

Abstract
Mycobacteria have been reported to cause a wide range of human diseases. We present the first whole-genome study of a Non-Tuberculous Mycobacterium, Mycobacterium sp. UM_CSW (referred to hereafter as UM_CSW), isolated from a patient diagnosed with bronchiectasis. Our data suggest that this clinical isolate is likely a novel mycobacterial species, supported by clear evidence from molecular phylogenetic, comparative genomic, ANI and AAI analyses. UM_CSW is closely related to the Mycobacterium avium complex. While it has characteristic features of an environmental bacterium, it also shows a high pathogenic potential with the presence of a wide variety of putative genes related to bacterial virulence and shares very similar pathogenomic profiles with the known pathogenic mycobacterial species. Thus, we conclude that this possible novel Mycobacterium species should be tightly monitored for its possible causative role in human infections.
Introduction
Mycobacteria show multiple modes of existence in nature, ranging from soil-dwelling saprophytes to animal and human pathogens [1]. The species associated with human infections have been increasing, particularly among the non-tuberculous mycobacteria (NTM) [2]. Although NTM species cause mostly opportunistic infections from an environmental source [3], potentially fatal infections [4] and human-to-human transmission have also been reported [5].
In our routine diagnostic service, a NTM was isolated from the sputum sample of a patient with a long history of bronchiectasis. As the results of our gene-based phylogenetic analysis suggested that it might be a novel species, we performed whole-genome sequencing to obtain more information on its taxonomic position, biology and pathogenicity. Here, we report the supporting evidence for its novelty and some interesting observations from our genomic analysis. The UM_CSW genome assembly has been deposited at GenBank under the accession number AUWQ00000000.
Materials and Methods
Library preparation and whole-genome sequencing
For DNA library preparation, the DNA samples were fragmented using Covaris S2 for 120 seconds at temperature of 5.5–6.0 degree Celsius. Agilent BioAnalyzer 2100 was used to examine the quantity and quality of the fragmented DNA. The sample was size selected using Invitrogen 2% agarose E-gels. Only the fragments having the adapter molecules at both ends underwent 10 cycles of PCR for the purpose of library construction. The validation of constructed genomic library was performed using Agilent BioAnalyzer 2100. The pool of 8pM was loaded onto 1 lane of Illumina HiSeq 2000 flow cell v3 for 100bp paired-end sequencing according to manufacturer’s recommended protocols.
Read preprocessing, genome assembly and annotation
Exact duplicates and exact reverse complement duplicates reads resulted from shot-gun sequencing were filtered by using standalone PRINSEQ lite version 0.20 [6]. Sequencing of UM_CSW at approximately 800x coverage (assuming genome size of 6Mbp) enabled us to prepare reads for assembly at higher quality by further filtering on reads with ambiguous nucleotides. The raw Illumina reads were trimmed at a threshold of 0.01 (Phred score of 20) and the sequences obtained were de novo assembled using CLC Genomic Workbench version 5.1 (CLC bio, Denmark). The assembly of the reads was carried out at length faction of 0.7 and similarity fraction of 0.9, accepting minimal size of contig at 500bp.
The assembled genome was annotated using the RAST pipeline, which is a fully automated annotation engine for complete or draft archaeal and bacterial genomes [7]. Of the 6,076 RAST-predicted protein-coding genes, 1,944 (32%) genes were successfully assigned to 376 subsystems/functional categories, whereas the remaining genes were not assigned to any subsystem. The presence of plasmid replicons was predicted by PlasmidFinder-1.2 [8]. Clusters of regularly interspaced repeats (CRISPR) were identified by CRISPR Finder [9].
Genome size estimation
To estimate the genome size of UM_CSW, k-mer was counted using the tool called Preqc from the SGA assembly program [10]. The tool counts and plots 31-mer histogram from 20,000 reads and estimates the genome size by identifying the peak of the Poisson distribution. The estimation is based on the principle as follows, where the mean number of times a unique genomic k-mer appears in the reads, λ_k is as follows:
Where n is the number of reads, l is the read length and G being the genome size.
Phylogenetic study
Three bacterial classification markers hsp65, rpoB, and 16S rRNA were selected for the single gene-based phylogenetic studies. To clarify the phylogenetic relationship between UM_CSW and members of the M. avium complex using whole-genome data, the core genome of M. intracellulare ATCC13950, M. indicus pranii MTCC9506, M. colombiense CECT3035, M. avium K10 along with M. tuberculosis CCDC5079, M. afficanum GM041182 and M. marinum M was identified using Panseq 2.0 [11]. M. tuberculosis CCDC5079, M. africanum GM041182, and M. marinum M were used as outgroups. The SNPs within each core genome were identified, extracted and concatenated into one large sequence.
Model testing was performed on each set of aligned sequence prior the phylogenetic reconstructions. Using the proposed substitution models based on corrected Akaike information criterion, maximum likelihood trees were inferred with 1,000 boostrapping replication. All the likelihood estimations were done in MEGA version 5.2 [12].
Amino Acid Identity (AAI) Analysis
The average amino acid identity (AAI) was calculated using the method described by Konstantinidis & Tiedje [13]. Mycobacterial genomes from GenBank were downloaded and annotated using RAST. The protein-coding sequences of the UM_CSW genome was used as the reference for comparison against other mycobacterial genomes using the BLAST search to determine the conserved genes. The cut-off for the BLAST search was set at ≥30% sequence identity and ≥70% sequence coverage at the amino acid level. The average of the amino acid identity of all conserved genes between a pair of genomes was computed to measure the genetic relatedness between them.
Whole-genome Average Nucleotide Identity (ANI) analysis
The ANI has been reported to be a reliable alternative to the use of classical housekeeping genes for the evaluation of bacterial genetic relatedness [13]. To categorize genomes into the same species, the genomes must show an ANI value > 95% which is equivalent to the 70% DNA-DNA Hybridization (DDH) level [14]. The ANI is calculated based on the average percentage sequence similarity between all conserved genes in a pair of different genomes. The ANI calculation were performed using JSpecies [15] using the Blast parameters of 150 as dropoff value for gapped alignment, -1 as the penalty for a nucleotide mismatch, F as the filter query sequence, 1e-15 as the expectation value (E) and 2 as the number of processors to use; ANI calculation parameters as, percentage of identity > = 30, percentage of alignment > = 70 and length as 1020.
