

Abstract
Peroxisome proliferator–activated receptor (PPAR)-α is a transcription factor that has been reported to inhibit gentamicin-induced apoptosis in renal tubular cells. However, the antiapoptotic mechanism of PPARα is still unknown. In this study, we found that PPARα overexpression induced Na+/H+ exchanger-1 (NHE1) expression in the rat renal tubular cells NRK-52E. Beraprost, a PPARα ligand, also increased NHE1 expression in the renal tubules in normal mice, but not in PPARα knockout mice. Chromatin immunoprecipitation assays revealed that two PPARα binding elements were located in the rat NHE1 promoter region. Na+/H+ exchanger activity also increased in the PPARα-overexpressed cells. Flow cytometry showed that the PPARα-overexpressed cells were resistant to apoptosis-induced shrinkage. Cariporide, a selective NHE1 inhibitor, inhibited the antiapoptotic effect of PPARα in the gentamicin-treated cells. The interaction between NHE1 and ezrin/radixin/moesin (ERM) and between ERM and phosphatidylinositol 4,5-bisphosphate in the PPARα-overexpressed cells was more than in the control cells. ERM short interfering RNA (siRNA) transfection inhibited the PPARα-induced antiapoptotic effect. PPARα overexpression also increased the phosphoinositide 3-kinase (PI3K) expression, which is dependent on NHE1 activity. Increased PI3K further increased the phosphorylation of the prosurvival kinase Akt in the PPARα-overexpressed cells. Wortmannin, a PI3K inhibitor, inhibited PPARα-induced Akt activity and the antiapoptotic effect. We conclude that PPARα induces NHE1 expression and then recruits ERM to promote PI3K/Akt-mediated cell survival in renal tubular cells. The application of PPARα activation reduces the nephrotoxicity of gentamicin and may expand the clinical use of gentamicin.
INTRODUCTION
PPARα is a nuclear receptor for long-chain fatty acids and various fatty acid–derived compounds (1,2). Ligand-activated PPARα heterodimerizes with the retinoic X receptor (RXR) to regulate the expression of certain lipid metabolism–associated genes, such as the malonyl-CoA decarboxylase gene, by binding PPAR response elements (PPREs) located in the regulatory regions (3–5). Recent studies have shown that some drugs and hormones, such as l-carnitine, pravastatin and urotensin II, exert an antiapoptotic effect on renal tubular cells through PPARα activation (6–8). The activation of PPARα by fibrate treatment was found to inhibit cisplatin-mediated renal tubular injury in renal epithelial cells (9). Prostacyclin, a PPARα ligand, protects renal tubular cells from gentamicin-induced apoptosis through a PPARα-dependent pathway (10). Beraprost, an analog of prostacyclin, also protects mice from acute renal failure induced by radiographic contrast media (11). PPARα overexpression in rat renal tubular cells significantly inhibits doxorubicin-induced apoptosis (12). These findings suggest that PPARα expresses a strong antiapoptotic effect on renal tubular cells.
Gentamicin, an aminoglycoside antibiotic, is one of the first-line antibiotics for a wide range of gram-negative bacterial infections because of its clinical effectiveness and low cost (13). However, gentamicin is also nephrotoxic and induces acute kidney injury (AKI) in about 30% of patients (13,14). The key cytotoxic mechanism of gentamicin in renal proximal tubular cells is apoptosis inducing (15). Gentamicin reduces Bcl-xL expression and causes the release of cytochrome c from the mitochondria to activate caspase-3 and consequently induces mitochondria-mediated apoptosis in renal tubular cells (16). Until now, there has not been an ideal clinical remedy to prevent gentamicin-induced AKI.
Because of the antiapoptotic effect, PPARα is supposed to be a potential therapeutic target of gentamicin-induced apoptotic injury in renal tubular cells. A full exploration of the protective mechanism of PPARα will help to develop an effective remedy for gentamicin-induced AKI. Some studies show that reactive oxygen species downregulation is involved in the PPARα protective function in brain and renal tubular cells (10,17). The protective effect of PPARα is associated with heme oxygenase-1 expression and nuclear factor (NF)-κB inhibition (6,12). However, these mechanisms do not fully explain the PPARα antiapoptotic effect in renal tubular cells.
Recently, we found that PPARα overexpression upregulated Na+/H+ exchanger-1 (NHE1) in renal tubular cells. NHE1, an isoform of the membrane sodium-hydrogen antiporter, mediates Na+/H+ transport to maintain the cytosolic pH and cellular volume in almost all cells (18). In renal tubular cells, mature NHE1 is localized almost exclusively to the basolateral membrane (19). Recent studies reveal that NHE1 counteracts apoptosis in the renal proximal tubule and other tissues (19,20). NHE1 activation is an important regulatory volume increase mechanism, causing renal tubular cells to become resistant to apoptosis-induced shrinkage (21). NHE1-dependent H+ extrusion also leads to intracellular alkalinization to defeat apoptosis-associated cytosol acidification (18). In addition to Na+/H+ transportation, the NHE1 cytosolic tail domain binds with ezrin/radixin/moesin (ERM) proteins and phosphatidylinositol 4,5-bisphosphate (PIP2) to induce phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, which resists initial apoptotic stress (20). Genetic NHE1 loss of function causes renal tubule epithelial cell apoptosis, which is reduced by NHE1 reconstitution (22). These findings indicate that NHE1 activity is critical for tubular epithelial cell survival.
The present study aims to investigate the antiapoptotic mechanism of PPARα in renal tubular cells. The regulatory role of PPARα in NHE1 expression implies a connection between the antiapoptotic function of NHE1 and the protective effect of PPARα in renal tubular cells. Therefore, we explored the role of NHE1 and its signaling transduction in the PPARα antiapoptotic mechanism in gentamicin-treated rat renal tubular cells.
MATERIALS AND METHODS
Cell Culture
We purchased rat proximal renal tubular cells (NRK-52E) from the Bioresource Collection and Research Center. Cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with antibiotic/antifungal solution and 10% fetal calf serum. When the cells became confluent, the medium for the cultured cells was changed to the serum-free medium for overnight incubation before the experiment. The NHE1 inhibitor cariporide was purchased from Sigma-Aldrich.
RNA Extraction and the Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was extracted from the NRK-52E cells by using a TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. The isolated total RNAs were then analyzed by Welgene Biotech with next-generation sequencing analysis. For quantitative real-time polymerase chain reaction (RT-PCR) of the NHE1, the total RNA were reverse-transcribed by using a TaqMan Reverse Transcription Kit (Life Technologies) and then applied in TaqMan gene expression assays with specific primers of rat NHE1 (Rn00561924_m1) according to the manufacturer’s instructions (Life Technologies). Reactions were run on an RT-PCR system (ABI PRISMH 7700, Applied Biosystems). Specific primers for GAPDH (Rn01775763_g1) were used to normalize the amount of sample added. Relative amounts of the NHE1 mRNA were quantitated using the comparative computed tomography (CT) method. All quantifications were performed on triplicate samples of three separate experiments.
