

Abstract
Melanoma is one of the most lethal skin cancers worldwide, primarily because of its propensity to metastasize. Thus, the elucidation of mechanisms that govern metastatic propensity is urgently needed. We found that protein kinase Cε (PKCε)–mediated activation of activating transcription factor 2 (ATF2) controls the migratory and invasive behaviors of melanoma cells. PKCε-dependent phosphorylation of ATF2 promoted its transcriptional repression of the gene encoding fucokinase (FUK), which mediates the fucose salvage pathway and thus global cellular protein fucosylation. In primary melanocytes and cell lines representing early-stage melanoma, the abundance of PKCε-phosphorylated ATF2 was low, thereby enabling the expression of FUK and cellular protein fucosylation, which promoted cellular adhesion and reduced motility. In contrast, increased expression of the gene encoding PKCε and abundance of phosphorylated, transcriptionally active ATF2 were observed in advanced-stage melanomas and correlated with decreased FUK expression, decreased cellular protein fucosylation, attenuated cell adhesion, and increased cell motility. Restoring fucosylation in mice either by dietary fucose supplementation or by genetic manipulation of murine Fuk expression attenuated primary melanoma growth, increased the number of intratumoral natural killer cells, and decreased distal metastasis in murine isograft models. Tumor microarray analysis of human melanoma specimens confirmed reduced fucosylation in metastatic tumors and a better prognosis for primary melanomas that had high abundance of fucosylation. Thus, inhibiting PKCε or ATF2 or increasing protein fucosylation in tumor cells may improve clinical outcome in melanoma patients.
INTRODUCTION
The lethality of melanoma is attributed to its ability to metastasize to distant sites of the body, a propensity that remains a clinical obstacle for sustained therapeutic efficacy (1). Increased incidence of metastasis is associated with worse overall survival outcomes (2). Thus, the elucidation of key molecular mechanisms that underlie the motility, the propensity to colonize at distant sites, and the ability to adapt to altered microenvironmental conditions is needed to create new opportunities for clinical management of melanoma. Rewired signal transduction pathways, primarily those mediated by mitogen-activated protein kinase (MAPK) [involving BRAFand NRAS (which are encoded by the most prevalent mutated driver genes in melanoma) and their downstream effector, extracellular signal-regulated kinase (ERK)] and the phosphoinositide–dependent protein kinase-1 (PDK1)/phosphatidylinositol 3-kinase (PI3K) family of protein kinases [involving the tumor suppressor phosphatase and tensin homolog (PTEN) and the kinases AKT and protein kinase C (PKC)] (3–5), underlie the development, progression, and propensity of melanoma to metastasize. Among the transcription factors that are affected by these up-regulated protein kinases is microphthalmia-associated transcription factor (MITF), a master regulator of melanocyte biogenesis that is implicated in key cellular programs, including pigmentation and cell death as well as migration and drug resistance (3, 6–10). The abundance and activity of MITF are often linked with the propensity of melanoma to withstand environmental pressure and acquire its plasticity—the ability to adjust to rough growth conditions, dictated by microenvironment (nutrient and oxygen) or chemotherapy-imposed stress (11–13). The mechanisms underlying the plasticity of melanoma also include the switch from a proliferative to an invasive state, as exemplified by vascular mimicry, whereby melanoma cells exhibit vascular antigens and altered signaling that enables melanoma cells to exploit the stromal vasculature. Among the rewired pathways that are implicated in melanoma metastasis are BRAF-mediated down-regulation of the cyclic guanosine monophosphate–specific phosphodiesterase PDE5A and MITF-mediated regulation of Rho–guanosine triphosphatase pathways, which include their downstream effectors P-rex1, Rab5, Rac1, and MDA9 (4, 5, 9, 10, 14–19). However, this knowledge has not yet benefited currently available therapies. Clinical trials using inhibitors of angiogenesis or matrix metalloproteinases have not yielded promising results (20), and melanoma resistance to therapies, including inhibitors of mutant BRAF, is associated with increased metastatic burden (21). Thus, further elucidation of the mechanistic underpinnings of melanoma metastasis is needed.
Among the transcription factors regulated by both the MAPK and PDK1 pathways is activating transcription factor 2 (ATF2), a member of the activator protein-1 (AP1) complex. Its phosphorylation on Thr69/71 by the p38, c-Jun N-terminal kinase (JNK), or ERK cascades enables its transcriptional activity, whereas its phosphorylation on Thr52 by PKCε directs its nuclear localization, thereby augmenting its overall transcriptional output. Enhanced ATF2 activity, seen in advanced melanoma, contributes to its development and metastatic capacity, in part by enhancing proliferation and attenuating apoptotic signaling, which are mediated by the activation of the AP1 transcriptional complex, in concert with the inactivation of other cellular networks such as interferon-β1 (IFNβ1) (22, 23). Correspondingly, increased abundance of both PKCε and nuclear ATF2 correlates with metastasis and poor clinical outcome (24, 25). Consistent with the tumorigenic roles of ATF2 in melanoma, inhibition of its transcriptional activity or nuclear localization using small molecules or peptides impairs melanoma development and attenuates metastasis in vivo (26, 27). We recently reported that ATF2 could impair therapeutic outcome by direct transcriptional repression of IFNB1 expression (28). Here, we investigated the precise ATF2 transcriptional targets that promote the metastatic capacity of melanoma and found that ATF2 transcriptionally represses the gene encoding fucokinase (FUK). FUK is the most upstream kinase of the fucose salvage pathway that facilitates the crucial phosphorylation of L-fucose that directs its conversion into guanosine diphosphate (GDP)-L-fucose, rendering it usable for fucosylation of proteins in the cell (29). FUK activity appears to account for up to 40% of overall fucosylation through the fucose salvage pathway, whereas the remaining 60% is produced by the de novo synthesis pathway, which converts intracellular GDP-D-mannose into GDP-L-fucose (30). Protein fucosylation is crucial for leukocyte adhesion to the vasculature, pointing to its role in cell adhesion and motility (31). Transcriptional silencing of FUK by ATF2 reduced global fucosylation dynamics, which results in enhanced metastatic propensity of melanoma.
RESULTS
PKCε-ATF2 signaling transcriptionally represses FUK and cellular fucosylation
To identify the ATF2-regulated transcriptional repertoire that drives melanoma resistance and metastasis, we analyzed previously performed gene expression profiling data from human elanoma cells that were depleted of endogenous ATF2 and reconstituted with either wild-type ATF2 (ATF2WT), a transcriptionally active, PKCε-phosphomimic mutant ATF2 (ATF2T52E), or a nonphosphorylatable and transcriptionally inactive mutant ATF2 (ATF2T52A) (28). From this analysis, we identified putative transcriptional targets of ATF2 that may underlie its tumorigenic effects. Among the top significantly altered canonical signaling pathways, Ingenuity Pathway Analysis of these targets suggested that ATF2T52E (the transcriptionally active mutant) attenuates GDP-L-fucose biosynthesis by transcriptionally down-regulating FUK, which controls the fucose salvage pathway.
Target validation by quantitative reverse transcription polymerase chain reaction (qRT-PCR) revealed an about 75% increase in FUK mRNA expression in WM793 cells after knocking down ATF2 using short hairpin RNA (shRNA) (Fig. 1A, left, and fig. S1A, upper left). Reconstitution of shRNA-transfected cells with ATF2T52A (the transcriptionally inactive mutant) further enhanced the induction of FUK, whereas reconstitution with ATF2T52E or ATF2WT suppressed the induction of FUK expression caused by ATF2 knockdown or expression of the inactive mutant (Fig. 1A and fig. S1A). Similar results were observed in WM1346 or WM1366 cells (fig. S1A). Consistent with these findings, knocking down PKCε resulted in an about twofold increase in FUK mRNA expression (Fig. 1A, right). In contrast, the expression of other genes in the fucose salvage pathway (including FPGT, FX, and GMD) was not significantly affected in ATF2-deficient WM793 cells by reexpression of active or inactive mutant ATF2 compared with cells reexpressing wild-type ATF2 (fig. S1B). These data would suggest that up-regulated PKCε, as observed in advanced melanomas, may increase the phosphorylation and activation of ATF2, resulting in decreased FUK mRNA expression. This inverse relationship was further supported by our analysis of The Cancer Genome Atlas (TCGA) expression data, in which ATF2 and FUK expression were inversely correlated in a 473-patient cohort (fig. S1C). qRT-PCR analysis of human primary melanocytes, vertical growth phase (VGP) melanoma cell lines (WM793, WM1346, and WM1366), metastatic melanoma cell lines (LU1205, 501Mel, and YUGASP), and even human embryonic kidney (HEK) 293 cells generally revealed a progressive reduction in FUK mRNA levels in cell lines with increasing protein abundance of PKCε and Thr52-phosphorylated ATF2 (Fig. 1B). Thus, these data suggest that PKCε-phosphorylated ATF2 suppresses FUK mRNA expression in the more advanced melanomas.