Genome Island prediction
Genomic islands in the genome of UM_CSW were predicted using IslandViewer, which is based on unique features in codon usage, dinucleotide sequence composition and presence of mobile element genes [16]. IslandViewer integrates few other genomic island prediction methods to predict the genomic islands. It includes the sequence composition base approaches SIGI-HMM [17] and IslandPath-DIMOB [18] that were shown to have highest specificity of around 86% to 92% and an accuracy of about 86%, as well as the comparative genomics approach IslandPick [19].
Estimation of pathogenicity of the mycobacterial genomes
PathogenFinder 1.1, a web-server for the prediction of bacterial pathogenicity [20] was used to estimate the pathogenicity of UM_CSW along with 19 other mycobacterial genomes. The genome sequences of type strains of 19 mycobacterial species were downloaded from NCBI in the fasta format for the analysis. Each genome was then uploaded to the PathogenFinder 1.1 web-server individually, selecting Automatic Model Section as Phylum and Assembled Genome/Contigs* for the Sequencing Platform as the parameters for the estimation.
Virulence gene and prophage prediction
The protein sequences predicted by RAST were BLAST searched against the virulence factor database (VFDB) [21]. Using our in-house Perl scripts, we selected the genes that were orthologous to virulence genes with at least 60% identity and at least 60% sequence coverage in query and subject.
Prophages in the genome of UM_CSW was identified using PHAST (PhAge Search Tool) web server [22]. Putative protein sequences in the genome of UM_CSW were BLAST searched against two prophage sequence databases to identify phage-related proteins. The two databases are the NCBI phage database and the prophage database that contains 159 prophage regions and 9,061 proteins that are not found in the NCBI phage database.
Comparative Pathogenomics Analysis
All Mycobacterium genomes used in this analysis were downloaded from NCBI and re-annotated using the RAST pipeline. For each genome, all RAST-predicted protein-coding gene sequences were searched against the VFDB database [21] through the use of BLASTP [23]. The genes that were orthologous to virulence genes must have at least 60% identity and at least 60% sequence coverage in query and subject. The output results were processed and passed to our in-house R scripts for clustering and visualization using a heat map with dendrograms showing clustered mycobacterial species with closely related sets of virulence genes, sorted according to similarities across the strains and genes. This graphical representation is useful and to allow for the spontaneous comparison of pathogenomic profiles between genomes.
Results
General Genome Overview
We sequenced the genome of UM_CSW using the Illumina HiSeq technology platform at high sequencing depth (approximately 737x, assuming genome size of 6.51Mbp). This yielded 48,832,804 raw reads. The de novo assembly of the pre-processed 48,173,293 reads resulted in 186 contigs with a total genomic size of 6.4Mbp. With the contig sizes ranging from 521 bp to 231,836 bp, the assembly had N75, N50 and N25 values of 39,170 bp, 66,558 bp and 123,976 bp respectively. This assembly covered 98.3% of the predicted genome size of UM_CSW (6.51Mbp), suggesting that the assembled genome is close to complete. As with other known mycobacteria, the genome of UM_CSW has a high G+C composition of 67.8%. It contains 6,076 putative coding sequences (CDSs), 47 tRNAs and a single 16S-5S-23S ribosomal RNA operon (S1 Table). No plasmid replicons, prophages or clusters of regularly interspaced repeats (CRISPR) were found in the genome. Circular representation of UM_CSW genome is shown in Fig 1.
Fig 1. Circular representation of UM_CSW genome.

The indication for each feature from the outermost layer: (a) The alternating brown and yellow coloured bars represent the odd and even numbered contigs respectively; (b) The dark and light grey coloured bars represent the protein-coding genes in the forward and the reverse strands respectively; (c) rRNAs (light blue) and tRNAs (dark blue); (d) genomic islands (green); (e) virulence genes (red); (f) GC plot (purple and light green).
Molecular and Phylogenetic Analyses
The taxonomic position of UM_CSW was initially examined by phylogenetic inferences of three global bacterial identification markers, the 16S rRNA, hsp65 and rpoB. Using type strains of recognized Mycobacterium species, the 16S rRNA-based tree showed the closest relatives of UM_CSW to be M. interjectum (99.5% sequence similarity; 100% sequence coverage) and M. saskatchewanense (99% sequence similarity; 100% sequence coverage) (S1 Fig), both of which are slow-growing schotochromogens. In the hsp65-based tree, however, the closest neighbors were two non-pigmented rapid growers, M. cosmeticum (95.7% sequence similarity) and M. canariasense (94.9% sequence similarity) (S2 Fig). Further incongruity was seen in the rpoB tree where another two slow-growing schotochomogens, M. arosiense and M. colombiense were seen as the closest relatives with 96.5% and 96.7% sequence similarities respectively (S3 Fig). The other species grouped along with UM_CSW in the rpoB tree were M. intracellulare, M. chimaera and M. timonense which, along with M. arosiense and M. colombiense, form a part of the M. avium complex (MAC) [24]. Interestingly, neither the 16S- nor the hsp65-based tree grouped UM_CSW with any of the members of the M. avium complex. Moreover, the closest relatives, M. interjectum and M. saskatchewanense in the 16S gene-based tree, and M. cosmeticum and M. canariasense in the hsp65-based tree, had more distant relationships with UM_CSW in the rpoB-based tree, with sequence similarities of 95.7%, 95.4%, 91.6%, and 90.3% respectively. The close relationship between UM_CSW and the members of the M. avium complex is also supported by a core-genome SNP-derived phylogenetic tree (Fig 2).
Fig 2. Phylogenetic relationship of UM_CSW with other mycobacterial species.

The phylogenetic tree was generated using core genome SNPs and the maximum likelihood method. Bootstrap numbers were generated in 1,000 runs. Nodes with bootstrap support values are indicated.
ANI and AAI Analyses
In an attempt to resolve the discrepancies from the single gene based phylogenetic inferences, we subjected our isolate to ANI and AAI analyses. Using UM_CSW as the reference genome, we calculated the ANI and AAI for 31 other mycobacterial species with genome sequences available. Two genomes with an ANI >95% and AAI >96% were considered to belong to the same species, while those with ANI and AAI below these thresholds were considered separate species [14].
