

Abstract
p300 is a multifunctional transcriptional coactivator that interacts with numerous transcription factors and exhibits protein/histone acetyltransferase activity. Loss of p300 function in humans and in mice leads to craniofacial defects. In this study, we demonstrated that inhibition of p300 histone acetyltransferase activity with the compound, C646, altered the expression of several genes, including Cdh1 (E-cadherin) in mouse maxillary mesenchyme cells, which are the cells that give rise to the secondary palate. The increased expression of plasma membrane-bound E-cadherin was associated with reduced cytosolic β-catenin, that led to attenuated signaling through the canonical Wnt pathway. Furthermore, C646 reduced both cell proliferation and the migratory ability of these cells. These results suggest that p300 histone acetyltransferase activity is critical for Wnt-dependent palate mesenchymal cell proliferation and migration, both processes that play a significant role in morphogenesis of the palate.
Keywords: p300, C646, embryo, histone acetyltransferase, palate, craniofacial, Wnt
INTRODUCTION
p300 and its paralog, Creb binding protein (CBP) are large, multifunctional proteins that participate in the regulation of gene expression by serving as a platform for the assembly and acetylation of a wide array of proteins, including many transcription factors and histones [1]. Indeed, it is documented that they can bind over 400 proteins and acetylate at least 100 of these (for a complete list, please see [1]). In our laboratory, we are focused on understanding the molecular underpinnings of craniofacial defects, including cleft palate, the most common human birth defect [2]. Perturbation of many signaling pathways that utilize CBP or p300, such as the TGFβ and Wnt signal transduction networks, can lead to palatal clefting [3,4]. Furthermore, human mutations in CBP/p300 result in Rubinstein-Taybi Syndrome (RTS) with affected individuals exhibiting defects in craniofacial/palate development [5]. This phenotype has been recapitulated in mice with a p300 mutation resulting in its underexpression [6]. We have previously demonstrated that both CBP and p300 are expressed in the developing embryonic mouse secondary palate [7] and that knockdown of p300 function in primary cells isolated from the developing secondary palate leads to the perturbation of several processes necessary for normal palatal ontogeny (e.g. proliferation and TGFβ-stimulated MMP9 synthesis [8]). Because of the increasing evidence that a critical functional domain of p300 confers histone acetyltransferase (HAT) activity [9], we sought to examine the effects of inhibition of p300 HAT activity in primary cells derived from the mouse secondary palate. These results extend our previous work and suggest a novel mechanism by which loss of p300 function perturbs cellular function and may alter normal craniofacial/palate development.
MATERIALS AND METHODS
A. Animals
Outbred Hsd:ICR (CD-1®) mice (referred to herein as ICR) were purchased from Harlan Laboratories, Inc. (Indianapolis, IN) and housed at 22°C with a 12 h on/12h off light cycle and access to food and water ad libitum. Timed pregnancies were obtained by overnight housing of a single mature male with two nulliparous females. The presence of a vaginal plug the following morning was taken as evidence of mating and this time point was referred to as gestation day (GD) 0.5. All procedures for the humane use and handling of mice were approved and overseen by the University of Louisville Institutional Animal Care and Use Committee and encompass guidelines as set out in the European Commission Directive 86/609/EEC for animal experimentation.
B. Establishment of primary mouse embryo maxillary mesenchyme (MEMM) cells
Pregnant mice were euthanized on GD13.5 by carbon dioxide asphyxiation followed by cervical dislocation. Embryos were removed and placed in ice-cold PBS. Palatal processes were microdissected, dissociated with 0.05% trypsin-EDTA (Life Technologies, Carlsbad, CA), and passed through a 70 μm filter to obtain a single-cell suspension. Cells were then seeded into culture dishes appropriate for each experiment so that they were approximately 60% confluent at the time of treatment. For all experiments described in this paper, the cultures were not passaged.
B. Antibodies
Antibodies to E-cadherin, acetylated histone H3 (Lys-9), total histone H3, and cleaved caspase-3 were purchased from Cell Signaling Technology (Beverly, MA). Anti-β-catenin was purchased from EMD Millipore (Billerica, MA) and HRP-linked secondary antibodies were purchased from Life Technologies.