Fig. 1. PKCε-ATF2 signaling transcriptionally represses FUK and cellular fucosylation.
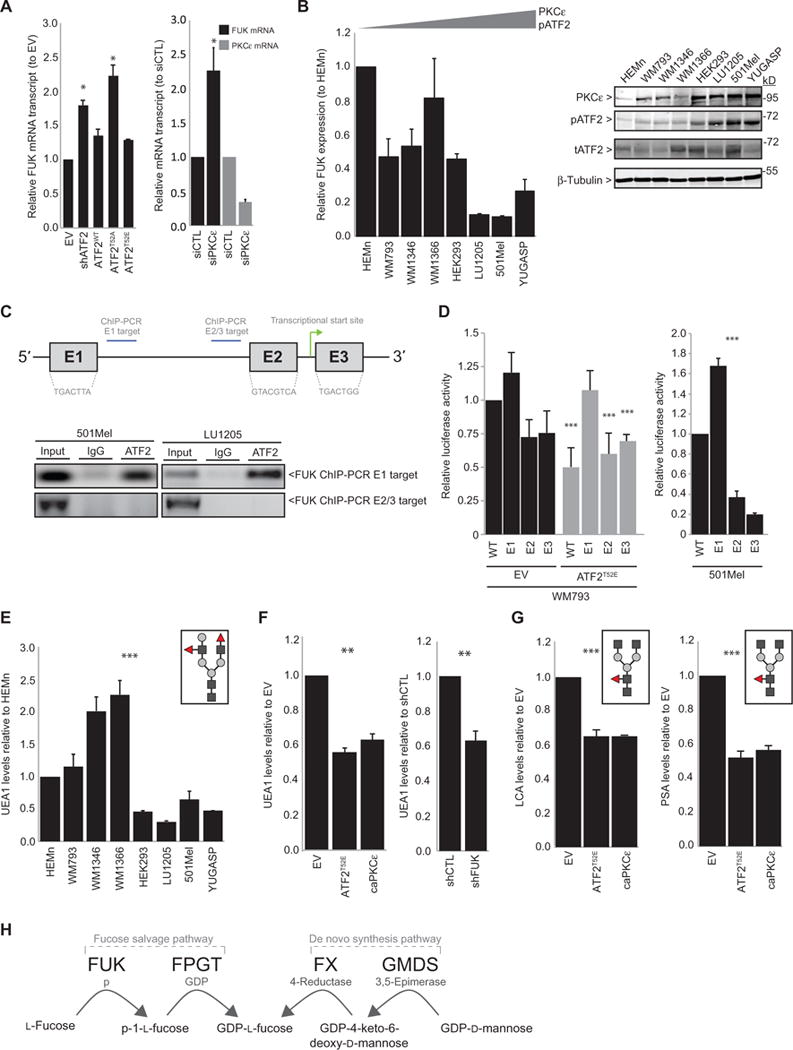
(A) qRT-PCR analysis for FUK or PKCε expression in WM793 cells transfected with (left) EV, ATF2-targeted shRNA (shATF2), or shATF2 and ATF2WT, ATF2T52A, orATF2T52E;or(right)control (siCTL) or PKCε-targeted siRNA (siPKCε). (B) qRT-PCR for FUK mRNA expression (left) and immunoblotting for FUK protein abundance (right) in primary human melanocytes (HEMn), VGP melanoma cell lines (WM793, WM1346, and WM1366), metastatic melanoma cell lines (LU1205, 501Mel, and YUGASP), and transformed HEK293 cells. pATF2, phosphorylated ATF2; tATF2, total ATF2. (C) Top: FUK 5′ promoter with ATF2 binding sites (E1, E2, and E3), ChIP targets, and transcriptional start site. Bottom: ATF2 ChIP of the FUK 5′ promoter E1 and E2/3 targets in 501Mel (left) and LU1205 (right) cells. IgG, immunoglobulin G. (D) Wild-type (WT) or E1, E2, or E3 single-mutant FUK promoter luciferase activities in (left) WM793 cells transfected with EV (black) or ATF2T52E (gray) or in (right) 501Mel cells. (E) Fluorescence-activated cell sorting (FACS) analysis of UEA1 lectin binding of cell lines in (B).Graphic inset represents a glycan recognized by UEA1 (red triangles: α-1,2/1,3-fucose; dark gray squares: N-acetylglucosamine; light gray circles: galactose). (F) UEA1 FACS analysis in WM793 cells transfected with (left) EV, ATF2T52E, or caPKCε or (right) control or FUK-targeted shRNA (shFUK). (G) FACS analysis of LCA and PSA in WM793 cells transfected as indicated. Graphic inset represents a glycan recognized by LCA or PSA (red triangles: α-1,4/1,6-fucose). (H) A schematic of the fucose salvage and de novo synthesis pathways. All data are means ± SD from three experiments. *P < 0.05, **P < 0.005, ***P < 0.0005 by a standard t test compared with controls.
Therefore, we set to determine how the PKCε-ATF2 axis controls FUK expression and therefore global protein fucosylation. To determine whether the down-regulation of FUK expression by PKCε-mediated phosphorylation of ATF2 is direct, we assessed the binding of ATF2 to the FUK 5′ promoter, which is predicted to contain three ATF2 consensus binding sites (E1, E2, and E3; Fig. 1C). Chromatin immunoprecipitation (ChIP) with an ATF2 antibody in melanoma cells with high abundance of phosphorylated ATF2 (501Mel and LU1205 cells) revealed that ATF2 does bind the FUK promoter at E1, whereas it was not detected at the E2- or E3-proximal regions (Fig. 1C), suggesting that the E1 binding site is required for ATF2-mediated transcriptional repression. To confirm which of the three possible sites are required for ATF2, we monitored luciferase activity driven by FUK 5′ promoter–luciferase constructs containing mutations in each individual binding site to abolish ATF2 recognition in WM793 cell lines that were transfected with either empty vector (EV) or ATF2T52E, the active mutant that should suppress FUK promoter activity. In “PKCε/phosphorylated ATF2-low” WM793 cells, the expression of ATF2T52E repressed the activity of the wild-type FUK promoter to ~50% of that in cells expressing the EV (Fig. 1D). Compared to the control, a similar degree of repression of promoter constructs in which sites E2 or E3 but not E1 were mutated was elicited by expression of ATF2T52E (Fig. 1D). When transfected into “PKCε/phosphorylated ATF2-high” 501Mel cells, the activity of the E1-mutant FUK promoter construct was increased (~1.6-fold) compared with that of either wild-type or E2- or E3-mutant promoter constructs (Fig. 1D, right). Together, these data suggest that the binding of PKCε-phosphorylated ATF2 to site E1 on the FUK 5′ promoter represses the transcription of FUK.
To confirm that PKCε-ATF2–modulated FUK expression correlates with cellular protein fucosylation, we probed melanocyte and melanoma cell lines with a lectin from Ulex europaeus (UEA1), which binds with affinity to α-1,2-linked fucose–containing glycoconjugates (32). Flow cytometric analysis of the amount of UEA1 binding revealed that cell surface protein fucosylation was decreased in the metastatic melanoma cell lines compared with both the VGP cell lines and primary (nonmalignant) melanocytes (Fig. 1E). The relative decrease in cell surface fucosylation in the metastatic cell lines compared with the VGP cell lines generally correlated with the FUK expression observed in those cell lines (Fig. 1B). Overexpression of a constitutively active PKCε mutant (caPKCε) or of ATF2T52E in “PKCε/ATF2-low” WM793 melanoma cells decreased the UEA1 signal by ~40%, similar to that observed after FUK knockdown by shRNA (Fig. 1F and fig. S1D). A similar reduction in cell surface fucosylation was observed when using two other lectins, Lens culinaris (LCA) and Pisum sativum (PSA), which exhibit specificities toward core-fucosylated N-glycans (33), in melanoma cells expressing caPKCε or ATF2T52E (Fig. 1G). Similar changes in fucosylation were observed in PKCε/ATF2-low expressing WM1346 and WM1366 cell lines transfected with caPKCε or ATF2T52E (fig. S1E). These data indicate that the increased presence of PKCε and PKCε-phosphorylated ATF2 is sufficient to reduce cellular fucosylation mediated by FUK. That the PKCε-ATF2–mediated loss of FUK results in about a 40% reduction in cellular fucosylation suggests that the FUK-mediated fucose salvage pathway contributes to about half of cellular fucosylation in melanoma cells, whereas the residual fucosylation might be provided by the de novo synthesis pathway (Fig. 1H).