Our analysis showed that the closest relatives of UM_CSW were all members of the M. avium complex such as M. parascrofulaceum ATCC25954, Mycobacterium sp. MOTT36Y, M. intracellulare ATCC13950, M. colombiense CECT3035, and M. avium 104 (Fig 3 and S4 Fig). The ANIs between UM_CSW and these species ranged between 81.6% and 82.1% (Fig 3), whereas the AAI values ranged between 81.7% and 82.2%. The ANI and AAI values between UM_CSW and M. cosmeticum which was the closest relative in the hsp65-based tree were only 75% and 68.2% respectively. These ANI (<95%) and AAI (<96%) values suggest that UM_CSW is genetically distinct from known members of the M. avium complex and other mycobacteria used in this analysis.
Fig 3. ANI analysis.

The top six relatives of Mycobacterium sp. UM_CSW are all members of the M. avium complex.
Functional Analysis
The RAST subsystem annotation revealed that a large portion of the genome is comprised of genes related to basic structures and functions of the bacterial cell (Fig 4). In addition, there were a large number (122) of stress response genes that may indicate the ability of UM_CSW to adapt to different environments. Interestingly, no genes were involved in phages, prophages, transposable elements and plasmids, suggesting that UM_CSW may have a strong defence system to prevent the invasion of foreign DNA into its genome. Among the 166 genes involved in virulence, disease and defence, the majority (78%) were involved in invasion and intracellular resistance, while the rest (22%) were involved in the resistance to antibiotics and toxic compounds. In this latter category, there were genes involved in copper homeostasis (7 genes), resistance to beta-lactam antibiotics (7 genes), cobalt-zinc-cadmium resistance (5 genes) and resistance to fluoroquinolones (2 genes), as well as two genes encoding mercuric reductase. However, it was arsenic resistance genes which led this category with 13 genes. The presence of genes responsible for metal and arsenic resistance suggests that UM_CSW might have an environmental origin.
Fig 4. Functional classification of RAST-predicted protein-coding genes in the genome of UM_CSW.

Arsenic Resistance Potential
Studies have shown that arsenic may occur in four oxidation states, of which, arsenate [As(V)] and arsenite [As(III)] are readily soluble in water and abundantly present in nature. As(V), being an analogue of phosphate, can enter bacterial cells via phosphate transport systems (Pst and Pit) [25]. We found in the UM_CSW genome, putative Pst genes coding for the phosphate transport system by which arsenic may enter the cell. Microbes are known to reduce As(V) by two distinct mechanisms: the ArsC system that confers resistance by reducing As(V) and another mechanism that involves dissimilatory reduction of arsenate, as part of a respiratory pathway. The UM_CSW genome houses genes encoding arsenical resistance protein (Acr3), arsenate reductase (EC 1.20.4.1), arsenic efflux pump protein, and arsenical pump-driving ATPase (EC 3.6.3.16), which indicates that UM_CSW may have the AsrC system of arsenic resistance [25].
Genomic Island Analysis
We predicted eight putative Genomic Islands (GIs) that are at least 10kb in size (Table 1; S2 Table). All GIs showed bias of G+C content (58%-62.8%) compared to the overall G+C content (67.8%) of the UM_CSW genome.
Table 1. Summary information of the GIs predicted in the genome of UM_CSW.
| GIs | Genomic Length (bp) | Number of CDSs | G+C content (%) | Selected Genes |
|---|---|---|---|---|
| GI1 | 11,040 | 15 | 62.3 | ThiJ/PfpI family protein |
| putative transcriptional regulator, AraC family | ||||
| low molecular weight T-cell antigen TB8.4 | ||||
| GI2 | 14,745 | 13 | 61.7 | Transcriptional regulator, TetR family |
| putative cytochrome P450 hydroxylase | ||||
| Hydrolase | ||||
| Transcriptional regulator KorSA, GntR family | ||||
| Transfer protein traSA | ||||
| GI3 | 10,251 | 10 | 62.8 | Coenzyme F420-dependent oxidoreductase |
| Transcriptional regulator, TetR family | ||||
| putative cytochrome P450 hydroxylase | ||||
| Long-chain-fatty-acid—CoA ligase (EC 6.2.1.3) | ||||
| GI4 | 11,329 | 15 | 61.3 | TesB-like acyl-CoA thioesterase 4 |
| Transcriptional regulator, TetR family | ||||
| PROBABLE CHOLESTEROL DEHYDROGENASE | ||||
| Transcriptional regulator, IclR family | ||||
| GI5 | 14057 | 12 | 58 | ATP-dependent DNA helicase UvrD/PcrA |
| putative ATPase | ||||
| GI6 | 12666 | 12 | 59.7 | NADH oxidase (putative) |
| dNTP triphosphohydrolase, broad substrate specificity, subgroup 2 | ||||
| DNA methylase N-4/N-6 | ||||
| GI7 | 12727 | 5 | 59.9 | Superfamily II DNA/RNA helicases, SNF2 family |
| Adenine-specific DNA methylase containing a Zn-ribbon | ||||
| GI8 | 37271 | 37 | 62.6 | Integrase |
| Death on curing protein, Doc toxin | ||||
| Long-chain-fatty-acid—CoA ligase (EC 6.2.1.3) | ||||
| Butyryl-CoA dehydrogenase (EC 1.3.99.2) | ||||
| Transcriptional regulator, GntR family | ||||
| L-carnitine dehydratase/bile acid-inducible protein F | ||||
| Alpha-methylacyl-CoA racemase (EC 5.1.99.4) | ||||
| MaoC family protein | ||||
| Alcohol dehydrogenase (EC 1.1.1.1) | ||||
| 3-ketoacyl-CoA thiolase (EC 2.3.1.16) @ Acetyl-CoA acetyltransferase (EC 2.3.1.9) | ||||
| Enoyl-CoA hydratase (EC 4.2.1.17) | ||||
| 3-ketoacyl-(acyl-carrier-protein) reductase (EC:1.-) | ||||
| putative acyl-CoA dehydrogenase (EC:1.3.99.-) | ||||
| acyl-CoA dehydrogenase domain protein | ||||
| Acyl-CoA thioesterase II (EC 3.1.2.-) | ||||
| probable oxidoreductase/Short-chain dehydrogenase | ||||
| putative cytochrome P450 hydroxylase |
To identify the origin of these GIs, we performed simple BLAST searches using the sequences of the GIs against the National Center for Biotechnology Information (NCBI) NR database [26]. Interestingly, we found that the whole sequence of GI2 aligned perfectly with a sequence segment in the genomes of M. sp. MOTT36Y and M. yongonense 05–1390 (100% sequence identity; 100% sequence coverage; e-value of 0). This suggests that G12 might have originated from M. sp. MOTT36Y or M. yongonense 05–1390 and was inserted into the genome of UM_CSW through horizontal transfer. It is also possible that all these segments originated from the same source. The horizontal transfer of G12 is further supported by the presence of an integrase and tRNA genes which were found ahead of this GI. There are 13 putative genes in G12 comprising seven hypothetical proteins, several transcriptional regulator proteins and a transfer protein traSA flanking it. One of the transcriptional regulator proteins is from the KorSA, GntR family, which has been reported to be a transcriptional repressor usually binding to a conserved 17-nucleotide sequence upstream of the genes korSA, and pra, a gene reported to positively control the replication, integration, and excision of the mobile integrative genetic element of Streptomyces pSAM2 [27, 28]. The traSA gene has been reported to be essential for pSAM2 intermycelial transfer and pock formation [28]. However, in-spite of being smaller, TraSA has similarity to Streptomyces plasmid transfer protein and more generally to double‑stranded DNA translocases [28] which usually possess a transmembrane domain in their N‑terminal part, an FtsK/SpoIIIE domain in their central part, and a winged‑Helix‑Turn‑Helix (wHTH) fold region. Sequence analysis indicates that the transmembrane domain in the N‑terminal part might associate traSA with the membrane while the FtsK/SpoIIIE domain acts as a pump which actively transports double‑stranded DNA [29]. The GI also has PPE proteins and proteins from the TetR family which might be essential for the survival of the organism inside a host and the maintenance of pathogenicity.