C. SDS-PAGE and Western blotting
Cytoplasmic fractions of MEMM cells were isolated with the NE-PER system (Life Technologies) in the presence of protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). For analysis of histones, MEMM cells were lysed with 0.5% Triton X-100 in PBS, nuclei pelleted and extracted overnight with 0.2N HCl. For the preparation of plasma membrane fractions, mitochondria and nuclei were first removed by differential centrifugation from the cell lysate. The remaining extract was centrifuged at 100,000 x g for 1h at 4°C. Protein assays were performed by the BCA method with reagents purchased from Life Technologies. Protein samples were separated on 4–12% SDS-PAGE gels (Life Technologies) and transferred to Immobilon-P nylon membranes (Millipore). Signals were developed with ECL-Plus chemiluminescence substrate (GE Healthcare Life Sciences, Pittsburgh, PA).
D. Reporter assays
Primary cultures of MEMM cells were cultured to ~60% confluence and transfected with the TOPflash vector as previously described in detail [10]. Twenty-four hours following transfection, cells were treated with 10 mM LiCl or 2 ng/ml recombinant Wnt-3a (R & D Systems, Minneapolis, MN) for 24h. Cells were scraped, lysed, and assayed using the Dual Luciferase reporter system (Promega Corp., Madison, WI).
E. Cell proliferation assay
Proliferation rates were assayed in 96-well plates using the CyQuant-NF DNA binding dye according to the manufacturer’s recommendations (Life Technologies). Cells were seeded at 2000 cells/well, allowed to adhere overnight, and then treated with 0–20 μM C646 or DMSO. Cell number was assayed 24h later with the VictorX3 plate reader (Perkin-Elmer, Waltham, MA).
F. RNA purification and cDNA synthesis
Total RNA was purified with the RNeasy kit (Qiagen, Inc., Valencia, CA), digested with DNase I and cDNAs prepared with the ViLo system (Life Technologies).
G. Taqman qRT-PCR and low-density array analysis
Gene expression assays were performed in a single-tube or 384-well format. For single-tube assays, gene-specific probe-primers were obtained from Life Technologies. cDNAs were assayed using the Taqman enzyme (Life Technologies). For the 384-well format, reactions consisting of cDNA and Taqman enzyme were loaded onto the custom plates and analyzed according to the manufacturer’s protocol (Life Technologies). The list of gene-specific probes is provided in Supplementary Table 1. The ViiA7 Real-Time PCR system was the instrument used for both assays (Life Technologies).
H. Wound assay
Primary cultures of MEMM cells were established and grown until confluent. A scratch was made with the tip of a sterile 200 μl disposable pipette tip, cultures washed 2X with fresh medium and replaced with the same. DMSO or C646 (final concentration of 15 μM) was added and photomicrographs taken at 0, 4, 6, 8, and 10 hours, using orientation marks on the underside of the culture dish to ensure precise alignment at each time point (see Supplemental Figure 1). The migration distance of one front was measured using Metamorph (Molecular Devices, Sunnyvale, CA). The experiment was performed three times.
I. p300 inhibitor
C646 was purchased from Sigma Chemical Co., (St. Louis, MO) and dissolved in tissue culture-grade DMSO (Sigma) to 10 mM and stored at −20°C in single-use aliquots. Additional details on C646 can be found in [11].
J. Statistical analyses
Statistical analyses were performed using GraphPad Instat, version 3.1a (GraphPad Software, Inc., La Jolla, CA). P-values less than 0.05 were considered to be significant.
RESULTS
To determine the effect of inhibiting p300 HAT activity in MEMM cells, we utilized C626, a cell membrane permeable, reversible inhibitor that exhibits high specificity and affinity for the HAT domain of p300 [13]. We first performed a preliminary assessment of the effect of increasing concentrations of C646 on the overall appearance of MEMM cell cultures. A 24h exposure to 0–20 μM C646 on cultures of MEMM cells is shown in Figure 1. There was a clear effect on the number of cells, with significant effects observed at concentrations above 15 μM.
Figure 1. C646 alters MEMM cell growth.