Construction for PPARα Overexpression
For PPARα overexpression, we amplified the rat PPARA gene fragment (GenBank: M88592.1) with BamH1 and Xba1 cuttings sites at the 5′ and 3′ ends from the total RNA by PCR with a 5′-primer (AAGGA TCCAT GGTGG ACACA GAGAG CCC) and 3′-primer (GGTCT AGATC AGTAC ATGTC TCTGT AGATC TC). The PCR product was inserted into pcDNA3 plasmid (Invitrogen) as a PPARα overexpression construct (pcPPAR). We transfected the NRK-52E cells with the plasmids using TurboFect Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Western Blot Analysis
A total of 20 μg NRK-52E lysate proteins were applied to each lane and analyzed by Western blotting. Anti-PPARα and anti-PIP2 antibodies were from Abcam (Cambridge, UK); the anti–NHE-1 (H-160) antibody was from Santa Cruz Biotechnology; the anti-PI3K p85, anti-Akt, anti–phosphate-Akt (Thr308), anti–Bcl-xL, anti–caspase-3, anti-ezrin/radixin/moesin and anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were from Cell Signaling Technology; and the anti-radixin antibody was from Thermo Fisher Scientific. Peroxidase-conjugated anti-rabbit or anti-goat IgG (1:5,000 dilution) was used as the second antibody to detect primary antibodies by enhanced chemiluminescence (Amersham Biosciences).
Apoptosis Detection
Cell apoptosis was detected by flow cytometry with fluorescein isothiocyanate–annexin V/propidium iodide (PI) double staining. Treated NRK-52E cells were harvested and washed twice with ice-cold phosphate-buffered saline (PBS) and stained using an apoptosis detection kit (Trevigen). The stained cells were analyzed by LSRFortessa™ Cell Analyzer (BD Biosciences, Becton Dickinson) and BD FACSDiva™ software.
Cytosolic pH Measurement
The cytosolic pH of the NRK-52E cells was monitored by using ratio-fluorometric pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5(6)-carboxy-fluorescein (BCECF, Invitrogen), as described previously (23). In brief, BCECF (10 μmol/L) was added to treated adherent cells, which were maintained in serum-free medium for 45 min. The BCECF fluorescence emission ratio of the 510-nm emission at 490- and 440-nm excitation was calibrated by exposing BCECF loaded cells to the six nigericin (10 μmol/L) calibration buffers, 2-(N-morpholino) ethanesulfonic acid (MES) (pH 5.5), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 6.5, 7.0, 7.5, 8.0) and N-cyclohexyl-2-hydroxyl-3-aminopropanesulfonic acid (CAPSO) (pH 9.5). Acute acid loading was induced by extracellular exposure to 20 mmol/L NH4Cl for 10 min. BCECF fluorescence was measured with a SpectraMax M5 spectrophotometer (Molecular Devices) at 490- and 440-nm excitation and 510-nm emission wavelengths. The cytosolic pH was converted from fluorescent ratio using the logarithmic equation described in the other report (23). Nigericin, MES, HEPES, CAPSO, and NH4Cl were purchased from Sigma-Aldrich.
Animals and Treatments
Male C57BL/6 mice and PPARα knockout mice (B6.129S4-Pparatm1Gonz N12) weighing 20–25 g and aged 7 wks were obtained from BioLasco Taiwan and Taconic Biosciences, respectively. The Taipei Medical University Committee of Experimental Animal Care and Use approved all animal experiments in this study. Mice were housed in a central facility, subjected to a 12-h light–dark cycle and provided with regular rat food and tap water. For monitoring NHE1 expression, mice received an intravenous (IV) injection of beraprost sodium (Cayman Chemical) dissolved in saline, at a dose of 100 μg/kg (n = 3), or received IV injections of saline (n = 3). The kidneys of the mice were harvested 3 h after the treatment with a surgical operation and homogenized for Western blotting analysis. For histological analysis, we fixed the harvested kidneys in 10% formalin and embedded in paraffin and sectioned at 4-μm thickness for hematoxylin/eosin staining or immunohistochemistry staining. Additionally, for detecting the antiapoptotic effect of beraprost on the kidneys, normal mice and PPARα knockout mice were separated into three groups, respectively. Mice (n = 3) for gentamicin treatment received an intraperitoneal (IP) injection of gentamicin (30 mg/kg/day) for 7 d. Mice (n = 3) for gentamicin and beraprost treatment received an IV injection of beraprost sodium at a dose of 100 μg/kg/day 30 min before each gentamicin injection. Control mice (n = 3) received IP and IV injections of saline. Treated and control mice were killed 24 h after the last treatment, and the kidneys of the mice were harvested and frozen for in situ terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay.
Immunohistochemistry
Slides were deparaffinized in xylene and rehydrated with 100% ethanol, 95% ethanol and water serially. We immersed the slides into antigen-retrieval buffer (10 mmol/L trisodium citrate dihydrate, 0.05% Tween-20, pH 6.0) boiled by water bath for 30 min. Slides were stained by using an UltraVision Quanto HRP Detection kit (Thermo Fisher Scientific) with an NHE1 antibody (sc-28758, Santa Cruz) dilution (1:500) according to the manufacturer’s instructions. The slides were observed by using an Axiovert 40CFL microscope (Carl Zeiss Microscopy) with a 20×/0.3 Ph1 objective lens. The images were acquired by a SPOT RT3 camera (SPOT Imaging Solutions) with SPOT Software.
In Situ Terminal Deoxynucleotidyl TUNEL Assay
We processed kidney slides with an ApopTag Fluorescein in situ apoptosis detection kit (Chemicon International) according to the manufacturer’s instructions. The slides were observed by using an Axiovert 40CFL microscope (Carl Zeiss Microscopy) with a 20×/0.3 Ph1 objective lens and HBO 50 Microscope Illuminator (Carl Zeiss Microscopy).
Chromatin Immunoprecipitation (ChIP) Assay
NRK-52E cells were cross-linked, lysed and applied in the ChIP assay using the Chromatin Immunoprecipitation Assay Kit and EZ-Zyme™ Chromatin Prep Kit (Millipore) according to the manufacturer’s instructions. The cross-linked DNA samples were amplified by PCR with specific primers, as shown in Figure 3. PCR products were analyzed by 8% polyacrylamide gels stained with SYBR Green.
Figure 3.