GI4 is another interesting GI with 15 putative protein-coding genes, 11 of which encode hypothetical proteins. Of the remaining 4 proteins, one is from the TetR family, while the others are a TesB-like acyl-CoA thioesterase 4, an IclR family transcriptional regulator and a probable cholesterol dehydrogenase. Of the 11 hypothetical proteins, three showed strong homology with the Emopamil-binding protein (EBP) of M. rhodesiae. The EBP protein has emopamil binding domains, including the sterol acceptor site and the catalytic centre, which show Delta8-Delta7 sterol isomerase activity. Human sterol isomerase, a homologue of mouse EBP, is suggested not only to play a role in cholesterol biosynthesis, but also to affect lipoprotein internalisation. It has already been shown that the utilization of host cholesterol is essential for the persistence of Mycobacterium within the IFN-γ-activated macrophages [30]. Moreover, cholesterol plays a key role in the uptake of mycobacteria by macrophages and has also been reported to mediate the phagosomal association of a coat protein TACO, which prevents the degradation of mycobacteria in lysosomes [31]. Other reports suggested that cholesterol oxidase is essential for the pathogenesis conferred by Mycobacterium, by interfering with the TLR2-mediated signaling pathway and thus enabling intracellular growth and survival in human macrophages [32]. Thus the presence of the homolog of EBP and a probable cholesterol dehydrogenase in the same GI indicates that this region may be essential for cholesterol metabolism by the organism, which in turn, might play a role in its pathogenicity.
In GI1, we predicted a possible Stage II sporulation protein along with the presence of a putative heat shock protein 70 (partial), transcriptional regulator of AraC family, ThiJ/PfpI family protein and a response regulator receiver/ANTAR domain-containing protein. The presence of these proteins in close vicinity, points to the possibility of them being part of the stress response system of the organism. The predicted region also hosts a low molecular weight T-cell antigen TB8.4, a possible transmembrane protein or permease and a probable antibiotic biosynthesis monooxygenase.
The largest island, GI8, has a genomic length of 37,271 bp, a G+C content of 62.6% and 37 genes. This predicted region is flanked by a couple of hypothetical proteins followed by 4 tRNA genes and an integrase. One of the hypothetical proteins showed homology with a dehydratase MaoC which was preceded by another MaoC family protein. MaoC family proteins normally have an oxidoreductase activity and dehydratase MaoC has been reported to be an enoyl-CoA hydratase that is involved in linking the β-oxidation and polyhydroxyalkanoate (PHA) biosynthetic pathways [33]. The region also has the transcriptional regulator of the GntR family and a possible TetR family transcriptional regulator as well. Most interesting is the presence of Death on curing protein, Doc toxin, in this region. Doc toxin is a part of a two protein operon, the Phd-Doc toxin–antitoxin (TA) system (suicide or ‘‘addiction modules”) where Doc is the toxin and Phd (prevents-host-death) is the antitoxin. TA systems usually play an important role in cell survival during intervals of stress, by inducing a state of reversible growth arrest. TA systems have also been associated with biofilm formation, bacterial persistence during antibiotic treatment, and bacterial pathogenesis [34]. We could not find the antitoxin Phd, the counterpart of the Doc toxin in TA systems within the GI8. We found the presence of another Doc toxin which was immediately followed by a hypothetical protein showing strong homology with the antitoxin Phd in the genome; however, they were not a part of any of the predicted GIs.
The presence of these GIs suggests that UM_CSW is likely to have acquired, through horizontal gene transfer, new capabilities which are important for the survival and adaptation of this bacterium in its ecology and are potentially contributory to its virulence.
UM_CSW Has a Pathogenic Potential
Although UM_CSW was isolated from a clinically derived sample, it could be just a colonizing bacterium. To examine its pathogenicity, we used the PathogenFinder, which is a web-server for the prediction of bacterial pathogenicity by analysing the genome, or raw reads provided by the user [20]. PathogenFinder uses an unbiased method which relies on groups of proteins, created without regard to their annotated function or known involvement in pathogenicity. The method has been tested with many groups of bacteria and had achieved an accuracy of 88.6%. In this analysis, we also compared the predicted pathogenicity of UM_CSW with 19 other well-studied mycobacterial species (comprising both pathogenic and the non-pathogenic mycobacteria).
Our results showed UM_CSW to be predicted as a human pathogen with a probability of 0.841. As anticipated, all the known pathogenic mycobacteria were classified as human pathogens with high probability of around 0.8 and above, while the known non-pathogenic mycobacteria much lower probability (S4 Table). These results further support our view that UM_CSW might be a human pathogen capable of causing disease in the source patient.