PKCε-ATF2 repression of FUK promotes melanoma motility, adhesion, and invasiveness
Fucosylation and glycosylation play roles in cell-cell interaction and motility. For example, fucosylation plays a crucial role in leukocyte adhesion and extravasation, whereas in lung and other cancers, altered glycosylation or fucosylation appears to correlate with metastasis (34–37). Thus, we assessed whether ATF2-repressed FUK expression affected melanoma motility and invasiveness using culture-based scratch assays. Ectopic expression of FUK in two metastatic melanoma cell lines (501Mel and LU1205), which exhibited endogenously high abundance of PKCε and phosphorylated ATF2 and low abundance of FUK (Fig. 1B), increased the amount of cell surface fucosylation by about three- and eightfold, respectively (fig. S1F), and attenuated motility by ~50% compared to control cells (Fig. 2A). Consistently, adding L-fucose to cultures of WM793, WM1346, or WM1366 cells (which had low abundance of PKCε and phosphorylated ATF2 and high abundance of FUK) also increased cell surface protein fucosylation and reduced cell motility to a similar extent (Fig. 2B and fig. S1G). To determine whether L-fucose uptake increases the extent of surface protein fucosylation, we performed radiolabeled L-fucose uptake assays in both “FUK-high” (WM793 and WM1346) and “FUK-low” (501Mel) melanoma cells, as well as HEK293 cells. Our data showed that fucose uptake rates were not significantly variable between these cell lines, indicating that the rate of L-fucose uptake does not significantly influence the amount of surface protein fucosylation (fig. S1H). In contrast, the amount of fucose-1-phosphate (phosphorylated L-fucose) detected was about three to five times higher in the FUK-high cultures compared with FUK-low cultures (fig. S1H), suggesting that FUK plays a determinant role in the production of fucose-1-phosphate in melanoma cells. Notably, treating 501Mel cells with a cell-penetrable, acetylated GDP-L-fucose (ac-GDP-L-fucose) (38), which bypasses the need for phosphorylation by FUK, was sufficient to increase surface protein fucosylation by about sixfold and to suppress motility in these cells by ~30 to 50% compared to controls (Fig. 2B and fig. S2A). Conversely, either overexpressing ATF2T52E (the active mutant) or knocking down FUK enhanced the motility of WM793 cells by ~50 to 80% (Fig. 2C and fig. S1D). Notably, overexpressing FUK and ATF2T52E in WM793 cells largely prevented the ATF2T52E-mediated increase in motility. Similar effects in motility were observed in WM1346 and WM1366 cells (fig. S2B). Because protein fucosylation is crucial for leukocyte adhesion to the vasculature, we assessed whether FUK might promote cellular adhesion. Indeed, ectopic expression of FUK was sufficient to double the number of that adhered to cell culture plates within 2 hours in cultures of HEK293 cells, which correlated with the effect of FUK expression to increase the abundance of fucosylated proteins by more than twofold in these cells as well as in cultures of 501Mel and LU1205 melanoma cells (Fig. 2D and figs. S1F and S2C). Furthermore, LU1205 or 501Mel melanoma cultures, which exhibit poor sphere-forming ability when grown in suspension, formed compact and uniform spheres upon FUK expression, further supporting a role of FUK in controlling cellular adhesion in melanoma cells (Fig. 2E). Finally, we assessed whether FUK-dependent adhesion of melanoma cells would affect their invasive properties. To this end, spheroid-grown 501Mel and LU1205 melanoma cells and HEK293 cells, each expressing either a control EVor FUK, were placed in Matrigel plugs and monitored for their invasion from the spheroids into the plugs. Expression of FUK reduced spheroid size by >30% and caused a corresponding decrease in invasion (Fig. 2F and fig. S2, D and E). A substantial loss of invasive behavior (by ~90%) in HEK293 cells was also observed in a Boyden chamber assay (fig. S2E). Together, these findings indicate that PKCε-ATF2-FUK axis–regulated cellular protein fucosylation regulates the motility and invasive capacities of melanoma, as well as HEK293 cells, consistent with our previous finding that perturbation of PKCε-mediated phosphorylation of ATF2 using small molecule compounds impairs melanoma motility (27).
Fig. 2. FUK expression reduces melanoma motility and invasiveness and increases adhesion.
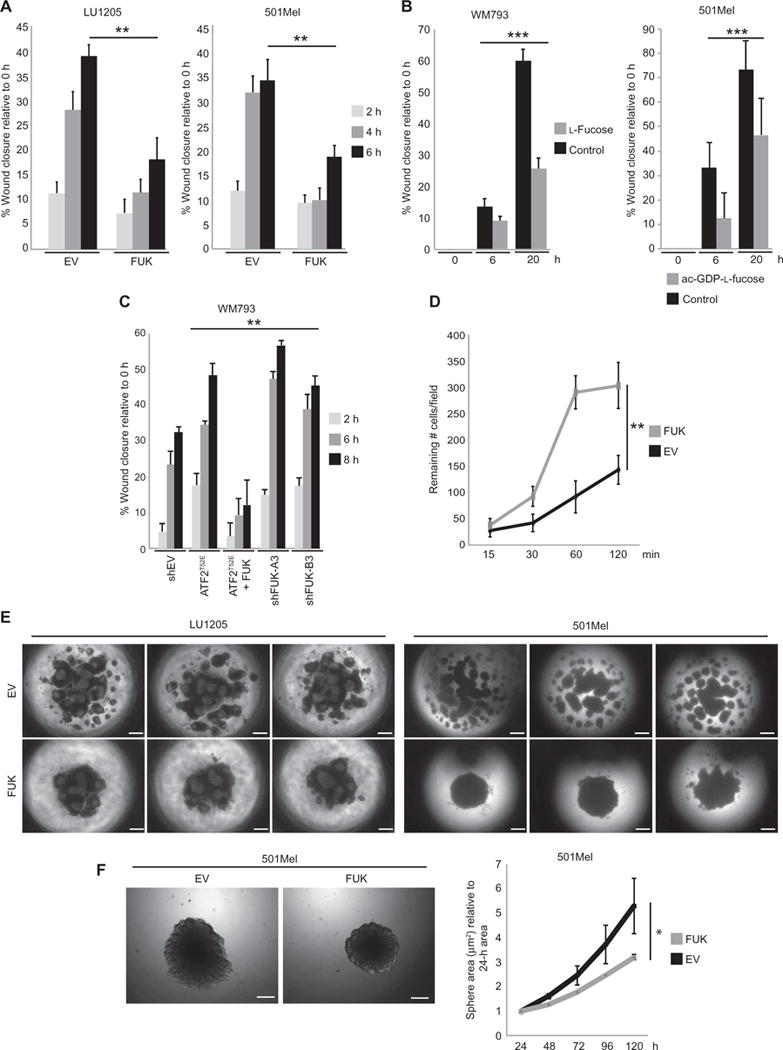
(A) Migration of LU1205 (left) or 501Mel (right) cells transfected with EV or FUK, assessed by scratch assays. (B) Migration of WM793 or 501Mel cells assessed by scratch assays in cultures supplemented with control water (5 μl) or 50 μM L-fucose (left) or 25 μM ac-GDP-L-fucose (right) for 36 hours. (C) Migration of WM793 cells transfected with control shRNA (shEV), one of two FUK-targeted shRNAs, ATF2T52E, or ATF2T52E and FUK, assessed by scratch assays. (D) The number of EV- or FUK-expressing HEK293 cells that remained attached (per 10× field) after wash-off at the indicated time points was quantified (four 10× fields pertime point were counted). (E) Representative EV-or FUK-expressing LU1205 (left) or 501Mel (right) spheroids from three independent experiments. (F) Representative image (left) and quantitation (right) of the area of GFP-EV– or GFP-FUK–expressing 501Mel spheroids grown in Matrigel plugs. Data in (A) to (D) and (F) are means ± SD from three experiments. *P < 0.05, **P < 0.005, ***P < 0.0005 by a standard t test compared with controls or starting time point. Scale bars, 100 μm.
