

Abstract
The phenotypic spectrum among girls with heterozygous mutations in the X-linked intellectual disability (XLID) gene CASK (calcium/calmodulin-dependent serine protein kinase) includes postnatal microcephaly, ponto-cerebellar hypoplasia, seizures, optic nerve hypoplasia, growth retardation and hypotonia. Although CASK knockout mice were previously reported to exhibit perinatal lethality and a 3-fold increased apoptotic rate in the brain, CASK deletion was not found to affect neuronal physiology and their electrical properties. The pathogenesis of CASK associated disorders and the potential function of CASK therefore remains unknown. Here, using Cre-LoxP mediated gene excision experiments; we demonstrate that deleting CASK specifically from mouse cerebellar neurons does not alter the cerebellar architecture or function. We demonstrate that the neuron-specific deletion of CASK in mice does not cause perinatal lethality but induces severe recurrent epileptic seizures and growth retardation before the onset of adulthood. Furthermore, we demonstrate that although neuron-specific haploinsufficiency of CASK is inconsequential, the CASK mutation associated human phenotypes are replicated with high fidelity in CASK heterozygous knockout female mice (CASK(+/-)). These data suggest that CASK-related phenotypes are not purely neuronal in origin. Surprisingly, the observed microcephaly in CASK(+/-) animals is not associated with a specific loss of CASK null brain cells indicating that CASK regulates postnatal brain growth in a non-cell autonomous manner. Using biochemical assay, we also demonstrate that CASK can interact with metabolic proteins. CASK knockdown in human cell lines cause reduced cellular respiration and CASK(+/-) mice display abnormalities in muscle and brain oxidative metabolism, suggesting a novel function of CASK in metabolism. Our data implies that some phenotypic components of CASK heterozygous deletion mutation associated disorders represent systemic manifestation of metabolic stress and therefore amenable to therapeutic intervention.
Electronic supplementary material
The online version of this article (doi:10.1186/s40478-016-0295-6) contains supplementary material, which is available to authorized users.
Keywords: CASK, MAGUK, Cerebellar hypoplasia, Non-cell autonomous, X-linked intellectual disability, Metabolism
Introduction
CASK is an evolutionarily conserved gene which encodes for a member of the membrane-associated guanylate kinase (MAGUK) protein family [31]. In mammals CASK was discovered due to its ability to bind to the cytosolic tail of neuronal adhesion molecules neurexins and therefore is primarily identified as a scaffolding protein at the neuronal synapse [21]. However, CASK deletion does not alter synapse formation in C. elegans [25], Drosophila [56] or mouse [3]. CASK ortholog lin-2 was identified in C. elegans as early as 1980 and was found in screens for cell lineage specificity rather than synaptic function [16, 24]. Although CASK is essential for survival in mouse, detailed electrophysiological analysis on CASK null mice failed to uncover changes in core neuronal functions such as membrane excitability, calcium-dependent pre-synaptic release or post-synaptic receptor organization [3]. In fact, CASK evolved before the emergence of the nervous system [31, 43] and is present in tissues from all three germ layers in mammals [21, 60]. Within the brain, CASK is expressed by neurons and non-neuronal cells such as oligodendrocytes [2] and astrocytes [36]. The phylogenetic data and tissue distribution of CASK, together with the phenotypes of CASK-mutated animal models, strongly suggest that CASK may have an important but currently ill-defined non-synaptic function(s). In fact, CASK has been noted to play a role in wide variety of cellular functions including transcription regulation [27], insulin signaling and secretion [67, 72], and cancer biology [68].
Subjects with mutations in CASK exhibit autistic traits, intellectual disability [20], Ohtahara syndrome [51], infantile spasms [40], FG syndrome [48], mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH) [45, 47, 64]. In addition, CASK mutations are associated with growth retardation, optic nerve hypoplasia/ atrophy, epilepsy, sensorineural deafness and hypotonia often resulting in scoliosis [8, 42]. This symptomatology is strikingly similar to that of metabolic diseases [38]. Mitochondrial respiratory chain defects and mutations in mitochondrial proteins are often associated with cerebellar hypoplasia [14, 34]. Mitochondrial diseases frequently affect the optic nerve and auditory sensory circuits (reviewed in [9, 55]). Moreover, epilepsy and hypotonia are primary manifestations in mitochondrial encephalopathies [30, 61]. Syndromes associated with CASK mutations, e.g. Ohtahara syndrome, can also arise from respiratory chain defects [10, 69]. Recently, we found that CASK interacts with mitochondrial proteins in Drosophila melanogaster, pointing to a potential role of CASK in the regulation of metabolism [44].