Virulence Genes in the UM_CSW Genome
To examine the virulence gene profile of UM_CSW, we used our in-house analysis pipeline. We predicted 141 virulence genes which are orthologous to the experimentally verified virulence genes in the virulence factor database (VFDB) (S5 Table). This number constitutes 73.8% of the 191 genes in M. tuberculosis and includes genes important for mycobacterial cell wall structure, cell entry, protein and lipid metabolism, inhibition of antimicrobial factors, gene expression regulation, and metal transportation. For instance, UM_CSW appeared to have all three fbp-encoded proteins (fbpA, fbpB and fbpC) in the antigen 85 complex required for maintaining the integrity of the mycobacterial cell wall [35], the whole set of nine mammalian cell entry (mce) genes found widely among mycobacteria and five sigma factors (transcription initiation factors) associated with virulence in M. tuberculosis (sigA, sigE, sigF, sigH and sigM) [36]. The number of PE (proline-glutamate) and PPE (proline-proline-glutamate) protein families, however, is much less than in other mycobacterial pathogens, forming only approximately 1.8% of the coding genome (as predicted by the RAST server), compared to about 10% in M. tuberculosis [37]. These proteins, unique for mycobacteria are involved in host immune evasion and cell entry [38]. Molecular evolutionary studies have revealed that various orthologues of PE/PPE were found to be deleted in avirulent mycobacterial strains [39], indicating that this family of proteins, owing to their importance in disease pathogenesis, may be specific to the pathogenic mycobacteria [40] including NTM. The PE-PGRS (PE_polimorphic GC-rich sequences) genes that encode cell surface proteins are closely linked with 5 esx gene clusters (ESX1-5) [40]. In UM_CSW, we found only 3 ESX clusters, ESX 3, ESX 4 and ESX 5. The presence of ESX-5 supports the classification of UM_CSW as a slow-growing Mycobacterium, as most rapid-growing mycobacteria appear to lack this cluster of secretion factors [40]. Genes of the Lpr family (Lpr-BCDEGHKLOQ) were also predicted. LprG has been reported to play a vital role in the expression of surface-exposed lipoarabinomannan (LAM), through which M. tuberculosis interacts with the mannose receptors on macrophages. This interaction is critical for cell entry, inhibition of phagosome-lysosome fusion and intracellular survival of M. tuberculosis [41]. Studies have shown that genetic deletion of LprG resulted in reduced virulence of M. tuberculosis [42].
Virulence genes that inhibit apoptosis and resistance to host toxic compounds
Bacteria are able to cause disease if they are able to counteract the antimicrobial responses of the host cell through increasing resistance to host toxic compounds, avoidance of the induction of apoptosis and inhibiting phagosome maturation [43]. We found virulence genes (hspX, ahpC, sodC and katG) in UM_CSW that have been reported to detoxify reactive oxygen species (ROS) and reactive nitrogen species (RNS) [44, 45]. UM_CSW also showed the presence of virulence genes like secA2 and nuoG which are reported to be involved in inhibition of apoptosis. Hinchey and colleagues showed that the secA2 is able to prevent apoptosis in bone marrow derived macrophage, while a M. tuberculosis secA2 mutant induced higher macrophages apoptosis compared to its parental strain [46].
Putative haemolysin
Haemolytic activity has been reported in M. tuberculosis [47], M. haemophilum [48] as well as in fast-growing mycobacteria [49]. The haemolysin-like genes we predicted in UM_CSW might play a role in altering the membrane encompassing the bacterium in macrophage phagosomes to allow molecules to pass through more readily and to facilitate interactions between the bacterium and the host cells’ killing mechanisms [50].
Mycobactin genes
Mycobactins are a class of salicylate containing siderophores produced by both pathogenic and saprophytic mycobacteria for iron internalization. They also act as a temporary iron-holding molecule in order to prevent sudden influx of excess iron in case of sudden availability of the metal after a period of iron limitation [51, 52]. Recent studies have shown that mycobactins are very important for the growth and the virulence of M. tuberculosis, and the disruption of mycobactin biosynthesis may lead to attenuation of growth and virulence [53]. The genome of UM_CSW showed the presence of most of the mycobactin genes (mbt) except for mbtJ, fadD33, fadD332 and fadE14. mbtJ, encoding for acetyl hydroxylase, has been reported to be involved in synthesis of the mycobactin core scaffold while fadD33 and fadE14, encoding for acyl-AMP ligase/acyl-CoA synthase and acyl-ACP dehydrogenase respectively, are responsible for the assembly of mycobactin side chains [54]. Based on the reports suggesting the importance of fadD33 in the growth of M. tuberculosis in the liver [55], it may be hypothesised that the absence of fadD33 in the genome of UM_CSW may affect its growth in the liver.
Other virulence genes
Two interesting genes found in UM_CSW, are the papA2 associated with the biosynthesis of the cell surface lipid Sulfolipid-1 which is unique to pathogenic mycobacteria, and sapM which encodes a lipid phosphatase. PapA2 is essential for the sequential acylation of trehalose-2-sulfate to form SL(1278), a diacylated intermediate in Sulfolipid-1 biosynthesis, which has been shown to activate the adaptive immune response in human patients [56]. The sapM gene has been reported to be essential for arresting phagosomal maturation and growth of the pathogen [57].
Also present were a set of genes related to cellular metabolism such as TBMG01014, Rv2957, RA2984, TBFG12971, MT3031, MCAN29771, JTY2973, BCG2978, Mb2981, MAP1234, MAF29620, which are present only in UM_CSW, M. africanum, M. canetti, M. tuberculosis H37Rv, M. avium 104, M. intracellulare ATCC13950 and M. indicus pranii (Fig 5); the sufBDC-conserved protein, a DUF59-SUF system FeS assembly protein, which, through its implication in the bacterial resistance to iron limitation and oxidative stress, may play an essential role in cell survival [58], and the mlsA2, which is normally a part of a large circular virulence plasmid called pMUM present in M. ulcerans, harbouring three large genes (mlsA1, mlsA2, and mlsB) [59]. These genes are responsible for encoding polyketide synthases that are required for the synthesis of the lipid toxin mycolactone, which is the primary virulence factor for the pathogens [59].
Fig 5. Comparison of virulence genes in UM_CSW and other mycobacterial species.