PPAR response elements (PPREs) for PPARα in the rat NHE1 promoter region. (A) A schematic representation of the rat NHE1 promoter (−3106 to −1). Transcription start site is marked by +1. The nucleotide positions of the black box indicate the localization of the putative PPREs. (B) Nucleotide sequences surrounding the rat malonyl-CoA decarboxylase (MCD) PPREs and putative NHE1 PPREs. We used two well-known PPREs in the rat MCD promoter region in the ChIP assay as positive controls. Nucleotide sequences of MCD PPREs are shown in bold. Nucleotide sequences of putative NHE1 PPREs have a gray background. Primers designed for ChIP are underlined. Two sets of primers specific for −322/−248 and −207/−127 regions (C1 and C2) containing PPREs in the rat MCD promoter and three sets of primers specific for −2889/−2802, −2388/−2289 and −615/−531 regions (N1, N2 and N3) containing putative PPREs in the rat NHE1 promoter were used in the PCR. (C) ChIP analysis of the rat NHE1 promoter DNA. ChIP analysis was performed with an anti-PPARα antibody. The PCR products were detected by agarose gel electrophoresis. M, DNA marker (base pair [bp]). (D) The luciferase reporter assay of the truncated rat NHE1 promoter in the pcPPAR-transfected NRK-52E cells. The pcPPAR-transfected cells were transfected with pGL3 vector, −2889/−1 NHE1-LUC, −2388/−1 NHE1-LUC or a −615/−1 NHE1-LUC construct as indicated. Results are means ± SD (n = 3). *P < 0.05 by Student t test.
Luciferase Reporter Assay
We extracted rat genomic DNA from the NRK-52E cells by using a Genomic DNA Mini Kit (Geneaid). Rat NHE1 promoter fragments (−2889/−1, −2388/−1, and −615/−1) were amplified by PCR from rat genomic DNA with the forward primer 1 (GGTAC CAACT AGGAA TCATG CTTCC TGTGA C), forward primer 2 (GGTAC CAGGA ACTGG ACACT CCCAG AGCT), forward primer 3 (GGTAC CCTGC GCCGA GGCGC CTTCC C) and reverse primer (GGATC CACTG ATCCC CCAGC CTAGG A). The PCR products were digested with KpnI and BamH1 and cloned into the reporter plasmid pGL3-Basic (Promega) to generate −2889/−1 NHE1-LUC, −2388/−1 NHE1-LUC and −615/−1 NHE1-LUC. Plasmids were verified by DNA sequencing and compared with the nucleotide sequences of the rat NHE1 promoter fragment (GenBank NW_001084844.1). Cells were transfected with 100 ng luciferase reporter plasmids as described above. Cell extracts were measured by using the Dual-Luciferase Reporter Assay system (Promega). Firefly luciferase activity values were divided by Renilla luciferase activity values to obtain the normalized luciferase activities.
Immunoprecipitation
The transfected cells were lysed at 4°C in lysis buffer (50 mmol/L Tris, pH 7.5, 1% Nonidet P-40, 0.5% sodium deoxycholate, 150 mmol/L NaCl and protease inhibitors). The immunocomplexes were precipitated by protein A agarose (Roche Molecular Biochemicals) with anti-NHE1 (Santa Cruz), anti-ERM (Cell Signaling Technology) or anti-PIP2 (Abcam) antibodies, following the manufacturer’s instructions.
Short Interfering RNA (siRNA) Transfection
The rat ezrin siRNA (item number s132627) was purchased from Life Technologies, and the rat radixin and moesin siRNA (item numbers M-101027-01-0005 and M-093613-01-0005, respectively) were purchased from Thermo Fisher Scientific. PPARα-overexpressed cells were transiently transfected with the siRNAs or mock control oligonucleotides by using a Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions. Twenty four hours after transfection, the cells were washed and resuspended in new culture media for an additional 24-h gentamicin treatment and Western blot analysis.
Statistics
Data were presented as the mean ± standard deviation (SD). Statistical differences between the two groups were determined using a Student t test. One-way analysis of variance (ANOVA) was used in cytosolic pH detection. Differences between the groups were considered significant if P values were smaller than 0.01.
RESULTS
PPARα Protects Renal Tubular Cells from Gentamicin-Induced Apoptosis
To investigate the antiapoptotic mechanism of PPARα, we transfected rat renal tubular cells (NRK-52E) with rat PPARα cDNA construct (pcPPAR) to overexpress PPARα protein. Compared with blank vector-transfected cells, the PPARα protein level increased more than four-fold in the pcPPAR-transfected cells (Figure 1A). We next evaluated the influence of PPARα overexpression on the gentamicin-induced apoptosis in NRK-52E cells by annexin V/PI staining and flow cytometry. As shown in Figures 1B and C, 3 mmol/L gentamicin induced ~24% apoptosis in the control cells, but only 4% apoptosis in the pcPPAR-transfected cells. This result shows that PPARα overexpression protects the NRK-52E cells from gentamicin-induced apoptosis. To investigate the antiapoptotic effect of PPARα in vivo, both normal and PPARα knockout mice were treated with gentamicin (30 mg/kg/day) or saline as controls; the experimental groups were treated with beraprost, a synthetic ligand of PPARα (100 μg/kg/day) in addition. After 7-d treatments, the kidneys were harvested and sectioned for in situ TUNEL assay. As shown in Figure 1D, the scattered and bright nuclei stained by TUNEL staining could be detected over the entire renal cortex from gentamicin-treated mice and beraprost/gentamicin-treated PPARα knockout mice, but they were rarely detected in the specimens of the controls and beraprost/gentamicin-treated normal mice. This result reveals that beraprost-induced PPARα activation inhibits the gentamicin-induced cell apoptosis in renal tubular cells in vivo.
Figure 1.

The inhibitory effect of PPARα overexpression on gentamicin-induced apoptosis in NRK-52E cells. NRK-52E cells were transfected with pcPPAR or blank vectors and then treated with or without 3 mmol/L gentamicin for 24 h. (A) PPARα expression levels in the pcPPAR transfected cells. The level of PPARα protein in each sample was determined by Western blot analysis. GAPDH was used as the loading control. Relative increases in the protein bands are also presented in a bar chart form. Results are expressed as mean ± SD (n = 3). C, blank vector control; P, pcPPAR transfection. *P < 0.01 versus the blank vector control group (by Student t test). (B) Representative flow cytometric data of gentamicin-induced apoptosis. Treated cells were stained with annexin V/PI and analyzed by flow cytometry. The sum of the cell percentages in quadrants 2 and 4 represents the percentage of apoptosis. G, gentamicin treatment. (C) The apoptotic percentage of gentamicin-treated cells. Results are means ± SD (n = 3). *P < 0.01 by Student t test. (D) The protective effect of beraprost against gentamicin-induced renal tubular cell apoptosis in vivo. Mice were injected with saline, gentamicin and beraprost as described in Materials and Methods. Apoptotic cells in kidneys of experimental mice were detected using in situ TUNEL staining. TUNEL-labeled nuclei were revealed as bright spots in cortex sections from gentamicin-treated mice. The identical fields stained for TUNEL were also stained using PI to reveal the positions of cell nuclei. PPARα KO, PPARα knockout mice. The number of TUNEL-labeled cells per millimeter-squared cortex area in each sample was also compiled and demonstrated. Results are expressed as the mean ± SD (n = 6). *P < 0.05 compared with the normal mice treated with gentamicin alone. Scale bar, 50 μm.