Primary cultures of MEMM cultures were established and treated with 0–20 μM C646 for 24h. Photomicrographs were taken at 200X power. Note the reduction in cell number at 15 and 20 μM.
To confirm that the effect of C646 on MEMM cells was due to inhibition of p300 HAT, we assayed the acetylation state of lysine-9 of histone H3 (H3K9-Ac), a known substrate for p300 [12]. Acid-extraction and Western blotting for H3K9-Ac and total H3 demonstrated that 15 μM C646 led to a significant decrease in the level of H3K9-Ac (Figure 2). Because other HATs also target H3K9, the large decreases (31%) in acetylation of this histone was unexpected and suggest that p300 HAT activity is a major contributor to histone H3 acetylation in these cells.
Figure 2. C646 decreases acetylation of histone H3 in MEMM cells.
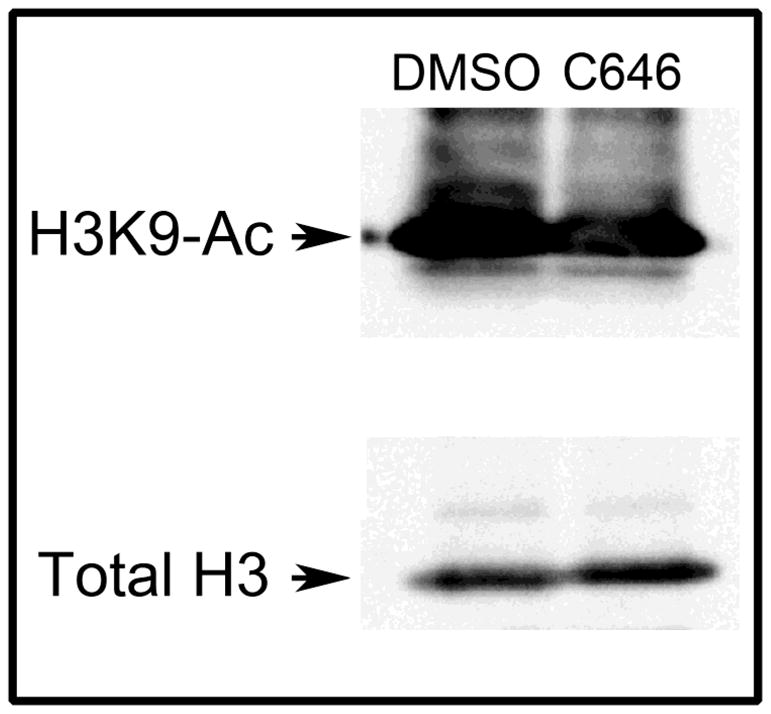
Subconfluent primary cultures of MEMM cells were incubated with DMSO or 15 μM C646 for 24h and then histones were isolated by acid-extraction of nuclei. Samples were then assayed for the presence of H3K9-Ac (top panel) or total H3 (bottom panel). Treatment with C646 led to a 31% decrease in H3K9-Ac.
Because fewer numbers of cells were seen at the higher doses of C646 compared to DMSO controls (Figure 1), we determined the rate of cell proliferation of MEMM cells following treatment for 24h with C646. Primary cultures of MEMM cells were treated with 5–20 μM C646 for 24h and the number of cells determined with a fluorescent DNA binding agent (CyQUANT). As shown in Figure 3, treatment of cells with C626 resulted in a significant, dose-dependent decrease in cell proliferation. At 48h and with 5 μM C646, cell growth was inhibited by 50% and reached 76% inhibition with 20 μM C646. Because effects on proliferation and/or apoptosis can alter the concentration of cells, we also determined the levels of cleaved Caspase-3. The data in Figure 4 demonstrate that treatment of cells with C626 also resulted in a dose-related increase in cleaved Caspase-3. Therefore, the C626-induced diminution of cell number seen in Figure 1 was likely the result of a combination of decreased cell proliferation and an increase in apoptosis.
Figure 3. C646 inhibits proliferation of MEMM cells.