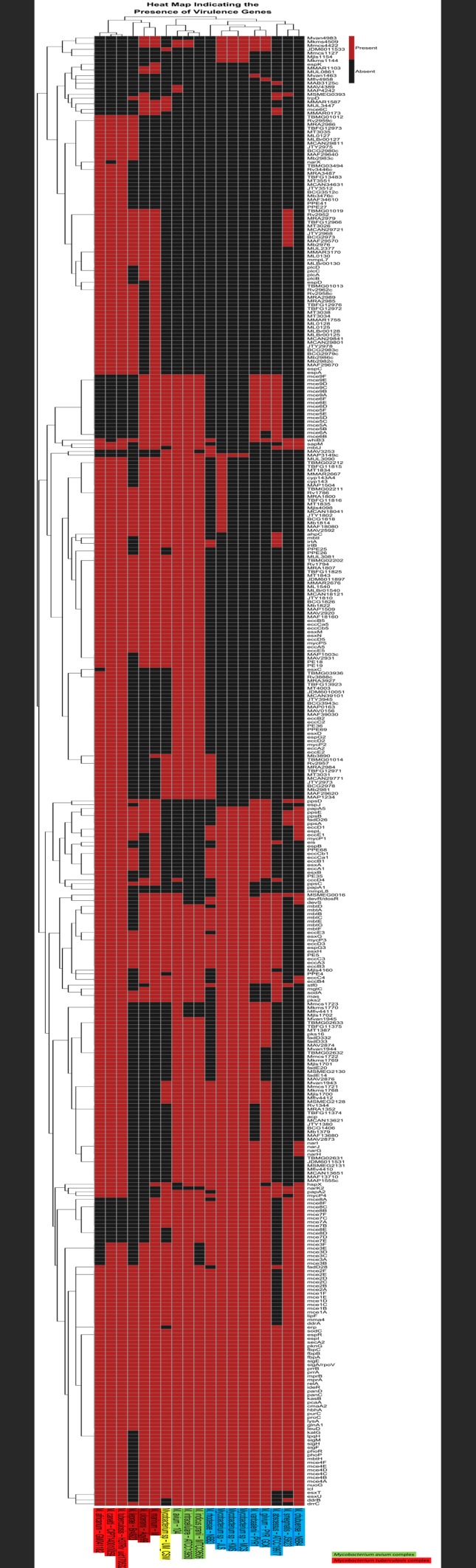
Mycobacterium species were grouped according to their virulence gene profiles. From right to left: group 1, rapid growers M. chelonae to M. rhodesiae; group 2, members of the M. avium complex and UM_CSW; group 3, slow growers M. marinum, M. ulcerans and M. leprae and group 4, members of the M. tuberculosis complex.
In addition, there were transporter proteins, transmembrane proteins, permease, multidrug resistant proteins (e.g. protein B, Drug efflux membrane protein, EmrB/QacA family drug resistance transporter) and a number of metal transporter proteins such as Znu, czcD and MnTH which may have potential roles in the pathogenicity of UM_CSW.
Comparative Pathogenomics Analysis
On the heat map (Fig 5), the putative gene ahpC which has been reported to be required for stress adaptation and drug resistance [60] is the only virulence gene that differentiated known pathogens (M. tuberculosis complex, M. avium complex, M. leprae Br4923, M. ulcerans Agy99, M. marinum M, and M. abscessus ATCC19977) from mycobacterial species that are not normally considered human pathogens (M. smegmatis JS623, M. chubuense NBB4, M. rhodesiae NBB3, Mycobacterium sp. JLS, Mycobacterium sp. KMS, Mycobacterium sp. MCS, M. vanbaalenii PYR1, and M. gilvum PYR_GCK). The gene profile of UM_CSW is most similar to that of the M. avium complex represented by M. avium 104, M. intracellulare ATCC13950, and M. indicus pranii MTCC9506. A number of mammalian cell entry (mce) operons, mce5, mce6 and mce9 were found in UM_CSW and M. avium complex but not in M. tuberculosis and some other pathogenic mycobacteria. On the other hand, the genes encoding phospholipses C (plcA, plcB, plcC and plcD) which enable intracellular pathogens to escape from phagosomal vacuoles by disrupting the host membrane [61] were all absent in UM_CSW and the M. avium complex but were in the M. tuberculosis complex and some other mycobacterial pathogens.
The genes found in both the M. tuberculosis complex and M. avium complex but not in UM_CSW included many related to the biosynthesis of PDIM (phthiocerol dimycocerosate) which is essential for the prevention of phagosomal acidification [62] and the genes for the biosynthesis and transport of PGL (phenolic glycolipid) which has been reported to reduce pro-inflammatory cytokine release in the host [63, 64]. Other important virulence genes found missing include nark 2 which encodes the nitrate/nitrite transporter responsible for inducing nitrate reductase activity in response to decreasing oxygen levels by regulating both its transcription and activity [65]; narG, narH, narI and narJ that encode the anaerobic nitrate reductase enzyme required for nitrate respiration in the absence of oxygen [66] and narX for fused nitrate reductase, the first transcriptionally induced gene in anaerobic dormant mycobacteria [67].
Discussion
We present here the genome sequence and analysis of a clinically derived isolate, Mycobacterium sp. UM_CSW. Using the three global bacterial identification markers, 16S rRNA, hsp65 and rpoB, we failed to resolve the taxonomic position of this isolate which was shown to cluster variously with both rapid- and slow-growing mycobacterial species. After further analyses with nucleotide and protein sequences from whole-genome and whole-proteome data as well as comparative pathogenomic analysis against other mycobacterial species, it became more apparent that UM_CSW is likely to be a novel Mycobacterium species closely related to the M. avium complex.
A thorough in-silico functional analysis revealed some interesting features of UM_CSW. This genome has arsenic- and arsenic resistance-related genes suggesting an environmental origin and the potential for the development of resistance to this chemical element. On the other hand, there is evidence for a high pathogenic potential with the presence of genes associated with the maintenance of cell wall integrity; cell entry; evasion of host immune system; resistance against host toxic compounds, iron limitation and oxidative stress; inhibition of apoptosis; and other virulence factors. The results of PathogenFinder also indicated that UM_CSW might be a human pathogen. However, since UM_CSW was isolated from a patient with a long history of bronchiectasis, we cannot discount the possibility of it being an opportunistic pathogen like most NTMs [2]. With the presence of all the potential virulence factors, whether UM_CSW is a human pathogen or just an opportunistic pathogen still needs to be experimentally verified.
Conclusion
We have successfully sequenced and analyzed the genome of a strain of Mycobacterium that might be a new mycobacterial species as we proposed in this study. Our analysis suggested an environmental origin but also a high potential for causing disease in human hosts. Future biological and epidemiological studies will determine its significance as a human pathogen.