L-Fucose administration or murine Fuk expression attenuates tumor growth and metastasis in mice
On the basis of the inhibitory effects of FUK and protein fucosylation on the motility and invasiveness of melanoma cells that we observed in cultured cells, we sought to determine whether the administration of L-fucose or the expression of FUK might facilitate these effects in vivo. To this end, we used the SW1 murine melanoma model, which harbors mutant Nras and is metastatic in syngeneic C3H mice. Similar to the human cell lines examined, the metastatic SW1 cell line exhibited ~80% increased expression of the transcript encoding PKCε and ~50% reduced expression of murine Fuk (mFuk) compared to its nonmetastatic parental K1735 cell line (Fig. 3A) (12). When L-fucose was added to the culture medium or when cells were engineered to ectopically express mFuk, SW1 cells grown in two-dimensional cultures exhibited impaired motility in an in vitro scratch assay (Fig. 3, B and C). Furthermore, mFuk-expressing SW1 cells formed more compact spheroids than EV-expressing cells when grown in hanging droplet culture (Fig. 3D), similar to our observations described above in human melanoma cell lines (Fig. 2E). Because the SW1 cell lines and human melanoma cells behaved similarly, we proceeded to use the SW1 cells in an in vivo model of tumor development and metastasis.
Fig. 3. Increased FUK expression attenuates melanoma development and metastasis and correlates with increased survival in human patient samples.
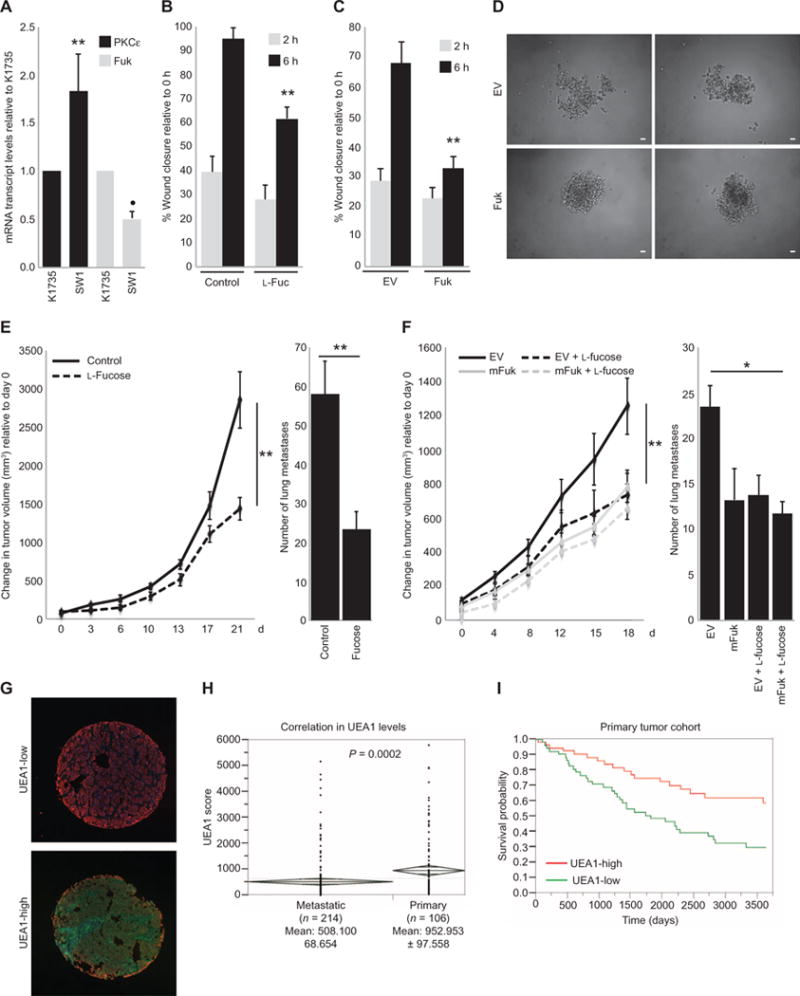
(A) qRT-PCR analysis of PKCε and mFuk expression in SW1 and K1735 cells. (B and C) Scratch assays assessing the migration of SW1 cells (B) in which cultures were supplemented with control water (Control; 5 μl) or L-fucose (L-Fuc; 50 μM) for 36 hours or (C) in which cells were transfected with EV or mFuk. (D) Two representative spheroids (n = 3 experiments) formed by EV- or FUK-expressing SW1 cells in culture. Scale bars, 10 μm. (E and F) Tumor volume curves (left) and lung metastasis counts (right) of (E) C3H/HeJ mice bearing implanted SW1 tumors or (F) mice bearing implanted EV-or mFuk-expressing SW1 tumors provided with control water (Control) or water supplemented with L-fucose (Fucose,100mM).Data are means ± SD from six mice per group. (G) Representative UEA1-low (upper) and UEA1-high (lower) histospots from a human melanoma TMA (n = 320) immunostained with UEA1 lectin (green), S100/HMB45 (red), and 4′,6-diamidino-2-phenylindole (DAPI, blue). (H) Distribution of UEA1 scores within S100/HMB45-positive melanoma cells of individual histospots from primary or metastatic melanoma samples. Means, SD, and n as indicated; P = 0.0002 by unpaired t test. Diamonds depict means ± 1 SD. (I) Kaplan-Meier survival curves according to UEA1 abundance in a melanoma patient cohort. Data in (A) to (C) are means ± SD from three experiments. *P < 0.05, **P < 0.005 by a standard t test compared with parental K1735 cell lines, controls, or starting time point.
In C3H/HeJ mice with established SW1 melanoma tumors, the administration of L-fucose–supplemented water was sufficient to increase protein fucosylation by more than twofold and attenuate melanoma primary tumor growth by ~60% compared to control mice (Fig. 3E and fig. S3A). Correspondingly, an ~60% reduction in the number of lung metastases was observed in the mice that were provided L-fucose–supplemented water compared to those provided normal drinking water (Fig. 3E).
We further performed an independent assessment of the effects elicited by mFuk expression on melanoma growth and metastasis using SW1 cells that were genetically manipulated to express mFuk (fig. S3B) and compared tumor growth and metastasis with control EV–expressing SW1 cells. Expressing mFuk in SW1 cells before implantation reduced the growth of primary tumors in mice by ~40% compared to those bearing EV-expressing tumors, and the addition of dietary fucose to the drinking water elicited similar extent of tumor growth suppression (Fig. 3F, left). Notably, mice with mFuk-expressing SW1-derived tumors also had a similarly reduced incidence of lung metastasis as was observed in the mice supplemented with dietary fucose (>40% reduction; Fig. 3F, right). Both dietary L-fucose supplementation and genetic manipulation of mFuk expression were sufficient to increase the abundance of fucosylated protein in the tumors by at least twofold (fig. S3C). Together, these data indicate that increasing cellular fucosylation by either L-fucose supplementation or increased mFuk expression effectively reduces metastatic burden in vivo.
Because fucosylation has been reported to play crucial roles in the development of immune cells and have potential effects on immune surveillance of tumor cells (30, 39), we assessed whether the presence of immune cells was affected in tumors of mice that received L-fucose supplementation or in tumors overexpressing mFuk. Immunostaining analysis revealed a significant increase in the numbers of CD45+ lymphocytes in the SW1-derived tumors of mice that were supplemented with L-fucose (by ~240% relative to those provided control water; fig. S3D). A similar increase in CD45+ lymphocytes was observed in mFuk-expressing tumors, regardless of further L-fucose supplementation (by ~290% relative to EV-expressing tumors; fig. S3F). Immunofluorescence staining for natural killer (NK) cells, which reportedly elicit fucosylation-dependent antitumor activity (40), revealed that L-fucose supplementation, mFuk expression in the tumor cells, or both increased intratumoral NK cell populations (>300% relative to control water or EV; fig. S3, E and G). Together, these findings indicate that the fucosylation state of melanoma tumor cells affects the presence or absence of intratumoral immune cell populations, including NK cells, which might contribute to tumor suppression.