PPARα Upregulates NHE1 Expression and Increases NHE1 Activity in Renal Tubular Cells
To investigate the gene expression influenced by PPARα overexpression, we used next-generation sequencing to analyze the mRNA profiles of the NRK-52E cells transfected with pcPPAR or the blank vector. NHE1 mRNA showed a significant increase in the pcPPAR-transfected cells and was further validated. Compared with the control cells, the NHE1 protein level increased more than three-fold in the pcPPAR-transfected cells (Figure 2A). The NHE1 mRNA level also increased more than four-fold in the pcPPAR-transfected cells as revealed by quantitative real-time PCR analysis (Figure 2B). We further observed NHE1 activity in the pcPPAR-transfected cells by measuring the intracellular pH values with pH-sensitive fluorophore BCECF. Ten minutes after an NH4Cl prepulse, the cytosol pH in the control cells rose from 7.01 to 7.66 in 180 s, whereas the cytosol pH in the pcPPAR-transfected cells rose from 7.03 to 7.67 in 140 s (Figure 2C). The pH recovery in the pcPPAR-transfected cells was faster than in the control cells. The NHE1 inhibitor cariporide significantly inhibited the cytosolic pH recovery in both the control and pcPPAR-transfected cells.
Figure 2.

NHE1 upregulation caused by PPARα overexpression and activation in renal tubular cells. (A) NHE1 expression levels in NRK-52E cells. The levels of NHE1 protein in pcPPAR- or blank vector–transfected cells were determined by Western blot analysis. GAPDH was used as the loading control. Relative increases in the protein bands are also presented in a bar chart form. Results are expressed as mean ± SD (n = 3). P, pcPPAR; C, blank vector. (B) NHE1 mRNA levels in NRK-52E cells. The mRNA quantity of NHE1 in each sample was detected by a quantitative RT-PCR. The relative mRNA level of NHE1 is shown as the mean ± SD from six experiments. (C) Cytosolic pH values. NRK-52E cells were transfected with pcPPAR or blank vectors (Control) and then treated with or without 1 μmol/L cariporide. For cytosolic pH recovery assay, the cells were incubated with 20 mmol/L NH4Cl for 10 min. The pH recovery in the pcPPAR-transfected cells was faster than in the control cells. Results are means ± SD (n = 3). *P < 0.01 by one-way ANOVA. (D) Cell volume detected by flow cytometry. NRK-52E cells were transfected with pcPPAR or blank vectors (Control), pretreated with or without 1 μmol/L cariporide for 30 min and then treated with or without 3 mmol/L gentamicin for 24 h. Cells were analyzed by flow cytometry with forward scatter-area (FSC-A) parameter measuring. FSC-A is proportional to cell volume. x Axis, the FSC-A scales; y axis, the cell numbers. Dotted lines indicate the means of FSC-A. (E) NHE1 expression level in renal tissue from beraprost-treated mice. Mice were injected with beraprost to activate PPARα as described in Materials and Methods. The levels of NHE1 protein in the kidneys of beraprost-treated mice were determined by Western blot analysis. Malonyl-CoA decarboxylase (MCD) was used as a positive control, and GAPDH was used as the loading control. Relative increases in the NHE1 protein bands are also presented in a bar chart form. Results are expressed as mean ± SD (n = 3). P, pcPPAR; C, blank vector. (F) Expression of NHE1 in renal tissue from beraprost-treated normal mice and PPARα knockout mice by immunohistochemistry. The white arrows indicate the positive label of NHE1 at the basolateral membrane of renal tubular cells in beraprost-treated normal mice. C, control; B, beraprost treatment. Scale bar, 50 μm.
On the other hand, we also monitored the cell volume of the pcPPAR-transfected cells by flow cytometry with forward scatter area (FSC-A) parameter measuring. Gentamicin treatment resulted in a significant reduction of the mean cell volume within 12 h in the control cells (Figure 2D). The cell volume of the pcPPAR-transfected cells obviously increased, and the gentamicin-induced cell volume decrease was limited in the pcPPAR-transfected cells. Similar to the pH changes, the pcPPAR transfection-induced volume increase and protective effect against gentamicin-induced volume decrease were inhibited by the cariporide treatment. These results suggest PPARα overexpression transcriptionally upregulates NHE1 expression and increases NHE1 activity in the NRK-52E cells.
We further treated mice with beraprost and found NHE1 expression level in the kidney of mice obviously increased (Figure 2E). As a positive control, the expression level of malonyl-CoA decarboxylase, a known PPARα-upregulated enzyme, also increased in the kidney of beraprost-treated mice. The immunohistochemistry data also showed that beraprost induced NHE1 expression in normal mice but not in PPARα knockout mice (Figure 2F). Beraprost-induced NHE1 was localized to the basolateral membrane of renal tubular cells in mice. These results reveal that beraprost-induced PPARα activation upregulates NHE1 expression in renal tubular cells in vivo.
PPARα Binding Sites of the Rat NHE1 Gene Promoter
The involvement of PPARα in the NHE1 transcription inspired us to identify the PPARα-binding sites in the −3106/−1 rat NHE1 gene promoter. We detected three putative peroxisome proliferator hormone response elements (PPREs) by computer-assisted analysis using TFBIND (http://www.ims.u-tokyo.ac.jp/imsut/en) and TRED (http://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home). These sites were named PPRE1, PPRE2 and PPRE3 as described in Figures 3A and B. We next clarified the binding capability of these PPREs to PPARα by ChIP analysis. The primers corresponding to the region spanning the three PPREs were designed as shown in Figure 3B. As shown in Figure 3C, the PPARα-immunoprecipitated sample exhibited positive regions containing PPRE1 and PPRE2, but not PPRE3. This result indicates that PPARα interacts with PPRE1 and PPRE2 of the NHE1 gene promoter in the NRK-52E cells.
To demonstrate further the functionality of these PPRE sites, 5′ deletions of the rat NHE1 gene promoter were linked to the luciferase reporter gene and analyzed in the pcPPAR-transfected NRK-52E cells. As shown in Figure 3D, the luciferase activity significantly increased in the pcPPAR-transfected cells cotransfected with −2889/−1 NHE1-LUC (containing PPRE1, PPRE2 and PPRE3) and −2388/−1 NHE1-LUC (containing PPRE2 and PPRE3), but not −615/−1 NHE1-LUC (containing PPRE3). The luciferase activity in the −2889/−1 NHE1-LUC cotransfected cells was significantly higher than in the −2388/−1 NHE1-LUC cotransfected cells. These results suggest that PPRE1 and PPRE2 are the transcriptional regulatory regions by PPARα in the rat NHE1 promoter.