Supporting Information
(TIF)
(TIF)
(TIF)
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
We would like to thank all members from the Genome Research Informatics Group for their IT or bioinformatics assistance and supports. Special thanks to Professor Dr. Ngeow Yun Fong from University of Malaya for her generosity in providing DNA material, valuable inputs and proofreading this manuscript. This research was funded by the UM High Impact Research grant (grant number: UM-MOHE UM.C/HIR/MOHE/08) from the Ministry of Higher Education Malaysia and the UMRG grant (Project code: RG541-13HTM) from the University of Malaya
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by University of Malaya (UM) High Impact Research grant (grant number: UM-MOHE UM.C/HIR/MOHE/08), and University of Malaya Research Grant (UMRG) grant (Project code: RG541-13HTM).
References
- 1.Roberts ED, Koneman EW, Kim YK. Mycobacterium. In: Balows A, Hausler J, W. J., Herrmann KL, Isenberg HD, Shadomy HJ, editors. Manual of clinical microbiology. 5 ed. Washington, D.C.: American Society for Microbiology; 1991. p. 304–39. [Google Scholar]
- 2.Cook JL. Nontuberculous mycobacteria: opportunistic environmental pathogens for predisposed hosts. British medical bulletin. 2010;96:45–59. 10.1093/bmb/ldq035 [DOI] [PubMed] [Google Scholar]
- 3.Falkinham JO 3rd. Ecology of nontuberculous mycobacteria—where do human infections come from? Seminars in respiratory and critical care medicine. 2013;34(1):95–102. 10.1055/s-0033-1333568 . [DOI] [PubMed] [Google Scholar]
- 4.Knoll BM, Kappagoda S, Gill RR, Goldberg HJ, Boyle K, Baden LR, et al. Non-tuberculous mycobacterial infection among lung transplant recipients: a 15-year cohort study. Transpl Infect Dis. 2012;14(5):452–60. 10.1111/j.1399-3062.2012.00753.x [DOI] [PubMed] [Google Scholar]
- 5.Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. The Lancet. 2013;381(9877):1551–60. S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics (Oxford, England). 2011;27(6):863–4. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics. 2008;9:75 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial agents and chemotherapy. 2014;58(7):3895–903. 10.1128/AAC.02412-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic acids research. 2007;35(Web Server issue):W52–7. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome research. 2012;22(3):549–56. 10.1101/gr.126953.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laing C, Buchanan C, Taboada EN, Zhang Y, Kropinski A, Villegas A, et al. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC bioinformatics. 2010;11:461 10.1186/1471-2105-11-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;28(10):2731–9. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2567–72. 10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. International journal of systematic and evolutionary microbiology. 2007;57(Pt 1):81–91. 10.1099/ijs.0.64483-0 . [DOI] [PubMed] [Google Scholar]
- 15.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(45):19126–31. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langille MG, Brinkman FS. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics (Oxford, England). 2009;25(5):664–5. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF, et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC bioinformatics. 2006;7:142 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao WW, Ung K, Aeschliman D, Bryan J, Finlay BB, Brinkman FS. Evidence of a large novel gene pool associated with prokaryotic genomic islands. PLoS genetics. 2005;1(5):e62 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langille MG, Hsiao WW, Brinkman FS. Evaluation of genomic island predictors using a comparative genomics approach. BMC bioinformatics. 2008;9:329 10.1186/1471-2105-9-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cosentino S, Voldby Larsen M, Moller Aarestrup F, Lund O. PathogenFinder—distinguishing friend from foe using bacterial whole genome sequence data. PloS one. 2013;8(10):e77302 10.1371/journal.pone.0077302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic acids research. 2005;33(Database issue):D325–8. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic acids research. 2011;39(Web Server issue):W347–52. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215(3):403–10. 10.1016/S0022-2836(05)80360-2 . [DOI] [PubMed] [Google Scholar]
- 24.Cayrou C, Turenne C, Behr MA, Drancourt M. Genotyping of Mycobacterium avium complex organisms using multispacer sequence typing. Microbiology. 2010;156(Pt 3):687–94. 10.1099/mic.0.033522-0 . [DOI] [PubMed] [Google Scholar]
- 25.Stolz JF, Basu P, Oremland RS. Microbial Arsenic Metabolism: New Twists on an Old Poison. Microbe. 2010. February 2010:53–9. [Google Scholar]
- 26.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research. 2007;35(Database issue):D61–5. 10.1093/nar/gkl842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sezonov G, Possoz C, Friedmann A, Pernodet JL, Guerineau M. KorSA from the Streptomyces integrative element pSAM2 is a central transcriptional repressor: target genes and binding sites. Journal of bacteriology. 2000;182(5):1243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagege J, Pernodet JL, Sezonov G, Gerbaud C, Friedmann A, Guerineau M. Transfer functions of the conjugative integrating element pSAM2 from Streptomyces ambofaciens: characterization of a kil-kor system associated with transfer. Journal of bacteriology. 1993;175(17):5529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bath J, Wu LJ, Errington J, Wang JC. Role of Bacillus subtilis SpoIIIE in DNA transport across the mother cell-prespore division septum. Science. 2000;290(5493):995–7. . [DOI] [PubMed] [Google Scholar]
- 30.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(11):4376–80. 10.1073/pnas.0711159105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288(5471):1647–50. . [DOI] [PubMed] [Google Scholar]
- 32.Klink M, Brzezinska M, Szulc I, Brzostek A, Kielbik M, Sulowska Z, et al. Cholesterol oxidase is indispensable in the pathogenesis of Mycobacterium tuberculosis. PloS one. 2013;8(9):e73333 10.1371/journal.pone.0073333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park SJ, Lee SY. Identification and characterization of a new enoyl coenzyme A hydratase involved in biosynthesis of medium-chain-length polyhydroxyalkanoates in recombinant Escherichia coli. Journal of bacteriology. 2003;185(18):5391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu M, Zhang Y, Inouye M, Woychik NA. Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(15):5885–90. 10.1073/pnas.0711949105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infection and immunity. 2000;68(2):767–78. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manganelli R, Provvedi R, Rodrigue S, Beaucher J, Gaudreau L, Smith I. Sigma factors and global gene regulation in Mycobacterium tuberculosis. Journal of bacteriology. 2004;186(4):895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukhopadhyay S, Balaji KN. The PE and PPE proteins of Mycobacterium tuberculosis. Tuberculosis. 2011;91(5):441–7. 10.1016/j.tube.2011.04.004 . [DOI] [PubMed] [Google Scholar]