A large component of CASK-associated pathology develops postnatally, which has been reported by some authors as postnatal microcephaly or even atrophy [41]. Consistent with this idea, the brain of CASK null mice is normal at birth and properly laminated, and no defect in synapse formation is detectable [3]; deletion of CASK does, however, lead to an increased apoptotic rate in the mouse brain by 3-folds [3], which might easily explain the postnatal microcephaly. Since CASK constitutive knockout mice die within a few hours after birth, further analysis has not been possible.
Here, we demonstrate that the cerebellar hypoplasia caused by CASK heterozygous deletion mutation is not due to the loss of CASK gene specifically in cerebellar neurons. In fact, some of the MICPCH phenotypes may be primarily due to a non-neuronal function of CASK. Mice that are heterozygous for CASK deletion in neurons appear normal, but the constitutive whole-body heterozygous knockout (CASK(+/-)) female mice phenocopy the human disease state, indicating that MICPCH occurs due to an overall haploinsufficiency of the CASK gene. Surprisingly, the observed microcephaly in CASK(+/-) mice is not simply due to loss of only the CASK null cells i.e. it occurs in a non-cell autonomous manner. We further demonstrate that CASK interacts with mitochondrial proteins and that CASK(+/-) mice exhibit metabolic defects. Knocking down CASK expression in a human cell line recapitulates these defects. These findings indicate that mammalian CASK has a novel role in metabolic regulation which is crucial for postnatal brain growth in mammals.
Materials and methods
Generation of CASK(+/-) heterozygous and CASK neuronal knockout mice
CASK(+/floxed) female mice were crossed with male mice carrying a transgenic Cre recombinase controlled by the zona pellucida 3 promoter [33]. The F1 generation offsprings were genotyped to identify female mice carrying the Cre recombinase and the floxed CASK gene. These female mice were crossed with wild-type C57Bl6 male mice to obtain female CASK(+/-) mice. Crossings were continued with wild-type male mice to eliminate the Cre transgene. Colonies were maintained as CASK(+/-) females. CASK neuronal knockout mice were generated by crossing the CASK(+/floxed) female mice with a mouse line expressing Cre recombinase under the control of synapsin 1 promoter [71]. Male mice from the F1 generation bearing the Cre transgene and floxed CASK were analyzed. A table providing information on all mice used is shown in Additional file 1: Figure S1A.
Generation of CASK knockdown cells
HEK 293 cells were transduced with PLKO lentiviral particles carrying CASK shRNA or empty lentiviral particles. Experiments were performed 96 h post-transduction. Cell homogenates were analyzed for CASK expression using quantitative immunoblotting.
Mitochondrial respiration measurements
Mitochondrial oxygen consumption was measured in whole brain homogenates using the conventional Clark electrode assay as previously described [11]. Briefly, total respiration was measured in a buffer containing 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, 10 mM KH2PO4 and 1 mg/ml BSA (pH 7.4) in a water-jacketed cell magnetically stirred at 37 °C (Hansatech instruments, Norfolk, UK). Oxygen consumption rates were measured both in the presence and absence of potassium cyanide (KCN) to assess the rate of KCN sensitive respiration.Oxygen consumption measurements from HEK 293 cells were done as previously described [58, 59]. Briefly, HEK 293 cells were trypsinized and harvested in a sucrose-containing buffer (25 mM Tris–HCl, 10 mM K2HPO4 and 150 mM sucrose (pH 7.4)). Oxygen consumption in cell suspensions were measured in a water-jacketed cell magnetically stirred at 37 °C (Hansatech instruments, Norfolk, UK).