MEMM cells were seeded into 96-well plates and then treated with 0–20 μM C646 for 0–48h. The number of cells was determined using the DNA binding agent, CyQUANT. Each point represents the average ± standard deviation of 5 replicates. At 48h, cell proliferation was inhibited by 50% (5 μM) to >75% (20 μM) and were all statistically significant (5 μM, P < 0.05; 10 μM P < 0.001; 15 μM and 20 μM, P < 0.0001; one-way ANOVA).
Figure 4. C646 induces the presence of the apoptosis protein, cleaved Caspase-3.

Subconfluent primary cultures of MEMM cells were treated with 0–15 μM C646 for 24 hours and then total cell lysates analyzed by Western blotting for active (cleaved) Caspase-3 (top panel), stripped, and then reprobed with anti-β-tubulin antibodies (bottom panel). There was no detectable cleaved Caspase-3 in control samples or in cultures treated with 10 μM C646. Low levels of cleaved Caspase-3 were seen following treatment of cells with 12.5 μM C626. Increased expression of cleaved Caspase-3 was seen following treatment of cells with 15 μM C646.
To explore the underlying mechanism of this effect, we assayed the expression of a panel of genes that 1) were selected based upon their known roles in cell proliferation, apoptosis, and differentiation of cells that give rise to the secondary palate, and 2) have been linked to palatal clefting. The complete list of these genes is provided in Supplementary Table 1. To assay the expression of these genes, we employed a low-density, qRT-PCR-based approach that we have successfully used for miRNA expression analysis during midfacial development in the mouse [13]. By using this approach, we were able to simultaneously analyze the expression of 384 genes. MEMM cells were treated with 15 μM C646 for 24h, total RNA purified, cDNAs synthesized, and levels of gene expression assayed by qRT-PCR. The results are presented in Table 1. The expression of 15 genes was significantly increased and 5 decreased following inhibition of p300 HAT. The gene with the largest fold-change was for E-cadherin (Cdh1), which showed a 122-fold increase in expression following C646 treatment. This increased expression was confirmed by independent qRT-PCR assays (data not shown) and by Western blotting of a plasma membrane fraction prepared from MEMM cells treated with 15 μM C646 (Figure 5), that revealed a greater than 3-fold increase in the level of E-cadherin. Because one of the main binding partners of E-cadherin is β-catenin, we also analyzed the same plasma membrane fraction for its expression. Indeed, there was a 25% increase in the amount of plasma membrane-bound β-catenin. Concomitant with the increased level of β-catenin in the plasma membrane was a 40% decrease in its expression in the cytosol (Figure 6). Combined, these data suggest that C646 increased the expression of E-cadherin, which led to a sequestration of β-catenin in the plasma membrane, away from the cytosol where it also mediates canonical Wnt signaling.
Table 1.
C646 alters gene expression in MEMM cells
|
Genes upregulated by 24 h exposure to C646
| ||
|---|---|---|
| Gene symbol | Average Fold Change | p-value |
| Axin1 | 2.85 ± 0.75 | 0.041 |
| Ccne2 | 5.18 ± 1.18 | 0.031 |
| Cdh1 | 122.0 ± 40.01 | 0.039 |
| Cflar | 2.44 ± 0.17 | 0.031 |
| Cntn1 | 3.10 ± 0.57 | 0.040 |
| E2f2 | 4.62 ± 0.49 | 0.003 |
| Fnta | 2.39 ± 0.32 | 0.020 |
| Gdf15 | 12.0 ± 2.33 | 0.010 |
| Has1 | 18.4 ± 5.81 | 0.042 |
| Hipk2 | 2.29 ± 0.28 | 0.024 |
| Lama3 | 2.95 ± 0.28 | 0.022 |
| Mmp12 | 2.28 ± 0.34 | 0.024 |
| Mstn | 5.75 ± 0.47 | 0.003 |
| Pitx2 | 4.67 ± 0.92 | 0.018 |
| Rela | 2.78 ± 0.50 | 0.040 |
|
Genes downregulated by 24 h exposure to C646
| ||
|---|---|---|
| Gene symbol | Average Fold Change | p-value |
| Adamts1 | 0.33 ± 0.12 | 0.012 |
| Fst | 0.03 ± 0.01 | 0.006 |
| Gdf10 | 0.06 ± 0.01 | 0.011 |
| Kcnj2 | 0.21 ± 0.07 | 0.004 |
| Tgfbr2 | 0.25 ± 0.04 | 0.014 |
The expression of a selected panel of genes was analyzed by semi-quantitative, real-time PCR (TaqMan®). The data was reduced by the ΔΔCt method [23] using Gapdh as an internal control. The data are expressed as fold change in C646-treated cells, relative to that in DMSO-exposed control samples. The data are reported as average fold-change of 3 independent experiments ± S.E.M. p values were determined by one-way ANOVA.