- 38.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie. 2012;94(1):110–6. 10.1016/j.biochi.2011.09.026 . [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Pelayo MC, Caimi KC, Inwald JK, Hinds J, Bigi F, Romano MI, et al. Microarray analysis of Mycobacterium microti reveals deletion of genes encoding PE-PPE proteins and ESAT-6 family antigens. Tuberculosis. 2004;84(3–4):159–66. 10.1016/j.tube.2003.12.002 . [DOI] [PubMed] [Google Scholar]
- 40.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC evolutionary biology. 2006;6:95 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaur RL, Ren K, Blumenthal A, Bhamidi S, Gibbs S, Jackson M, et al. LprG-mediated surface expression of lipoarabinomannan is essential for virulence of Mycobacterium tuberculosis. PLoS pathogens. 2014;10(9):e1004376 10.1371/journal.ppat.1004376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla S, Richardson ET, Athman JJ, Shi L, Wearsch PA, McDonald D, et al. Mycobacterium tuberculosis Lipoprotein LprG Binds Lipoarabinomannan and Determines Its Cell Envelope Localization to Control Phagolysosomal Fusion. PLoS pathogens. 2014;10(10):e1004471 10.1371/journal.ppat.1004471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berthet FX, Lagranderie M, Gounon P, Laurent-Winter C, Ensergueix D, Chavarot P, et al. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science. 1998;282(5389):759–62. . [DOI] [PubMed] [Google Scholar]
- 44.Shi L, Sohaskey CD, North RJ, Gennaro ML. Transcriptional characterization of the antioxidant response of Mycobacterium tuberculosis in vivo and during adaptation to hypoxia in vitro. Tuberculosis. 2008;88(1):1–6. 10.1016/j.tube.2007.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voskuil MI, Bartek IL, Visconti K, Schoolnik GK. The response of mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbiol. 2011;2:105 10.3389/fmicb.2011.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. The Journal of clinical investigation. 2007;117(8):2279–88. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.King CH, Mundayoor S, Crawford JT, Shinnick TM. Expression of contact-dependent cytolytic activity by Mycobacterium tuberculosis and isolation of the genomic locus that encodes the activity. Infection and immunity. 1993;61(6):2708–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer LJ, Quinn FD, White EH, King CH. Intracellular growth and cytotoxicity of Mycobacterium haemophilum in a human epithelial cell line (Hec-1-B). Infection and immunity. 1996;64(1):269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Udou T. Extracellular hemolytic activity in rapidly growing mycobacteria. Canadian journal of microbiology. 1994;40(4):318–21. . [DOI] [PubMed] [Google Scholar]
- 50.Wren BW, Stabler RA, Das SS, Butcher PD, Mangan JA, Clarke JD, et al. Characterization of a haemolysin from Mycobacterium tuberculosis with homology to a virulence factor of Serpulina hyodysenteriae. Microbiology. 1998;144 (Pt 5):1205–11. . [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez GM, Smith I. Mechanisms of iron regulation in mycobacteria: role in physiology and virulence. Molecular microbiology. 2003;47(6):1485–94. . [DOI] [PubMed] [Google Scholar]
- 52.Ratledge C. Iron, mycobacteria and tuberculosis. Tuberculosis. 2004;84(1–2):110–30. . [DOI] [PubMed] [Google Scholar]
- 53.Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, et al. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. The Journal of infectious diseases. 2013;208(8):1255–65. 10.1093/infdis/jit250 [DOI] [PubMed] [Google Scholar]
- 54.Li W, He J, Xie L, Chen T, Xie J. Comparative genomic insights into the biosynthesis and regulation of mycobacterial siderophores. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;31(1):1–13. 10.1159/000343343 . [DOI] [PubMed] [Google Scholar]
- 55.Rindi L, Fattorini L, Bonanni D, Iona E, Freer G, Tan D, et al. Involvement of the fadD33 gene in the growth of Mycobacterium tuberculosis in the liver of BALB/c mice. Microbiology. 2002;148(Pt 12):3873–80. . [DOI] [PubMed] [Google Scholar]
- 56.Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, et al. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(27):11221–6. 10.1073/pnas.0611649104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puri RV, Reddy PV, Tyagi AK. Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PloS one. 2013;8(7):e70514 10.1371/journal.pone.0070514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huet G, Daffe M, Saves I. Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: evidence for its implication in the pathogen's survival. Journal of bacteriology. 2005;187(17):6137–46. 10.1128/JB.187.17.6137-6146.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R, et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(5):1345–9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherman DR, Mdluli K, Hickey MJ, Barry CE, 3rd, Stover CK. AhpC, oxidative stress and drug resistance in Mycobacterium tuberculosis. BioFactors. 1999;10(2–3):211–7. . [DOI] [PubMed] [Google Scholar]
- 61.Titball RW. Bacterial phospholipases C. Microbiological reviews. 1993;57(2):347–66. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Astarie-Dequeker C, Le Guyader L, Malaga W, Seaphanh FK, Chalut C, Lopez A, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS pathogens. 2009;5(2):e1000289 10.1371/journal.ppat.1000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sinsimer D, Huet G, Manca C, Tsenova L, Koo MS, Kurepina N, et al. The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infection and immunity. 2008;76(7):3027–36. 10.1128/IAI.01663-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, et al. Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chemistry & biology. 2008;15(1):51–61. 10.1016/j.chembiol.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giffin MM, Raab RW, Morganstern M, Sohaskey CD. Mutational analysis of the respiratory nitrate transporter NarK2 of Mycobacterium tuberculosis. PloS one. 2012;7(9):e45459 10.1371/journal.pone.0045459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weber I, Fritz C, Ruttkowski S, Kreft A, Bange FC. Anaerobic nitrate reductase (narGHJI) activity of Mycobacterium bovis BCG in vitro and its contribution to virulence in immunodeficient mice. Molecular microbiology. 2000;35(5):1017–25. . [DOI] [PubMed] [Google Scholar]
- 67.Hutter B, Dick T. Up-regulation of narX, encoding a putative 'fused nitrate reductase' in anaerobic dormant Mycobacterium bovis BCG. FEMS microbiology letters. 1999;178(1):63–9. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(TIF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.