Involvement of NHE1 Activity in PPARα Antiapoptotic Effect
We evaluated the involvement of NHE1 activity in the PPARα antiapoptotic effect in the NRK-52E cells by annexin V/PI staining and flow cytometry. As shown in Figures 4A and B, 3 mmol/L gentamicin did not induce significant apoptosis in the pcPPAR-transfected cells, but induced ~28% apoptosis in the cariporide-treated pcPPAR-transfected cells. Compared with Figure 1, cariporide significantly inhibited the antiapoptotic effect caused by PPARα overexpression in the NRK-52E cells.
Figure 4.

The involvement of NHE1 activity in the PPARα antiapoptotic effect. NRK-52E cells were transfected with pcPPAR or blank vectors, pretreated with or without 1 μmol/L cariporide for 30 min and then treated with or without 3 mmol/L gentamicin for 24 h. (A) Representative flow cytometric data of gentamicin-induced apoptosis in the pcPPAR-transfected cells. Treated cells were stained with annexin V/PI and analyzed by flow cytometry. The sum of cell percentages in quadrants 2 and 4 represents the percentage of apoptosis. P, pcPPAR transfection; G, gentamicin treatment; Cari, cariporide treatment. (B) The gentamicin-induced apoptotic percentage in the pcPPAR-transfected cells. Results are means ± SD (n = 3). *P < 0.05 by Student t test. (C) The influence of cariporide on gentamicin-induced apoptotic signals in the pcPPAR-transfected NRK-52E cells. Total proteins of NRK-52E cells were analyzed by Western blotting with the specific antibodies as indicated. GAPDH was used as a loading control. Relative increases in the protein bands are also presented in a bar chart form. Results are means ± SD (n = 3). *P < 0.05 versus the blank vector group without any treatment. #P < 0.05 versus the blank vector group with gentamicin treatment. **P < 0.05 versus the pcPPAR group with gentamicin treatment. Statistical differences between the two groups were determined using a Student t test.
The influence of PPARα overexpression on the apoptotic signaling pathways was investigated by Western blot analysis. As shown in Figure 4C, the cariporide treatment did not influence PPARα expression, but inhibited the PPARα-induced NHE1 expression. Gentamicin significantly reduced the NHE1 and Bcl-xL expression as well as induced cleaved caspase-3 in the normal and cariporide-treated cells, but not in the pcPPAR-transfected cells without cariporide treatment. Cariporide treatment even reduced the basal level of Bcl-xL in the control and pcPPAR-transfected cells. These results suggest that NHE1 plays a critical role in the PPARα antiapoptotic effect in the NRK-52E cells.
Association of ERM and PIP2 with PPARα Antiapoptotic Effect
Because NHE1 was reported to bind with ERM and PIP2 as a cell survival signal complex in the renal tubule epithelial cells, we next monitored the interaction between NHE1 and ERM as well as PIP2 in the PPARα-overexpressed NRK-52E cells by coimmunoprecipitation. As shown in Figure 5A, the interaction between NHE1 and ERM in the pcPPAR-transfected cells was more than in the control cells. The interaction between ERM and PIP2 was also obvious in the pcPPAR-transfected cells (Figure 5B), although the interaction between NHE1 and PIP2 was undetectable in our system (data not shown). ERM and PIP2 might be involved in the protective effect of PPARα overexpression. To further determine the involvement of ERM in the PPARα-induced protective effect, the siRNAs of ezrin, radixin and moesin were used for ERM knockdown in the pcPPAR-transfected NRK-52E cells. Compared to the mock controls, ezrin siRNA significantly reduced ezrin/radixin expression, radixin siRNA significantly reduced radixin expression and moesin siRNA significantly reduced moesin expression in the pcPPAR-transfected cells (Figure 5C). Gentamicin significantly reduced Bcl-xL expression and induced cleaved caspase-3 in the pcPPAR-transfected cells with ERM siRNA transfection (Figure 5C). These results suggest that ERM is involved in the PPARα antiapoptotic effect in the NRK-52E cells.
Figure 5.

The involvement of ERM and PIP2 in the PPARα-induced NHE1 signaling pathway. (A) The interaction between NHE1 and ERM. NRK-52E cells were transfected with pcPPAR or blank vectors. NHE1 and ERM in transfected cells were immunoprecipitated with anti-NHE1 and anti-ERM antibodies, respectively. ERM and NHE1 in the immunocomplexes were detected by using Western blotting. IgG was detected as a loading control. Relative increases in the protein bands are also presented in bar chart form. Results are means ± SD (n = 3). *P < 0.05 by Student t test. (B) Interaction between ERM and PIP2. ERM and PIP2 in the transfected cells were immunoprecipitated with anti-ERM and anti-PIP2 antibodies, respectively. PIP2 and ERM in the immunocomplexes were detected by using Western blotting. Relative increases in the protein bands are also presented in bar chart form. Results are means ± SD (n = 3). *P < 0.05 by Student t test. (C) Influence of ERM siRNA transfection on apoptotic signals in the pcPPAR-transfected cells. The pcPPAR-transfected cells were further transfected with ezrin (Ez), radixin (Ra), moesin (Mo) or mock control (Mk) siRNA and then treated with or without 3 mmol/L gentamicin for 24 h. Total proteins of treated cells were analyzed by Western blotting with the specific antibodies as indicated. GAPDH was used as a loading control. Relative increases in the protein bands are also presented in a bar chart form. Results are means ± SD (n = 3). *P < 0.05 versus the mock control group. #P < 0.05 versus the mock control group with gentamicin treatment. Statistical differences between the two groups were determined using the Student t test.
PI3K Upregulation Caused by PPARα Overexpression
To investigate the involvement of ERM-mediated PI3K/Akt signaling in the PPARα antiapoptotic effect, we monitored the expression of PI3K and phospho-Akt by Western blot analysis. As shown in Figure 6A, gentamicin reduced the expression of Bcl-xL and phospho-Akt in the vector control cells, but not in the pcPPAR-transfected cells. PPARα overexpression significantly increased the PI3K p85 subunit and phospho-Akt expression. Wortmannin, a specific inhibitor for PI3K, inhibited Akt phosphorylation and blocked the influence of PPARα overexpression on gentamicin-reduced Bcl-xL and gentamicin-induced cleaved caspase-3. These results suggest that the PI3K/Akt signaling pathway is involved in the PPARα antiapoptotic effect. Additionally, PI3K increase in the pcPPAR-transfected cells was inhibited by the cariporide treatment (Figure 6B), indicating that NHE1 activation is necessary for PPARα overexpression-induced PI3K expression.
Figure 6.