Figure 5. Co-expression of E-cadherin and β-catenin in the plasma membrane.

MEMM cells were treated with DMSO or 15 μM C646 for 24 hours and plasma membrane fractions isolated and analyzed by Western blotting for E-cadherin and non-phosphorylated β-catenin. Exposure of cells to C646 resulted in increased expression of both E-cadherin and β-catenin in the plasma membrane.
Figure 6. C646 reduces the levels of cytoplasmic β-catenin.
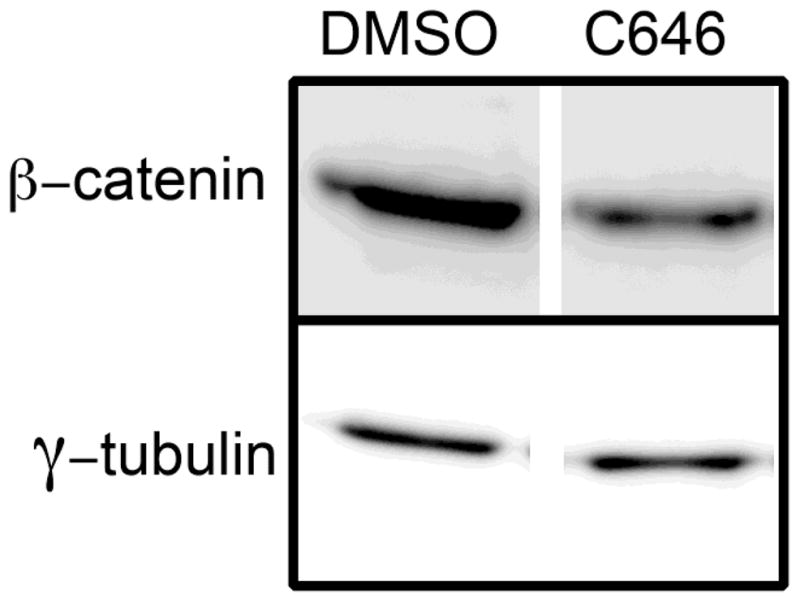
MEMM cells were treated with DMSO or 15 μM C646 for 24 hours and cytoplasmic extracts prepared. Samples were analyzed by Western blotting for non-phosphorylated β-catenin.
In order to determine if increased expression of E-cadherin perturbed canonical Wnt signaling, MEMM cells were transfected with the Wnt reporter plasmid, TOPflash, then treated with 100 ng/ml Wnt-3a in the presence or absence of 15 μM C646. The results are shown in Figure 7. C646 significantly inhibited the ability of Wnt-3a to stimulate transcription of the TOPflash reporter. The fold increase in Wnt-3a/β-catenin stimulated Topflash luciferase activity — when compared to control levels of activity — was significantly reduced in the presence of C626. These data are consistent with our hypothesis that increased expression of E-cadherin binds to and sequesters β-catenin away from downstream Wnt signaling proteins. We also assayed the expression of Axin2, a direct endogenous target of canonical Wnts. The expression of Axin2 was also decreased when cells were co-cultured with C646 and 10 mM LiCl compared to 10 mM LiCl alone (data not shown).
Figure 7. C646 attenuates canonical Wnt signaling.

Primary cultures of MEMM cells were cotransfected with the TOPflash plasmid and a control plasmid that constitutively expresses Renilla luciferase. After 24h, transfected cells were stimulated with 100 ng/ml Wnt-3a in the presence or absence of 15 μM C646 for an additional 24h. The levels of both Firefly (TOPflash) and Renilla were determined by luminometry and the data expressed as the ratio from 3 replicate samples. C646 led to a statistically significant decrease (P < 0.001, one-way ANOVA) in Wnt-3a stimulation of TOPflash activity.