Immunoblotting
Samples were separated by 10 % SDS-PAGE, transferred to nitrocellulose, blocked in 5 % skimmed milk for 2 h and incubated with primary antibody for 1 h followed by incubation with secondary antibody (1:5000 dilution) for 30 min at room temperature. The chemiluminescent signal was detected via the enzymatic reaction using ECL detection reagents (Amersham) and visualized on ChemiDoc (Biorad). For quantitative immunodetection, the blots were incubated with a fluorescent secondary antibody (Alexa 488) for 30 min at room temperature. The primary antibodies used included anti-ATP synthase subunit β (MitoSciences MS503, 1:1000), anti-synaptophysin (Sigma, 1:1000), anti-tubulin (DSHB, 1:1000), anti-CASK (Neuromab, 1:1000), and anti IDH (AssayBiotech, 1:1000).
Energy expenditure and respiratory exchange ratio measurements
The energy expenditure and respiratory exchange ratios were measured over 48 h using the indirect calorimetry (TSE Systems, Chesterfield, MO). Energy expenditure data were expressed relative to free fat mass and presented as mean ± SEM. The TSE system is equipped with beams pointing in the horizontal (X,Y) and vertical (Z) directions. As mice break these beams, the LabMaster software sums this movement, which is translated into cage activity expressed as meters/hr.
Protein quantitation
All protein quantitations were done using the Coomassie Bradford reagent from Biorad, following the manufacturer’s instruction.
Brain sectioning and immunofluorescent staining
Animals were deeply anesthetized to avoid any pain and sacrificed mechanically either by decapitation or exsanguination. All animal procedures were performed in accordance with the Virginia Tech guidelines for use and care of laboratory animals. Three-month-old mature adult mice were deeply anaesthetized using isoflurane, and mice hearts were cannulated and perfused with PBS (exsanguination) followed by 4 % paraformaldehyde (PFA). The perfused mice were decapitated and brains were dissected out and fixed in 4 % PFA overnight. Brains were cryopreserved by incubation in 30 % sucrose solution for 48 h. For sectioning, brains were embedded in CryoTek™ and 20 μm-thick cortical sections were generated using a cryostat (IEC). The cortical sections were immunostained as floating sections. Sections were permeabilized with 0.025 % Triton X-100 followed by blocking with 5 % goat serum. Sections were stained with primary antibodies for two hours followed by secondary antibody (1:500) incubation for 30 min. Finally, sections were mounted on slides using PermaFluor™ mounting medium (Thermo Scientific) and coverslips were sealed using nail polish.
Fatty-acid and glucose oxidation assays
Fatty-acid oxidation was assessed in the mouse brain, red and white gastrocnemius muscle and quadriceps femoris muscle by measuring and summing 14CO2 production and 14C-labeled acid-soluble metabolites from the oxidation of [1-14C]-palmitic acid (American Radiolabeled Chemicals, St. Louis MO) respectively. Briefly, samples were incubated in 0.5 μCi/ml of [1-14C]-palmitic acid for 3 h. Media was then removed and exposed to 45 % perchloric acid for 1 h to liberate 14CO2, which was trapped in a tube containing 1 M sodium hydroxide (NaOH). The NaOH was then placed into a scintillation vial with 5 ml scintillation fluid added. The vial was then placed on a scintillation counter (LS 4500, Beckman Coulter) and counted for the presence of 14C. Acid soluble metabolites were determined by collecting the acidified media and measuring 14C content. Glucose oxidation was assessed by measuring 14CO2 production in a similar manner to fatty acid oxidation with the exception that [U-14C]-glucose was substituted for [1-14C]-palmitic acid.
Body composition measurement
Body composition was measured using Burker mini spec LF90, as previously described [39].
Glutathione-S-transferase (GST) pull down assay
GST-fusion CASK and GST were expressed in the BL21-DE3 strain of E. coli and purified by affinity chromatography on a glutathione–Sepharose column (Amersham) [18]. GST-pulldowns from rat brain were performed essentially as described [37].