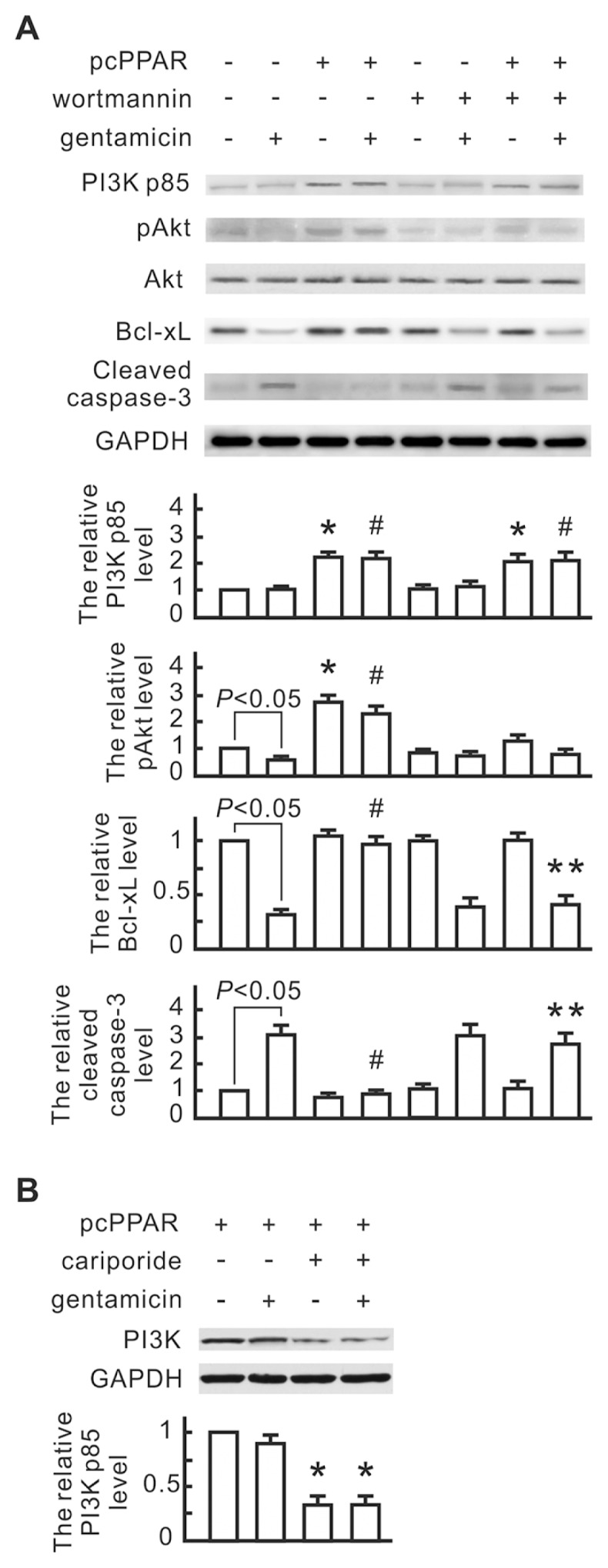
The involvement of PI3K and phospho-Akt in the PPARα-induced NHE1 signaling pathway. (A) The influence of wortmannin on gentamicin-induced apoptotic signals in the pcPPAR-transfected NRK-52E cells. NRK-52E cells were transfected with pcPPAR or blank vectors, pretreated with or without 1 nmol/L wortmannin (a PI3K inhibitor) for 30 min and then treated with or without 3 mmol/L gentamicin for 24 h. Total proteins of the transfected cells were analyzed by Western blotting with the specific antibodies as indicated. GAPDH was used as a loading control. Relative increases in the protein bands are also presented in a bar chart form. Results are means ± SD (n = 3). *P < 0.05 versus the blank vector group without any treatment. #P < 0.05 versus the blank vector group with gentamicin treatment. **P < 0.05 versus the pcPPAR group with gentamicin treatment. Statistical differences between the two groups were determined using the Student t test. (B) The influence of cariporide on PI3K expression in the pcPPAR-transfected cells. The pcPPAR-transfected cells were pretreated with 1 μmol/L cariporide for 30 min and then treated with or without 3 mmol/L gentamicin. PI3K in each sample was detected by Western blotting. Relative increases in the protein bands are also presented in a bar chart form. Results are means ± SD (n = 3). *P < 0.05 versus the blank vector group without any treatment (by Student t test).
Involvement of ERM and PI3K/Akt in PPARα Antiapoptotic Effect
The influence of ERM siRNA transfection and wortmannin on the PPARα antiapoptotic effect was further monitored by annexin V/PI staining and flow cytometry. Gentamicin induced ~30% apoptosis in the mock control cells, which was inhibited by pcPPAR transfection (Figure 7). Ezrin, radixin and moesin siRNA transfection individually reduced the PPARα antiapoptotic effect. Wortmannin also significantly inhibited the PPARα antiapoptotic effect. These results suggest that ezrin, radixin, moesin and PI3K/Akt are involved in the PPARα antiapoptotic effect.
Figure 7.

The involvement of ERM and PI3K in the PPARα antiapoptotic effect. The pcPPAR-transfected NRK-52E cells were further transfected with ezrin, radixin, moesin or mock control siRNA, or pretreated with or without 1 nmol/L wortmannin for 30 min, and then treated with or without 3 mmol/L gentamicin for 24 h. (A) Representative flow cytometric data of gentamicin-induced apoptosis. Treated cells were stained with annexin V/PI and analyzed by flow cytometry. The sum of the cell percentages in quadrants 2 and 4 represents the percentage of apoptosis. C, mock control; G, gentamicin treatment; P, pcPPAR transfection; siE, ezrin siRNA; siR, radixin siRNA; siM, moesin siRNA; wort, wortmannin treatment. (B) The apoptotic percentage of gentamicin-treated cells. Results are means ± SD (n = 3). *P < 0.05 versus the pcPPAR group with gentamicin treatment (by Student t test).
DISCUSSION
The detailed mechanism of the PPARα antiapoptotic effect in renal tubular cells is not yet clear. Some studies show that the cross-talk of PPARα with mitogen-activated protein kinases results in a coordinated suppression of apoptosis (24). PPARα has also been found to inhibit the transcription factor necrosis factor-κB in renal tubular cells (25). However, these mechanisms are not sufficient to explain the strong antiapoptotic effect of PPARα in renal tubular cells. In the present study, we demonstrated that PPARα bound the NHE1 promoter region and upregulated NHE1 expression in the rat renal tubular cells (Figures 2, 3). PPARα overexpression increased antiacidification and regulatory volume increase ability in the NRK-52E cells via NHE1 activity (Figure 2). NHE1 activity was necessary for the protective effect of PPARα against gentamicin-induced apoptosis (Figure 4). In the animal study, beraprost-induced PPARα activation also increased NHE1 expression in renal tubules and inhibited gentamicin-induced apoptosis (Figure 2). Gentamicin is a widely used antibiotic to treat gram-negative bacterial infection because of its low cost. But gentamicin can cause acute kidney injury in about 20% of patients (26). This potential nephrotoxicity seriously limits the use of gentamicin. On the basis of our results, the application of PPARα activation, such as beraprost treatment, could reduce the nephrotoxicity of gentamicin and expand the clinical use of gentamicin.