Previous studies have demonstrated that perturbation of palate mesenchymal cell migration is associated with palatal clefting [14]. Because increased expression of E-cadherin, could lead to greater cell-cell contact, we assayed the migratory potential of MEMM cells by performing a scratch assay. As shown in Figure 8, the application of 15 μM C646 significantly decreased the migratory ability of MEMM cells.
Figure 8. C646 reduces the rate of MEMM cell migration.

Confluent cultures of primary MEMM cells were established and then a scratch made of equal size. One culture was treated with DMSO and the other with 15 μM C646. Photomicrographs were taken at 4, 6, 8, and 10h and the distance of one migrating front measured with Metamorph. The top panels depict the appearance of the cultures at the end of the experiment (10h). The bottom graph represents the time course of migration of MEMM cells the average of 3 independent experiments. At each time point, C646 significantly inhibited the migration of MEMM cells (P < 0.05, unpaired t-test).
DISCUSSION
CBP and p300 are multifunctional coactivator proteins that mediate gene expression and are essential for normal embryonic development and adult tissue homeostasis. We have previously demonstrated that both CBP and p300 are expressed in the developing mouse secondary palate [7], and that knockdown of total p300 or CBP expression in murine embryonic maxillary mesenchymal (MEMM) cells with siRNAs perturbed several cellular processes critical for normal palate development, including proliferation and TGFβ-mediated matrix metalloproteinase synthesis [8]. To further explore distinct functions of p300 in MEMM cells, we took advantage of a recently reported HAT inhibitor, C646, that was developed based on extensive knowledge of the p300 active site, which differs from that for CBP [11]. Studies have shown that p300 HAT functions in a cell-specific manner [15,16,17]. The current study utilized C646 to test cellular outcomes in primary (MEMM) cells derived from tissue that will give rise to the maxilla and secondary palate. This inhibitor has been successfully used to test the function of the p300 HAT domain in a variety of cell lines [15,16,17]. MEMM cells are obtained from the embryonic secondary palate and maxilla and have been extensively used to test gene function with relevance to development of the orofacial region [18,19,20].
The most revealing result from the RT-PCR analysis of MEMM cells following treatment with C646 was that the expression of E-cadherin was significantly increased. The expression of E-cadherin in conjunction with β-catenin, is typically considered to mediate cell to cell contact. This phenomenon has several implications for cell function. First, because E-cadherin interacts with β-catenin, itself a central protein in canonical Wnt signaling, such interaction may reduce the available pool of β-catenin for signaling. Indeed, our data demonstrating C626-associated reduction in Wnt-3a stimulated transcription of the TOPflash reporter, and decreased Axin2 expression is consistent with such an interpretation. An increase in the amount of β-catenin associated with the plasma membrane of C626-treated MEMM cells lends further support to this interpretation.
We propose a model whereby p300 HAT activity is driving a partial mesenchymal to epithelial transition. Consistent with this hypothesis is a recent report demonstrating that human peritoneal mesothelial cells treated with high glucose, which promotes epithelial to mesenchymal transition, maintain their expression of E-cadherin when treated with C646 [21]. This was correlated with decreased TGFβ/Smad3 signaling. Because TGFβ is a major regulator of epithelial to mesenchymal transition, these results suggest that the HAT activity of p300 is required to mediate this effect. Interestingly, emodin, a resin found in many plants, and that acts as a laxative, has recently been shown to decrease canonical Wnt signaling through decreased β-catenin expression in colorectal cancer cell lines [22]. Concomitant with a diminution of Wnt signaling was a decrease in the expression of Snai1 and vimentin (both mesenchyme cell markers), an increase in expression of E-cadherin, and a conversion to an epithelial-like in morphology [22]. We also observed reduced expression of Snai1 in MEMM cells treated with C646 (data not shown). This loss of Snai1 expression alone could explain aberrant E-cadherin expression. Because C646 inhibited cell proliferation, increased apoptosis, and decreased cell migration, this is potentially a useful inhibitor that can be utilized to ameliorate the hallmarks of metastatic cancer cells (loss of epithelial character and the assumption of a highly motile, mesenchyme phenotype).