Reduced cell surface protein fucosylation correlates with metastasis and reduced survival in melanoma patient samples
To determine the association of fucosylation in human clinical melanoma samples with patient survival, we assessed fucosylation status in a large-cohort human melanoma tissue microarray (TMA; 320 patient samples). Fluorescein isothiocyanate (FITC)–conjugated UEA1 lectin and a tetramethyl rhodamine isothiocyanate (TRITC)–conjugated cocktail of antibodies for the melanoma-specific markers S100 and HMB45 were used to assess the abundance UEA1 lectin staining in melanoma histospots. UEA1 staining revealed a relatively homogeneous cytoplasmic or membranous staining pattern within the S100/HMB45-stained melanoma cells in each histospot (Fig. 3G). One-way analysis of variance (ANOVA) revealed that the UEA1 signals in S100/HMB45-positive melanoma cells within the primary melanoma histospots (n = 106) were about twice those detected within the metastatic tumor histospots (n = 214) (P = 0.0002; Fig. 3H). The finding of decreased cellular fucosylation in metastatic melanoma is consistent with our earlier TMA analyses in which increased abundance of PKCε and nuclear ATF2 was found in progressive stages of melanoma (24, 25). Although no correlation was found with age or ulceration, UEA1 abundance was higher in thinner lesions (Breslow thickness <2 mm; P = 0.043) and, surprisingly, in male patients (P = 0.026). By Cox univariate analysis of dichotomized scores of the primary tumor specimens, high UEA1 abundance was associated with improved overall survival and a 34% reduction in risk for metastasis [risk ratio, 0.664; lower confidence limit (CL), 0.494; upper CL, 0.879; P = 0.004; Fig. 3I). These observations indicate the physiological importance of fucosylation in suppressing the metastatic capacity of melanoma.
DISCUSSION
Here, we identified the regulation and importance of protein fucosylation in melanoma development and metastasis. Our data demonstrate that global protein fucosylation is negatively regulated by ATF2-mediated transcriptional repression of FUK, encoding the key rate-limiting kinase of the fucose salvage pathway. ATF2-mediated repression of FUK and reduced cellular protein fucosylation effectively decreased adhesion and increased cell motility and the invasive capacity of melanoma cells. That the overall amount of fucosylated protein was reduced only by ~40 to 50% by activation of the PKCε-ATF2 axis suggests that the auxiliary de novo synthesis pathway, which converts cellular GDP-D-mannose to GDP-L-fucose with the GMD/FX enzymes (30), likely accounts for the residual amount of fucosylation observed. That we did not observe an additive effect on tumor growth or immune cell presence when L-fucose was supplemented in addition to mFuk expression suggests the possibility of an as yet unidentified rate-limiting step downstream of mFuk in protein fucosylation in these cells. Nonetheless, the ability to modulate half of cellular fucosylation by L-fucose supplementation or by increasing FUK expression to reduce melanoma tumor growth and metastatic burden points to the importance of this regulatory axis in melanoma metastasis. In agreement, a B16 murine melanoma variant exhibiting poor metastatic capacity exhibits increased protein fucosylation compared to its highly metastatic variant (41, 42).
Protein fucosylation affects immune cell recognition, interactions, and general biology (30, 43–45). Mice deleted of the FX enzyme impairs the de novo fucose synthesis pathway, resulting in homing defects and impaired self-renewal capacity in bone marrow hematopoietic stem cells that are partially rescued by dietary L-fucose supplementation (46). Impaired protein fucosylation in leukocytes results in leukocyte adhesion deficiency type II, which is also partially rescued by dietary fucose supplementation (37). Notably, these scenarios result from complete loss of the de novo synthetic pathway in immune cells, and dietary L-fucose supplementation only rescued loss of the de novo pathway in mice or cell cultures. Our in vivo models suggest that partial change in salvage pathway–dependent fucosylated protein abundance affects melanoma recognition by immune cells. Indeed, we observed an increased presence of CD45+ immune cells in the L-fucose–supplemented mice and in mFuk-expressing tumors compared to controls. Although these data are insufficient to distinguish between the infiltration of immune cells due to enhanced tumor recognition and recruitment and that caused by the proliferation of resident intratumoral immune cells, as well as the precise type of immune cell subset responsible, they suggest that reduced fucosylation in melanoma cells might impair immune surveillance and responsiveness to the tumors. Consistent with the reported induction of NK cell–mediated tumor-lytic activity in melanoma cells that were engineered to express high levels of α-1,3-fucosyltransferase III (40), we found that NK cells were increased in both L-fucose–supplemented and mFuk-positive tumors. Thus, the tumor suppression, as observed in our study, might be mediated by immune or other stromal components. How altered fucosylation affects immune cell interactions with melanoma tumors warrants further investigation.
Our cellular and in vivo findings were corroborated by analysis of a large-cohort human TMA and assessment of TCGA patient expression data sets, suggesting that the mechanism identified in this study is conserved in melanomas and is of clinical importance. Our findings indicate a previously undisclosed therapeutic potential because this mechanism appears to be targetable by the application of L-fucose. The coadministration of L-fucose (or L-fucose analogs) with current therapies is among feasible considerations, which would be expected to provide immediate improvement of therapeutic efficacy. Notably, the melanoma side/stem cell population, characterized by its significant chemotherapeutic resistance, was previously reported to exhibit substantial down-regulation of FUK (47), suggesting that fucosylation in progressive melanomas might also affect chemotherapeutic responsiveness. Our findings establish the regulation and importance of protein fucosylation in melanoma and point to the importance of studying fucose analogs as part of combination therapy of this and other tumor types.
MATERIALS AND METHODS
Cell lines
Generally, the melanoma cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillinstreptomycin antibiotics. YUGASP melanoma cells were maintained in Opti-modified Eagle’s medium (Opti-MEM) supplemented with 10% bovine serum and antibiotics. Primary human neonatal melanocytes were maintained in melanocyte growth medium. All media were purchased from Life Technologies. L-fucose was purchased from Fisher Scientific.
Antibodies and immunostaining reagents
Antibodies used were purchased as follows: ATF2 (C-19 for immunoblotting and C-19X for ChIP assays), PKCε (C15), and heat shock protein 90α/β (H-114) from Santa Cruz Biotechnology; pT52-ATF2 from PhosphoSolutions; β-tubulin (E7-s) from the Developmental Studies Hybridoma Bank; CD45 (30-F11) from eBiosciences; and NKp46 (29A1.4) from Fisher Scientific. The FITC-conjugated lectins UEA1, LCA, and PSA were purchased from Vector Laboratories. The general membrane stain was purchased from Sigma-Aldrich.
DNA constructs and transfection
DNA plasmids were all transfected using JetPrime (Polyplus) or Lipofectamine 2000 (Invitrogen) according to the manufacturers’ protocols. The constitutively active His-tagged PKCε construct was a gift from J. Moscat. Other plasmids and siRNAs were previously described (25). EV and human FUK (pLENTI-GFP-EV and pLENTI-GFP-FUK) constructs were acquired from the Genomics Core facility of the Sanford Burnham Prebys Medical Discovery Institute (SBP). The mFuk construct was generated by cloning of mouse Fuk from murine neural progenitor complementary DNA (cDNA) (a gift from the Terskikh laboratory at SBP; primers used in table S1) and insertion into the pLENTI-GFP-EV construct at the Xba I and Nhe I sites within the pLENTI-GFP-EV multiple cloning site.
qRT-PCR analyses
RNA from cells treated as indicated was extracted using the Gene Elute Mammalian Total RNA Extraction System (Sigma-Aldrich). With the High-Capacity Reverse Transcriptase System (Life Technologies), the RNA was reverse-transcribed to cDNA, and qRT-PCR analysis was performed using the SYBR Green RT-PCR reagent (Invitrogen) on a Bio-Rad CFX Connect Real-Time System. The qRT-PCR cycles were designated as follows: 95°C for 10 min; 50 cycles of 95°C for 15 s, 55°C for 60 s, 72°C for 30 s; 95°C for 15 s, 55°C for 5 s; and 95°C for 30 s. Expression of the indicated genes was normalized to that of histone H3A. Primers for qRT-PCR analyses (table S1) were designed using Primer-BLAST (National Center for Biotechnology Information).