NHE1 is often thought to be a housekeeping protein because its major functions include maintenance of intracellular pH and volume (20). This study presents the first evidence that NHE1 expression is regulated directly by PPARα through PPREs in the NHE1 promoter region. The functional PPREs are located at −2863/−2847 and −2358/−2345 in the rat NHE1 promoter. Sequence analysis shows that these two PPREs are similar to the PPRE (−2979/−2967) in the human apolipoprotein A-IV gene (27), and the similarities are 10/17 and 10/14, respectively. Human hepatic apolipoprotein A-IV is a lipid metabolism–associated protein and regulated directly by PPARα. Most PPARα-regulated genes are associated with lipid metabolism, but NHE1 activity is independent of lipid metabolism. However, the NHE1 cytosolic tail is supposed to be a caspase-3 substrate in renal tubular cells (22). We also found gentamicin reduced NHE1 expression in the NRK-52E cells (Figure 4C). The NHE1 degradation is adverse to the maintenance of cell volume constancy and survival. Thus, PPARα-mediated NHE1 upregulation is an important survival mechanism in renal tubular cells.
Many studies have suggested that NHE1 functions directly influence apoptosis in renal tubular cells. NHE1 activation results in a Na+ and H2O influx that causes cytoplasmic volume expansion. Cells undergoing apoptosis exhibit persistent cell shrinkage, even under isotonic environments. NHE1-mediated regulatory volume increases protect the renal tubule cells against apoptotic volume decreases (21). Additionally, NHE1-induced Na+ entry also accompanies H+ extrusion and intracellular alkalinization, and apoptosis accompanies cytosol acidification (28). NHE1-mediated alkalinization can resist apoptosis by inhibiting caspase or acidic endonuclease catalysis (29–31), preventing location changes of the proapoptotic Bcl-2 family members (32), or removing mitochondrion-released H+ in apoptotic cells (33). Therefore, NHE1-dependent defense against cell volume reduction and intracellular acidification plays a role in the PPARα protective effect in the early stages of apoptosis (Figure 8).
Figure 8.

A working model addressing the NHE1 role in the PPARα antiapoptotic effect in renal tubular cells.
NHE1 is reported to act as a scaffold by binding with ERM proteins and PIP2, which initiates formation of a signaling complex that culminates in Akt activation and opposition to initial apoptotic stress (20). The interaction between NHE1 and ERM and between ERM and PIP2 significantly increased in the PPARα-overexpressed NRK-52E cells (Figure 5). ERM siRNA transfection reduced the PPARα antiapoptotic effect (Figures 5, 7), indicating that ERM played a role in the protective effect. PPARα overexpression also induced PI3K expression and Akt phosphorylation, which depended on NHE1 activity (Figure 6). Functional blockage of PI3K by wortmannin inhibited Akt phosphorylation and the PPARα antiapoptotic effect (Figures 6, 7). Our results suggest that NHE1 uses ERM to bind PIP2 in a signalplex, which induces PI3K expression. PI3K converts PIP2 into phosphatidylinositol (3,4,5)-phosphate [PI(3,4,5)P3] and results in the phosphorylation of Akt (34). The PI3K/Akt pathway is a well-known antiapoptotic pathway. Phosphorylated Akt activates antiapoptotic proteins such as Bcl-2 and Bcl-xL (35) and prevents cytochrome c leakage from the mitochondria. The blockage of cytochrome c leakage also inhibits proapoptotic protein caspase-9 and -3 activities (36). Our data show that PPARα upregulates NHE1expression and then recruits ERM and PIP2 to transmit the cell survival signal via the PI3K/Akt signaling pathway. Our study first provides the evidence that NHE1, ERM and PI3K are involved in the protective effect of PPARα against gentamicin-induced apoptosis in renal tubular cells.
Our results showed that cariporide inhibited PPARα-induced NHE1 and PI3K expression in the NRK-52E cells (Figures 4C and 6B). The detailed mechanism is still unclear. Recent studies reveal that PI3K expression is regulated by the promyelocytic leukemia zinc finger protein (PLZF) (37,38). PLZF is a sequence-specific DNA-binding transcriptional factor. The nuclear translocation of PLZF induces the expressions of PI3K p85-α and p85-β subunits at the transcriptional level. Naray-Fejes-Toth et al. (39) demonstrated that PLZF is a negative regulator of the epithelial Na channel in renal epithelial cells. This finding implies that there is a negative correlation between PLZF activation and Na transport. PLZF might be part of a negative feedback mechanism that inhibits PPARα-induced NHE1 and PI3K upregulation. However, more studies are necessary to investigate the mechanism of the inhibitory effect of cariporide on NHE1 expression in PPARα-overexpressed renal tubular cells.
CONCLUSION
This study indicated that NHE1 upregulated by PPARα protected the renal tubular cells from gentamicin-induced apoptosis via NHE1-associated pathways, including regulatory volume increase, alkalinization and PI3K/Akt activation. Besides gentamicin, some other important clinical drugs, such as cisplatin, cyclosporine A and radiocontrast agents, also induce apoptosis in renal tubular cells to cause nephrotoxicity. Therefore, the application of PPARα activation may reduce nephrotoxic drug-induced acute kidney injury and is helpful to expand the clinical use of the nephrotoxic drugs.
ACKNOWLEDGMENTS
This study was sponsored by National Science Council grant NSC101-2314-B-038-020-MY3 to YH Hsu.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Chen C-H, et al. (2015) Peroxisome proliferator–activated receptor α protects renal tubular cells from gentamicin-induced apoptosis via upregulating Na+/H+ exchanger NHE1. Mol. Med. 21:886–99.