Triton X-114 phase separation of homogenized mouse brain
Triton X-114 phase separation protocol was based on a previously published method [7]. Briefly, 4 % Triton X-114 (Sigma) in PBS containing protease inhibitors was mixed 1:1 with brain homogenate. The mixture was first incubated on ice for 10 min and then incubated at 37 °C for 10 min to promote separation of two phases. The sample was then centrifuged at 25 °C for 10 min at 13,000 g for separation into 1) aqueous phase on top, 2) a detergent phase in the middle, and 3) a pellet at the bottom.
Linear glycerol gradient centrifugation
A continuous glycerol gradient (10 % - 40 %) was generated using a Gradient Master™ (BioComp Instruments, Inc.) containing 25 mM HEPES-NaOH, pH 7.2, 150 mM NaCl, 5 mM DTT, 2 mM EDTA and protease inhibitors On a 10 mL continuous glycerol gradient buffer in ultracentrifuge tubes used with SW41 Ti Rotor (Beckman Coulter), 0.5 mL of the aqueous phase from the Triton X-114 phase separation of homogenized brain was carefully layered and then centrifuged for 20 h, at 35,000 rpm at 4 °C. A total of 9 fractions (1.1 mL each) were collected using the Piston Gradient Fractionator (BioComp Instruments, Inc.). Separate tubes were used for recombinant CASK protein and protein standards in the same experiment.
Brain subcellular fractionation and solubilization
Subcellular fractionation was performed similar to previous publication [11]. Briefly, dissected brains were rapidly transferred to ice cold homogenization buffer (i.e. 0.32 M sucrose and 20 mM HEPES pH 7.4 with protease inhibitors). The brain was homogenized in a motorized homogenizer (~20 strokes). Brain homogenates were centrifuged at 1000 g for 10 min to generate post nuclear supernatant (PNS). The PNS was centrifuged at 17,000 g for 15 min to obtain a pellet, which was subsequently washed once with homogenization buffer and then resuspended in homogenization buffer to obtain crude synaptosomes. Crude synaptosomes were subjected to ultracentrifugation (120,000 g). The supernatant was filtered through a 200 μm filter to obtain cytosolic fraction. The pellet was first solubilized in PBS containing 1 % Triton X-100 and protease inhibitors for two hours to obtain a Triton X-100-soluble fraction. The remaining pellet was further solubilized in PBS containing 1 % deoxycholic acid and protease inhibitors at 4 °C.
Force plate actometry and rotarod treadmill experiments
For force plate analyses, the adult littermate mice per genotype performed on a force plate (Bioseb) for 5 min as described previously [22]. Ataxic index was calculated as the area traveled over the distance covered on the force plate. We also tested the ability of mice to balance on a fixed speed rotarod test (10RPM) (Ugobasile). Each mouse was given three runs each day and an average was calculated.
Results
Cerebellar neuron-specific deletion of CASK does not produce cerebellar hypoplasia
Heterozygous deletion mutations in CASK produce cerebellar hypoplasia [8, 45]. It has also been demonstrated that mice carrying a floxed CASK gene (CASKfloxed) express ~ 33 % CASK compared to the wildtype littermates, and exhibit hypoplasia of cerebellar vermis [3, 45]. These data indicate that CASK may play a crucial role in the cerebellum. We therefore decided to use the Cre-loxP method to specifically delete CASK from cerebellar neurons. In order to ascertain the validity of this methodology, we first produced neuron-glia mixed cultures from P1.5 (postnatal day 1.5) CASKfloxed mice and transduced them with Cre-expressing lentivirus. Immunoblotting revealed that Cre expression specifically and completely eliminated CASK expression in these cultures within 96 h of virus addition. (Additional file 1: Figure S1B,C).
First, CASK expression was deleted from Purkinje cells -the major output neurons involved in motor coordination by crossing a CASKfloxed mouse line to a PCP2 (Purkinje cell protein 2)-Cre transgenic line [4]. Deleting CASK from Purkinje cells did not alter the arrangement or morphology of these cells (Fig. 1a, b, c). We analyzed the locomotion of these mice both in their home cage as well as on a rotarod. Mice lacking CASK in Purkinje cells exhibited no locomotory phenotype that could be interpreted as ataxic, suggesting that CASK is not required for the survival and functioning of Purkinje cells (Fig. 1d, e, f, g).