Flow cytometric analyses
Cell surface lectin analysis was done as follows. Cells were seeded at 1 × 105 cells per well into six-well tissue culture plates and treated the next day as indicated in the figure legends. After treatment, cells were harvested using 10mM EDTA (pH 8.0), washed once with phosphate-buffered saline (PBS), and incubated in 1× PKH26 general membrane stain (Sigma-Aldrich) for 10 min at room temperature and washed in an excess of PBS. The cells were then fixed in 2% formaldehyde (in PBS) for 45 min at room temperature in the dark. The cells were then washed in PBS and blocked with 0.2% IgG- and protease-free bovine serum albumin (Jackson ImmunoResearch) for 30 min at room temperature, further washed twice with PBS, and stained with the indicated lectins (0.2 μg/ml) for an additional hour. After two further washes with PBS, the cells were analyzed by FACS, with n = 10,000 cells (within the forward scatter/side scatter whole-cell gate) per replicate over three independent experiments. The FACS data were then analyzed using FlowJo software (Tree Star).
FUK ChiP and PCR
Cells were washed in room-temperature PBS and fixed in 1% formaldehyde in PBS for 10 min at room temperature. The cells were washed and resuspended in 10 ml of neutralization buffer (0.125 M glycine in PBS) and incubated for 5 min at room temperature. The cells were washed twice with 10 ml of cold PBS, lysed in 1 ml of lysis buffer [1% SDS, 10 mM EDTA, and 50 mM tris-HCl (pH 8.0)], and sonicated on ice to shear the DNA to ~500–base pair fragments. The sonicated fragments were cleared of debris by centrifugation at 16,000g at 4°C for 5 min, then diluted 1:10 in dilution buffer [1% Triton X-100, 0.01% SDS, 150 mM NaCl, 2 mM EDTA, and 20 mM tris-HCl (pH 8.0)] and precleared by rotating incubation with protein A/G beads (Santa Cruz Biotechnology) at 4°C for 2 hours. Equal concentrations of sonicated chromatin were incubated with 2 μg of control IgG or ATF2 (C-19X, Santa Cruz Biotechnology) antibody against ATF2 and allowed to incubate overnight at 4°C. Next, 25 μl of protein A/G beads was incubated with the samples for 4 hours with rotation at 4°C. The fragment-bound beads were then washed three times in wash buffer 1 [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM tris-HCl (pH 8.0)], followed by one wash in wash buffer 2 [0.1% SDS, 1% Triton X-100, 2mMEDTA, 500mM NaCl, and 20 mM tris-HCl (pH8.0)]. Next, the ATF2-bound chromatin was eluted from the beads in 300 μl of elution buffer (1% SDS and 100 mM NaHCO3) for 15 min with constant agitation. Six microliters of protease K (10 mg/ml) and 3 μl of ribonuclease A (20 mg/ml) were added to each eluted sample and incubated at 37°C for 30 min with agitation. The beads were pelleted by centrifugation at 16,000g and then transferred to new tubes; 12 μl of 5 M NaCl was added to the supernatants and incubated further at 65°C overnight to reverse the formaldehyde cross-link. The chromatin was then purified using a QIAquick PCR Purification Kit (Qiagen). Finally, 2 μl of eluted chromatin fragments per sample was used in a PCR to detect ATF2 binding to the FUK promoter as follows: 95°C for 10 min; 50 cycles of 95°C for 15 s, 55°C for 60 s, 72°C for 30 s. Primers used to detect the FUK promoter are shown in table S1.
Secreted luciferase assays
Secreted promoter luciferase constructs were generated as depicted in Fig. 1D. The wild-type FUK promoter was subcloned from WM793 cDNA into the pMCS–Gaussia Luc vector (Thermo Fisher Scientific) using Eco RI and Hind III restriction enzyme sites. ATF2 binding site mutant promoter constructs (E1, E2, and E3) were generated using the Lightning Mutagenesis Kit (Agilent) according to the manufacturer’s protocols. Primers are provided in table S1.
Cells were transfected as indicated, and media samples were collected and measured for Gaussia and Cypridina luciferase activity using a standard luminometer. Gaussia luciferase activity values were normalized to Cypridina luciferase activity values (from pCMV–Cypridina luciferase construct, which exhibits a constitutively cytomegalovirus (CMV) promoter–driven secreted Cypridina luciferase). The Cypridina luciferase–normalized values were plotted relative to the values indicated in the figure legends. The data are representative of three independent experiments.
Quantitation of L-fucose uptake and phosphorylated L-fucose (fucose-1-phosphate)
[3H]-L-Fucose metabolic labeling and analysis was performed as previously described, with minor modifications (48) as follows: WM793, WM1346, 501Mel, and HEK293 cells were seeded at ~60 to 70% confluence in six-well plates. An extra plate was seeded at the exact same density for normalization of protein concentration. The next day, the cells were labeled for 2 hours using 1 μCi [3H]-L-fucose + 10 μM unlabeled L-fucose. After 2 hours, the radiolabeled medium was gently removed, and cells were quickly washed with 1× PBS. Next, 0.5 ml of 50% methanol [1:1 methanol/water (v/v)] was added to each well. The six-well plateswere covered with lids and placed on dry ice (dry ice was also placed on top of the lids) for 30 min. After 30 min, the lysates were scraped into Eppendorf tubes, and 0.25 ml of chloroform was added to each tube. The tubes were carefully vortexed and centrifuged (13,000 rpm, 4°C, 20 min). The aqueous (top) layer was removed and split equally: one-half for scintillation counting of total aqueous L-fucose and the other half for quaternary aminoethyl (QAE) binding and scintillation counting (to assess fucose-1-phosphate).
The QAE binding was next performed to measure intracellular soluble fucose-1-phosphate levels (QAE resin binds to charged molecules; thus, phosphorylated [3H]-L-fucose would bind, whereas nonphosphorylated [3H]-L-fucose would not). For the control, QAE beads that were incubated for the same 15-min period as other conditions in extracted fraction, except that the control beads were incubated with media that contained only [3H]-L-fucose (no cells).
Using the other half of the extracted aqueous fractions, 25 μl of washed, packed QAE resin was added to the aqueous extracts, to which an additional 500 μl of 2 mM tris (pH 8.0) was added. The tubes were rotated at room temperature for 15 min. The tubes were then centrifuged (13,000 rpm, 4°C, 1 min) to pellet the resin, and the resins were washed three times using 1 ml of 2 mM tris (pH 8.0). After washing, the resins were resuspended in 250 μl of 2 mM tris (pH 8.0) and transferred to scintillation vials with 5 ml of EcoLume each. The samples were then subjected to scintillation counting. Triplicates of 1 μCi L-fucose in EcoLume alone were used for starting disintegrations per minute (DPM) controls. The algorithm used for calculating free L-fucose was as follows: [(final DPM/starting DPM) × 10 nmol]/(total μg protein) × 1000 = pmol L-fucose/μg protein.
Hanging droplet culture and scratch, Matrigel invasion, and Boyden chamber assays
For hanging droplet culture, 2.5 × 102 cells of the indicated cell lines were cultured upside down in 30 μl of standard culture medium. After 3 days, the hanging droplet cultures were imaged. For scratch assays, the indicated cell types were seeded into 12-well plates and grown to confluence. Twenty-four hours before scratch and imaging at the indicated time points, the wells were refreshed with new media. In the case of L-fucose or ac-GDP-L-fucose treatment, the cells were seeded and incubated in the presence of 50 μM L-fucose or 25 μM ac-GDP-L-fucose for 48 hours, followed by refreshing of media with L-fucose or ac-GDP-L-fucose and an additional overnight culture before wounding and imaging. All imaging in the above assays was performed using a standard light microscope. For the Matrigel invasion assay, GFP-coexpressing EV- or FUK-expressing 501Mel, LU1205, and HEK293 cells were cultured as spheroids in hanging droplets for 2 days and then transferred into 24-well plates containing 100 μl of solidified Matrigel (BD Biosciences) and then overlaid with an additional 150 μl of Matrigel. The spheroids were imaged as indicated using a standard light microscope. Boyden chamber assays were performed using standard tissue culture migration chambers (Corning) according to the manufacturer’s protocols and imaged at the indicated time points using a fluorescence microscope.