REFERENCES
- 1.Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 2014;53:124–44. doi: 10.1016/j.plipres.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Vane JR, Botting RM. Pharmacodynamic profile of prostacyclin. Am J Cardiol. 1995;75:3A–10A. doi: 10.1016/s0002-9149(99)80377-4. [DOI] [PubMed] [Google Scholar]
- 3.Smirnov AN. Nuclear receptors: nomenclature, ligands, mechanisms of their effects on gene expression. Biochemistry (Mosc) 2002;67:957–77. doi: 10.1023/a:1020545200302. [DOI] [PubMed] [Google Scholar]
- 4.Campbell FM, et al. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 5.Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: a key regulation of malonyl-CoA level. Biochem J. 2004;378:983–90. doi: 10.1042/BJ20031565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen HH, Chen TW, Lin H. Pravastatin attenuates carboplatin-induced nephrotoxicity in rodents via peroxisome proliferator-activated receptor alpha-regulated heme oxygenase-1. Mol Pharmacol. 2010;78:36–45. doi: 10.1124/mol.109.061101. [DOI] [PubMed] [Google Scholar]
- 7.Hsu YH, et al. Urotensin II exerts antiapoptotic effect on NRK-52E cells through prostacyclin-mediated peroxisome proliferator-activated receptor alpha and Akt activation. Mol Cell Endocrinol. 2013;381:168–74. doi: 10.1016/j.mce.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 8.Chen HH, et al. Peroxisome proliferator-activated receptor alpha plays a crucial role in L-carnitine anti-apoptosis effect in renal tubular cells. Nephrol Dial Transplant. 2009;24:3042–9. doi: 10.1093/ndt/gfp258. [DOI] [PubMed] [Google Scholar]
- 9.Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005;68:2680–93. doi: 10.1111/j.1523-1755.2005.00739.x. [DOI] [PubMed] [Google Scholar]
- 10.Hsu YH, et al. Prostacyclin protects renal tubular cells from gentamicin-induced apoptosis via a PPARalpha-dependent pathway. Kidney Int. 2008;73:578–87. doi: 10.1038/sj.ki.5002704. [DOI] [PubMed] [Google Scholar]
- 11.Itoh Y, Yano T, Sendo T, Oishi R. Clinical and experimental evidence for prevention of acute renal failure induced by radiographic contrast media. J Pharmacol Sci. 2005;97:473–88. doi: 10.1254/jphs.crj05002x. [DOI] [PubMed] [Google Scholar]
- 12.Lin H, et al. Peroxisomal proliferator-activated receptor-alpha protects renal tubular cells from doxorubicin-induced apoptosis. Mol Pharmacol. 2007;72:1238–45. doi: 10.1124/mol.107.037523. [DOI] [PubMed] [Google Scholar]
- 13.Ali BH, Al Za’abi M, Blunden G, Nemmar A. Experimental gentamicin nephrotoxicity and agents that modify it: a mini-review of recent research. Basic Clin Pharmacol Toxicol. 2011;109:225–32. doi: 10.1111/j.1742-7843.2011.00728.x. [DOI] [PubMed] [Google Scholar]
- 14.Singh AP, et al. Animal models of acute renal failure. Pharmacol Rep. 2012;64:31–44. doi: 10.1016/s1734-1140(12)70728-4. [DOI] [PubMed] [Google Scholar]
- 15.Servais H, Jossin Y, Van Bambeke F, Tulkens PM, Mingeot-Leclercq MP. Gentamicin causes apoptosis at low concentrations in renal LLC-PK1 cells subjected to electroporation. Antimicrob Agents Chemother. 2006;50:1213–21. doi: 10.1128/AAC.50.4.1213-1221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quiros Y, Vicente-Vicente L, Morales AI, Lopez-Novoa JM, Lopez-Hernandez FJ. An integrative overview on the mechanisms underlying the renal tubular cytotoxicity of gentamicin. Toxicol Sci. 2011;119:245–56. doi: 10.1093/toxsci/kfq267. [DOI] [PubMed] [Google Scholar]
- 17.Aleshin S, Reiser G. Role of the peroxisome proliferator-activated receptors (PPAR)-alpha, beta/delta and gamma triad in regulation of reactive oxygen species signaling in brain. Biol Chem. 2013;394:1553–70. doi: 10.1515/hsz-2013-0215. [DOI] [PubMed] [Google Scholar]
- 18.Orlowski J, Grinstein S. Na+/H+ exchangers of mammalian cells. J Biol Chem. 1997;272:22373–6. doi: 10.1074/jbc.272.36.22373. [DOI] [PubMed] [Google Scholar]
- 19.Parker MD, Myers EJ, Schelling JR. Na+-H+ exchanger-1 (NHE1) regulation in kidney proximal tubule. Cel Mol Life Sci. 2015;72:2061–74. doi: 10.1007/s00018-015-1848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schelling JR, Abu Jawdeh BG. Regulation of cell survival by Na+/H+ exchanger-1. Am J Physiol Renal Physiol. 2008;295:F625–32. doi: 10.1152/ajprenal.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada Y, et al. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu KL, et al. Renal tubular epithelial cell apoptosis is associated with caspase cleavage of the NHE1 Na+/H+ exchanger. Am J Physiol Renal Physiol. 2003;284:F829–39. doi: 10.1152/ajprenal.00314.2002. [DOI] [PubMed] [Google Scholar]
- 23.Loh SH, et al. Intracellular acid-extruding regulators and the effect of lipopolysaccharide in cultured human renal artery smooth muscle cells. PLoS One. 2014;9:e90273. doi: 10.1371/journal.pone.0090273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts RA, et al. PPAR alpha and the regulation of cell division and apoptosis. Toxicology. 2002;181–182:167–170. doi: 10.1016/s0300-483x(02)00275-5. [DOI] [PubMed] [Google Scholar]
- 25.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 26.Leehey DJ, et al. Can pharmacokinetic dosing decrease nephrotoxicity associated with aminoglycoside therapy. J Am Soc Nephrol. 1993;4:81–90. doi: 10.1681/ASN.V4181. [DOI] [PubMed] [Google Scholar]
- 27.Nagasawa M, et al. Identification of a functional peroxisome proliferator-activated receptor (PPAR) response element (PPRE) in the human apolipoprotein A-IV gene. Biochem Pharmacol. 2009;78:523–30. doi: 10.1016/j.bcp.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 2004;11:953–61. doi: 10.1038/sj.cdd.4401466. [DOI] [PubMed] [Google Scholar]
- 29.Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol. 2000;2:318–25. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 30.Rebillard A, et al. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007;67:7865–74. doi: 10.1158/0008-5472.CAN-07-0353. [DOI] [PubMed] [Google Scholar]
- 31.Altairac S, Zeggai S, Perani P, Courtois Y, Torriglia A. Apoptosis induced by Na+/H+ antiport inhibition activates the LEI/L-DNase II pathway. Cell Death Differ. 2003;10:548–57. doi: 10.1038/sj.cdd.4401195. [DOI] [PubMed] [Google Scholar]
- 32.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caroppi P, Sinibaldi F, Fiorucci L, Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr Med Chem. 2009;16:4058–65. doi: 10.2174/092986709789378206. [DOI] [PubMed] [Google Scholar]
- 34.Chang F, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 35.Qian J, Zou Y, Rahman JS, Lu B, Massion PP. Synergy between phosphatidylinositol 3-kinase/Akt pathway and Bcl-xL in the control of apoptosis in adenocarcinoma cells of the lung. Mol Cancer Ther. 2009;8:101–9. doi: 10.1158/1535-7163.MCT-08-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carthy CM, et al. Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology. 2003;313:147–57. doi: 10.1016/s0042-6822(03)00242-3. [DOI] [PubMed] [Google Scholar]
- 37.Hsu YH, et al. Far-infrared therapy induces the nuclear translocation of PLZF which inhibits VEGF-induced proliferation in human umbilical vein endothelial cells. PLoS One. 2012;7:e30674. doi: 10.1371/journal.pone.0030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schefe JH, et al. A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein. Circ Res. 2006;99:1355–66. doi: 10.1161/01.RES.0000251700.00994.0d. [DOI] [PubMed] [Google Scholar]
- 39.Naray-Fejes-Toth A, Boyd C, Fejes-Toth G. Regulation of epithelial sodium transport by promyelocytic leukemia zinc finger protein. Am J Physiol Renal Physiol. 2008;295:F18–26. doi: 10.1152/ajprenal.00573.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]