Development of the secondary palate requires precise control over cell proliferation, differentiation, apoptosis, and migration. Perturbations of any of these processes can result in palatal clefts. In the present study we have demonstrated that loss of p300 HAT activity alters at least three cellular processes that could contribute to craniofacial defects: cell proliferation, apoptosis, and cell migration. p300 (and CBP) are “hubs” that integrate signaling inputs from a variety of pathways (notably TGFβ and WNT for craniofacial development) through the assembly of cell-specific transcription factor complexes and through acetylation of a plethora of other proteins. The studies we present demonstrate, for the first time, that perturbation of this latter function can result in aberrant cell signaling leading to decreased proliferation and migration, both of which can cause significant craniofacial anomalies. That C646 perturbed WNT signaling in MEMM cells is consistent with a recent report by Gaddis et al. which demonstrated that inhibition of p300 by C646 in several cancer cell types also had a profound effect on WNT signaling (24). Those results, and ours presented in this paper, support the hypothesis that p300 is an essential component of epigenetic regulation of WNT signaling and loss of p300 function may lead to craniofacial defects because of its role in regulating this pathway.
CONCLUSIONS
p300 is an important mediator of growth, development, and differentiation. Specific inhibition of the HAT activity in MEMM cells led to an increased expression of several genes, including Cdh1. We have provided evidence that supports a model by which this aberrant expression of E-cadherin may sequester and thus inhibit signaling through the canonical Wnt signaling pathway, and secondarily, altering important processes for normal palate development.
Supplementary Material
The data in Figure 8 was generated by taking photomicrographs of precisely the same area of the tissue culture dish at specific intervals over a 10 hour time period and measuring the migration distance into a “scratch”. By creating orientation marks on the bottom of the tissue culture dish, we were able to photograph the same position at each time point. A direct comparison of these images enabled us to mark the initial position (red line) and then the advancing migration front (yellow line). By using Metamorph software we could measure (in microns) the distance between these two lines. Control- and C646-treated cells are represented in the left and right columns, respectively, with increasing time points from top to bottom.
{kind=link}
Highlights.
Inhibition of p300 HAT in cultured palate cells increased E-cadherin expression
Inhibition of p300 HAT perturbed canonical Wnt signaling in cultured palate cells
Blocking p300 HAT in palate cells altered rates of apoptosis, proliferation, and migration.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DE018215, HD053509, and P20 RR017702 from the COBRE program of the National Center for Research Resources and the NIGMS).
Abbreviations
- HAT
histone acetyltransferase
- MEMM
mouse embryo maxillary mesenchyme
Footnotes
CONFLICT OF INTEREST
None of the authors report any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dancy BM, Cole PA. Protein Lysine Acetylation by p300/CBP. Chem Rev. 2015 doi: 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stuppia L, Capogreco M, Marzo G, La Rovere D, Antonucci I, et al. Genetics of syndromic and nonsyndromic cleft lip and palate. J Craniofac Surg. 2011;22:1722–1726. doi: 10.1097/SCS.0b013e31822e5e4d. [DOI] [PubMed] [Google Scholar]
- 3.Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Jr, et al. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Ye X, Guo N, Nathans J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development. 2012;139:4383–4394. doi: 10.1242/dev.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–580. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viosca J, Lopez-Atalaya JP, Olivares R, Eckner R, Barco A. Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: Differential contribution of p300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol Dis. 2010;37:186–194. doi: 10.1016/j.nbd.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Warner DR, Pisano MM, Greene RM. Expression of the nuclear coactivators CBP and p300 in developing craniofacial tissue. In Vitro Cell Dev Biol Anim. 2002;38:48–53. doi: 10.1290/1071-2690(2002)038<0048:EOTNCC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 8.Warner DR, Pisano MM, Greene RM. Functional analysis of CBP/p300 in embryonic orofacial mesenchymal cells. J Cell Biochem. 2006;99:1374–1379. doi: 10.1002/jcb.20974. [DOI] [PubMed] [Google Scholar]