Fuk mouse models
Six mice were assigned to each of the conditions indicated in Fig. 3. For Fig. 3 (E and F), 1.25 ×106 SW1 murine melanoma cells were subcutaneously implanted into the rear right flanks of syngeneic C3H/HeJ mice. After about 10 days, when the primary tumors reached ~50 to 100 mm3, the mice were fed with either control water or 100 mM L-fucose–supplemented water, which was refreshed every 3 days. Tumor volume was measured at the indicated time points. When tumors reached ~2 cm3, the animals were sacrificed, and the tumors and lungs were harvested for subsequent analysis. Lung metastases were quantitated in >12 progressive sections through the lungs of three representative mice per group. For Fig. 3F, EV-and mFuk-expressing SW1 cells were implanted into four groups of C3H/HeJ mice (two groups each for EV and mFuk; one group of EV and mFuk tumor-bearing mice were fed with control water, one group of EV and mFuk tumor-bearing mice were fed with 100 mM L-fucose–supplemented water, which was refreshed every 3 days), analyzed, and harvested as above.
TMA analysis
Melanoma TMA analysis has been previously described (25) with minor modifications for immunofluorescence staining. UEA1-FITC lectin (EY Laboratories Inc.) and a 1:1 cocktail of S100 and HMB45 antibodies (DAKO) were applied at 0.2 μg/ml and 1:500 dilution, respectively. The TMA was counterstained with DAPI. Individual images of each histospot were captured by fluorescence microscopic scanning and image extraction using Aperio ScanScope. Briefly, to measure the UEA1 signals within S100/HMB45-positive melanoma cells in each histospot, each histospot image was subjected to the following measurement process using ImageJ software [National Institutes of Health (NIH)] by a blinded evaluator (E.L.): (i) each raw image was split into individual red (S100/HMB45), green (UEA1-FITC lectin), and blue (DAPI) channels; (ii) S100/HMB45-positive cells were gated by signal thresholding in the red channel; (iii) a selection mask was designated by the thresholded areas; (iv) integrated intensity and area of the selected red area were measured; (v) the selection mask was transferred to the green channel, and integrated intensity was measured; “UEA1 scores” were generated by the following equation: UEA1-FITC intensity/(S100 or HMB45 intensity × area). The blinded scores were then submitted to a second evaluator (L.J.) (who was unblinded to the information for each patient sample) for analysis of UEA1 abundance in primary and metastatic cohorts, as well as survival analysis and correlation with clinical parameters, using JMP version 5.0 software (SAS Institute). The difference in UEA1 abundance between primary and metastatic tissues was evaluated using unpaired t tests. The prognostic significance of UEA1 was first assessed using the Cox proportional hazards model with survival as an end point. For the association between UEA1 and survival, scores were dichotomized into “high” and “low” by the median value. Kaplan-Meier curves were generated; P values corresponding to these curves were obtained by the Mantel-Cox log-rank method. The association between scores and other clinical parameters was assessed by t tests or analysis of variance.
FUK and ATF2 expression correlation analysis of TCGA cutaneous melanoma
Briefly, using Illumina HiSeq data from a TCGA cutaneous melanoma study (473 samples), we computed the correlation between FUK and ATF2 expression and visualized the expression patterns in the UCSC Cancer Genome browser (https://genome-cancer.ucsc.edu/proj/site/hgHeatmap/; filename: TCGA_SKCM_exp_HiSeqV2-2015-02-24.tgz; version: 2015-02-24). The data were acquired from the UCSC Genome browser and analyzed as detailed in the Supplementary Materials.
Statistical analysis
Calculation of SDs was performed using the STDEV function in Microsoft Excel. SEMs were calculated by dividing the SD by the square root of n. P values were calculated using standard t test in GraphPad (GraphPad Software Inc.). Statistical methods were verified by a biostatistician (Y. Kim, H. Lee Moffitt Cancer Center, Tampa, FL).
Supplementary Material
Fig. S1. Transcriptional regulation of FUK by ATF2 and L-fucose uptake in melanoma cells.
Fig. S2. Increasing melanoma fucosylation slows motility and invasiveness.
Fig. S3. Increasing melanoma tumor fucosylation increases intratumoral CD45+ and NKp46+ immune cell infiltrates.
Table S1. Primers.
Acknowledgments
We thank members of the Ronai laboratory for critical scientific discussions and reading of this manuscript. We are grateful to the Sanford Burnham Prebys (SBP) bioinformatics, flow cytometry, and histology core and animal facilities for technical support. We are grateful to B. Ng and D. Scott (SBP) for guidance on the [3H]-L-fucose labeling and quantitation of L-fucose and fucose-1-phosphate experiments. We thank J. Moscat for providing the constitutively active His-tagged PKCε construct and the Terskikh laboratory (SBP) for providing murine neural progenitor cDNA (for cloning of mFUK). We thank Y. Kim (H. Lee Moffitt Cancer Center, Tampa, FL) for reviewing our statistical methods.
Funding: We would like to acknowledge support from the NIH (R01DK99551, to H.H.F.), the National Cancer Institute [PO1 (CA128814) and RO1 (CA179170)], the Hervey Family Non-Endowment Fund at the San Diego Foundation, a Melanoma Research Foundation grant to Z.A.R., and K99 (CA172705) grant and T32 (CA121949) fellowship to E.L. This work was also supported by the Assistant Secretary of Defense for Health Affairs through the Peer-Reviewed Cancer Program under award no. W81XWH-14-1-0127 to Z.A.R.
Footnotes
Author contributions: E.L. contributed to the experimental design and execution, data analyses, manuscript writing, and figure preparation; Y.F., D.D., and G.C. performed the animal experiments; M.N.F. designed the ac-GDP-L-fucose preparation and fucosylation experiments; A.P. performed the bioinformatics; L.J. performed the unblinded TMA data analysis; H.K. was our melanoma oncology collaborator and contributed to the TMA experiments; H.H.F. contributed to the fucosylation experimental design and provided technical advice; Z.A.R. contributed to the experimental design, data analyses, manuscript writing, and figure preparation.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The RNA sequencing data sets obtained from TCGA can be found at https://genome-cancer.ucsc.edu/proj/site/hgHeatmap, following the instructions provided in the Supplementary Materials.
