chem. 2009, 694, 2825. 10.1016/j.jorganchem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dugan T. R.; Bill E.; MacLeod K. C.; Brennessel W. W.; Holland P. L. Inorg. Chem. 2014, 53, 2370. 10.1021/ic4013137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vela J.; Stoian S.; Flaschenriem C. J.; Münck E.; Holland P. L. J. Am. Chem. Soc. 2004, 126, 4522. 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]; b Rodriguez M. M.; Stubbert B. D.; Scarborough C. C.; Brennessel W. W.; Bill; Eckhard E.; Holland P. L. Angew. Chem., Int. Ed. 2012, 51, 8247. 10.1002/anie.201202211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Use of thiolate C–S cleavage for sulfide complexes:; a Fujisawa K.; Moro-oka Y.; Kitajima N. J. Chem. Soc., Chem. Commun. 1994, 623. 10.1039/c39940000623. [DOI] [Google Scholar]; b Smiles D. E.; Wu G.; Hayton T. W. J. Am. Chem. Soc. 2014, 136, 96. 10.1021/ja411423a. [DOI] [PubMed] [Google Scholar]
- Theopold K. H.; Silvestre J.; Byrne E. K.; Richeson D. S. Organometallics 1989, 8, 2001. 10.1021/om00110a027. [DOI] [Google Scholar]
- Beinert H.; Holm R. H.; Münck E. Science 1997, 277, 653. 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- a Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Acc. Chem. Res. 2013, 46, 587. 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Chem. Rev. 2014, 114, 4041. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang Z.-Y.; Moure V. R.; Dean D. R.; Seefeldt L. C. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 19644. 10.1073/pnas.1213159109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rebelein J. G.; Hu Y.; Ribbe M. W. Angew. Chem., Int. Ed. 2014, 53, 11543. 10.1002/anie.201406863. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee C. C.; Hu Y.; Ribbe M. W. Angew. Chem., Int. Ed. 2015, 54, 1219. 10.1002/anie.201410412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge Crystallographic Database:Allen F. H. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 380. 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. J. Am. Chem. Soc. 2006, 128, 756. 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.