- 9.Yan G, Eller MS, Elm C, Larocca CA, Ryu B, et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J Invest Dermatol. 2013;133:2444–2452. doi: 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warner DR, Greene RM, Pisano MM. Cross-talk between the TGFbeta and Wnt signaling pathways in murine embryonic maxillary mesenchymal cells. FEBS Lett. 2005;579:3539–3546. doi: 10.1016/j.febslet.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 11.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crump NT, Hazzalin CA, Bowers EM, Alani RM, Cole PA, et al. Dynamic acetylation of all lysine-4 trimethylated histone H3 is evolutionarily conserved and mediated by p300/CBP. Proc Natl Acad Sci U S A. 2011;108:7814–7819. doi: 10.1073/pnas.1100099108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warner DR, Mukhopadhyay P, Brock G, Webb CL, Pisano MM, et al. microRNA expression profiling in the developing murine upper lip. Development, Growth, & Differentiation. 2014;56:434–447. doi: 10.1111/dgd.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He F, Xiong W, Yu X, Espinoza-Lewis R, Liu C, et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 2008;135:3871–3879. doi: 10.1242/dev.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao XN, Lin J, Ning QY, Gao L, Yao YS, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One. 2013;8:e55481. doi: 10.1371/journal.pone.0055481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Wang W, Liu C, Li W, Shu Q, et al. Critical role of histone acetylation by p300 in human placental 11beta-HSD2 expression. J Clin Endocrinol Metab. 2013;98:E1189–1197. doi: 10.1210/jc.2012-4291. [DOI] [PubMed] [Google Scholar]
- 17.Oike T, Komachi M, Ogiwara H, Amornwichet N, Saitoh Y, et al. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiother Oncol. 2014;111:222–227. doi: 10.1016/j.radonc.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 18.D’Angelo M, Greene RM. Transforming growth factor-beta modulation of glycosaminoglycan production by mesenchymal cells of the developing murine secondary palate. Dev Biol. 1991;145:374–378. doi: 10.1016/0012-1606(91)90136-q. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Zhang H, Gao L, Yin Y, Pan X, et al. Negative interplay of retinoic acid and TGF-beta signaling mediated by TG-interacting factor to modulate mouse embryonic palate mesenchymal-cell proliferation. Birth Defects Res B Dev Reprod Toxicol. 2014;101:403–409. doi: 10.1002/bdrb.21130. [DOI] [PubMed] [Google Scholar]
- 20.Feng C, Xu Z, Li Z, Zhang D, Liu Q, et al. Down-regulation of Wnt10a by RNA interference inhibits proliferation and promotes apoptosis in mouse embryonic palatal mesenchymal cells through Wnt/beta-catenin signaling pathway. J Physiol Biochem. 2013;69:855–863. doi: 10.1007/s13105-013-0262-7. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Liu K, Liang Y, Chen Y, Gong Y. Histone acetyltransferase inhibitor C646 reverses epithelial to mesenchymal transition of human peritoneal mesothelial cells via blocking TGF-beta1/Smad3 signaling pathway in vitro. Int J Clin Exp Pathol. 2015;8:2746–2754. [PMC free article] [PubMed] [Google Scholar]
- 22.Pooja T, Karunagaran D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur J Pharmacol. 2014;742:55–64. doi: 10.1016/j.ejphar.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Gaddis M, Gerrard D, Frietze S, Farnham PJ. Altering cancer transcriptomes using epigenomic inhibitors. Epigenetics & Chromatin. 2015;8:9–20. doi: 10.1186/1756-8935-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The data in Figure 8 was generated by taking photomicrographs of precisely the same area of the tissue culture dish at specific intervals over a 10 hour time period and measuring the migration distance into a “scratch”. By creating orientation marks on the bottom of the tissue culture dish, we were able to photograph the same position at each time point. A direct comparison of these images enabled us to mark the initial position (red line) and then the advancing migration front (yellow line). By using Metamorph software we could measure (in microns) the distance between these two lines. Control- and C646-treated cells are represented in the left and right columns, respectively, with increasing time points from top to bottom.