REFERENCES AND NOTES
- 1.Damsky WE, Theodosakis N, Bosenberg M. Melanoma metastasis: New concepts and evolving paradigms. Oncogene. 2014;33:2413–2422. doi: 10.1038/onc.2013.194. [DOI] [PubMed] [Google Scholar]
- 2.Balch CM, Gershenwald JE, Soong S-j, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr, Morton DL, Ross MI, Sober AJ, Sondak VK. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 4.Haass NK, Smalley KSM, Li L, Herlyn M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005;18:150–159. doi: 10.1111/j.1600-0749.2005.00235.x. [DOI] [PubMed] [Google Scholar]
- 5.Kirschmann DA, Seftor EA, Hardy KM, Seftor REB, Hendrix MJC. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726–2732. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferguson J, Arozarena I, Ehrhardt M, Wellbrock C. Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene. 2013;32:86–96. doi: 10.1038/onc.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, Weinberg RA. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. 2005;37:1047–1054. doi: 10.1038/ng1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, Testori A, Larue L, Goding CR. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nogueira C, Kim KH, Sung H, Paraiso KHT, Dannenberg JH, Bosenberg M, Chin L, Kim M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene. 2010;29:6222–6232. doi: 10.1038/onc.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015;28:390–406. doi: 10.1111/pcmr.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang S, Yang Y, Raycraft J, Zhang H, Kanan S, Guo Y, Ronai Z, Hellstrom I, Hellstrom KE. Melanoma cells transfected to express CD83 induce antitumor immunity that can be increased by also engaging CD137. Proc Natl Acad Sci USA. 2004;101:4990–4995. doi: 10.1073/pnas.0400880101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, Bahadoran P, Lacour JP, Tartare-Deckert S, Bertolotto C, Ballotti R. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene. 2012;31:2461–2470. doi: 10.1038/onc.2011.425. [DOI] [PubMed] [Google Scholar]
- 14.Arozarena I, Sanchez-Laorden B, Packer L, Hidalgo-Carcedo C, Hayward R, Viros A, Sahai E, Marais R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45–57. doi: 10.1016/j.ccr.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 15.Arozarena I, Bischof H, Gilby D, Belloni B, Dummer R, Wellbrock C. In melanoma, β-catenin is a suppressor of invasion. Oncogene. 2011;30:4531–4543. doi: 10.1038/onc.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindsay CR, Lawn S, Campbell AD, Faller WJ, Rambow F, Mort RL, Timpson P, Li A, Cammareri P, Ridgway RA, Morton JP, Doyle B, Hegarty S, Rafferty M, Murphy IG, McDermott EW, Sheahan K, Pedone K, Finn AJ, Groben PA, Thomas NE, Hao H, Carson C, Norman JC, Machesky LM, Gallagher WM, Jackson IJ, Van Kempen L, Beermann F, Der C, Larue L, Welch HC, Ozanne BW, Sansom OJ. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat Commun. 2011;2:555. doi: 10.1038/ncomms1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Díaz J, Mendoza P, Ortiz R, Díaz N, Leyton L, Stupack D, Quest AFG, Torres VA. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J Cell Sci. 2014;127:2401–2406. doi: 10.1242/jcs.141689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 19.Das SK, Bhutia SK, Kegelman TP, Peachy L, Oyesanya RA, Dasgupta S, Sokhi UK, Azab B, Dash R, Quinn BA, Kim K, Barral PM, Su ZZ, Boukerche H, Sarkar D, Fisher PB. MDA-9/syntenin: A positive gatekeeper of melanoma metastasis. Front Biosci. 2012;17:1–15. doi: 10.2741/3911. [DOI] [PubMed] [Google Scholar]
- 20.Zaki KA, Basu B, Corrie P. The role of angiogenesis inhibitors in the management of melanoma. Curr Top Med Chem. 2012;12:32–49. doi: 10.2174/156802612798919240. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, Niculescu-Duvaz D, Turajlic S, Hayes A, Gore M, Larkin J, Lorigan P, Cook M, Springer C, Marais R. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7:ra30. doi: 10.1126/scisignal.2004815. [DOI] [PubMed] [Google Scholar]
- 22.Bhoumik A, Gangi L, Ronai Z. Inhibition of melanoma growth and metastasis by ATF2-derived peptides. Cancer Res. 2004;64:8222–8230. doi: 10.1158/0008-5472.CAN-04-0714. [DOI] [PubMed] [Google Scholar]
- 23.Ronai Z, Yang YM, Fuchs SY, Adler V, Sardana M, Herlyn M. ATF2 confers radiation resistance to human melanoma cells. Oncogene. 1998;16:523–531. doi: 10.1038/sj.onc.1201566. [DOI] [PubMed] [Google Scholar]
- 24.Berger AJ, Kluger HM, Li N, Kielhorn E, Halaban R, Ronai Z, Rimm DL. Subcellular localization of activating transcription factor 2 in melanoma specimens predicts patient survival. Cancer Res. 2003;63:8103–8107. [PubMed] [Google Scholar]
- 25.Lau E, Kluger H, Varsano T, Lee K, Scheffler I, Rimm DL, Ideker T, Ronai ZA. PKCε promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Cell. 2012;148:543–555. doi: 10.1016/j.cell.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhoumik A, Jones N, Ronai Z. Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci USA. 2004;101:4222–4227. doi: 10.1073/pnas.0400195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varsano T, Lau E, Feng Y, Garrido M, Milan L, Heynen-Genel S, Hassig CA, Ronai ZA. Inhibition of melanoma growth by small molecules that promote the mitochondrial localization of ATF2. Clin Cancer Res. 2013;19:2710–2722. doi: 10.1158/1078-0432.CCR-12-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau E, Sedy J, Sander C, Shaw MA, Feng Y, Scortegagna M, Claps G, Robinson S, Cheng P, Srivas R, Soonthornvacharin S, Ideker T, Bosenberg M, Gonzalez R, Robinson W, Chanda SK, Ware C, Dummer R, Hoon D, Kirkwood JM, Ronai ZA. Transcriptional repression of IFNβ1 by ATF2 confers melanoma resistance to therapy. Oncogene. 2015;34:5739–5748. doi: 10.1038/onc.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker DJ, Lowe JB. Fucose: Biosynthesis and biological function in mammals. Glycobiology. 2003;13:41R–53R. doi: 10.1093/glycob/cwg054. [DOI] [PubMed] [Google Scholar]
- 30.Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, Homeister JW, Lowe JB. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vestweber D, Lühn K, Marquardt T, Wild M. The role of fucosylation in leukocyte adhesion deficiency II. Ernst Schering Res Found Workshop. 2004;44:53–74. doi: 10.1007/978-3-662-05397-3_4. [DOI] [PubMed] [Google Scholar]
- 32.Macartney JC. Fucose-containing antigens in normal and neoplastic human gastric mucosa: A comparative study using lectin histochemistry and blood group immunohistochemistry. J Pathol. 1987;152:23–30. doi: 10.1002/path.1711520104. [DOI] [PubMed] [Google Scholar]
- 33.Tateno H, Nakamura-Tsuruta S, Hirabayashi J. Comparative analysis of core-fucose-binding lectins from Lens culinaris and Pisum sativum using frontal affinity chromatography. Glycobiology. 2009;19:527–536. doi: 10.1093/glycob/cwp016. [DOI] [PubMed] [Google Scholar]
- 34.Häuselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol. 2014;4:28. doi: 10.3389/fonc.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004;95:377–384. doi: 10.1111/j.1349-7006.2004.tb03219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, Hsu TL, Wong CH. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci USA. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquardt T, Lühn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976–3985. [PubMed] [Google Scholar]
- 38.Sarkar AK, Brown JR, Esko JD. Synthesis and glycan priming activity of acetylated disaccharides. Carbohydr Res. 2000;329:287–300. doi: 10.1016/s0008-6215(00)00200-7. [DOI] [PubMed] [Google Scholar]
- 39.Moriwaki K, Noda K, Furukawa Y, Ohshima K, Uchiyama A, Nakagawa T, Taniguchi N, Daigo Y, Nakamura Y, Hayashi N, Miyoshi E. Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology. 2009;137:188–198. doi: 10.1053/j.gastro.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Ohyama C, Kanto S, Kato K, Nakano O, Arai Y, Kato T, Chen S, Fukuda MN, Fukuda M. Natural killer cells attack tumor cells expressing high levels of sialyl Lewis x oligosaccharides. Proc Natl Acad Sci USA. 2002;99:13789–13794. doi: 10.1073/pnas.212456599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finne J, Tao TW, Burger MM. Carbohydrate changes in glycoproteins of a poorly metastasizing wheat germ agglutinin-resistant melanoma clone. Cancer Res. 1980;40:2580–2587. [PubMed] [Google Scholar]
- 42.Kawano T, Takasaki S, Tao TW, Kobata A. N-linked sugar chains of mouse B16 melanoma cells and their low-metastasizing variant selected by wheat germ agglutinin. Glycobiology. 1991;1:375–385. doi: 10.1093/glycob/1.4.375. [DOI] [PubMed] [Google Scholar]
- 43.Feizi T. Carbohydrate-mediated recognition systems in innate immunity. Immunol Rev. 2000;173:79–88. doi: 10.1034/j.1600-065x.2000.917310.x. [DOI] [PubMed] [Google Scholar]
- 44.Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG, Turnbaugh PJ, Chervonsky AV. Rapid fucosylation of intestinal epithelium sustains host–commensal symbiosis in sickness. Nature. 2014;514:638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myers J, Huang Y, Wei L, Yan Q, Huang A, Zhou L. Fucose-deficient hematopoietic stem cells have decreased self-renewal and aberrant marrow niche occupancy. Transfusion. 2010;50:2660–2669. doi: 10.1111/j.1537-2995.2010.02745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo Y, Ellis LZ, Dallaglio K, Takeda M, Robinson WA, Robinson SE, Liu W, Lewis KD, McCarter MD, Gonzalez R, Norris DA, Roop DR, Spritz RA, Ahn NG, Fujita M. Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J Invest Dermatol. 2012;132:2440–2450. doi: 10.1038/jid.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiese TJ, Dunlap JA, Yorek MA. Effect of L-fucose and D-glucose concentration on L-fucoprotein metabolism in human Hep G2 cells and changes in fucosyltransferase and α-L-fucosidase activity in liver of diabetic rats. Biochim Biophys Acta. 1997;1335:61–72. doi: 10.1016/s0304-4165(96)00123-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Transcriptional regulation of FUK by ATF2 and L-fucose uptake in melanoma cells.
Fig. S2. Increasing melanoma fucosylation slows motility and invasiveness.
Fig. S3. Increasing melanoma tumor fucosylation increases intratumoral CD45+ and NKp46+ immune cell infiltrates.
Table S1. Primers.