Fig. 1.
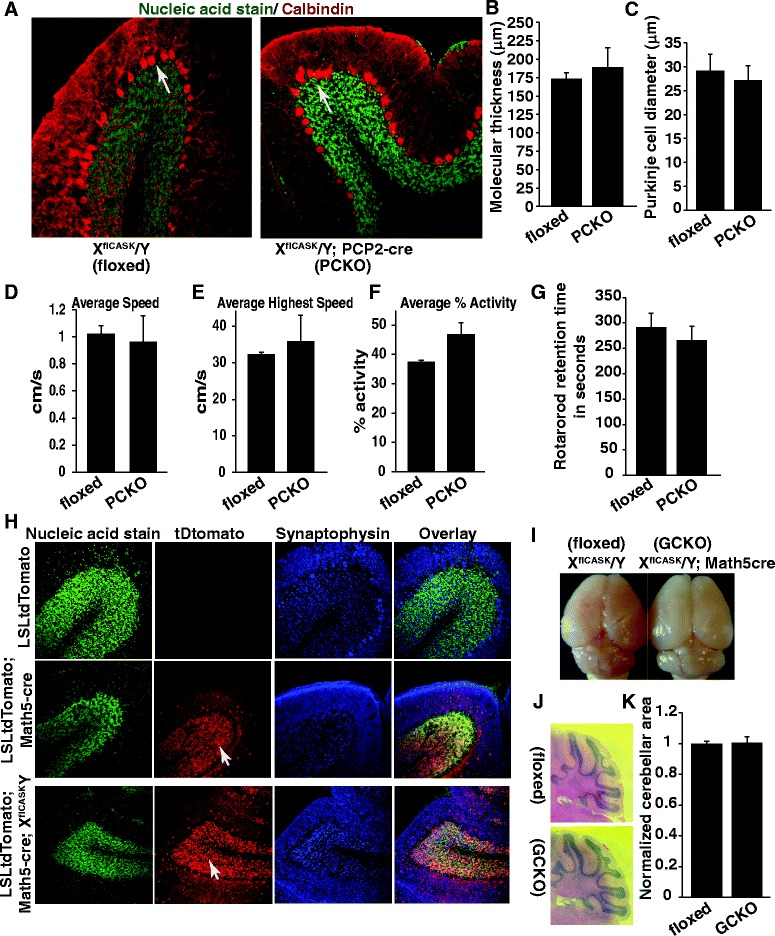
CASK deletion from Purkinje cells or granule cells does not alter cerebellar development. a Representative brain sections from two months old control mice carrying a floxed CASK gene (XflCASK/Y) and mice with CASK knocked out from Purkinje cells (XflCASK/Y; PCP2-cre); sections are stained with green (nucleic acid stain) and red (calbindin). Arrows indicate Purkinje cells. b Quantitation of molecular layer thickness and (c) Quantitation of Purkinje cell diameter; data are plotted as mean ± SEM, n = 3 mice. d, e, f Locomotion of mice analyzed in home cage using an overnight force plate experiment indicating no obvious change in locomotion. Average speed, average highest speed and average % activity are quantified and data are plotted as mean ± SEM, n = 3 mice. g Quantitation of retention time of mice on fixed speed rotarod, data are plotted as mean ± SEM, n = 4 mice. Floxed indicates CASK floxed mice and PCKO indicates Purkinje cell-specific CASK knockout mice. h Representative brain sections from two month old mice with indicated genotypes showing that CASK deletion from cerebellar granule cells does not lead to neuronal death or alteration in layering. LSL-tdTomato is a Cre indicator line in which a loxP-flanked stop cassette prevents expression of a red fluorescent protein; Math5-Cre mouse line has Cre recombinase expressed under the Math5 promoter. Arrows indicate inner granular layer where Cre-recombinase is expressed. Please note that the top two panels are cerebellar sections from mice with unperturbed CASK gene. i Representative brain images of two month old mice with indicated genotype. j Parasaggital sections of cerebellum showing that CASK deletion from granule cells does not lead to cerebellar hypoplasia. k Quantitation of cerebellar area in parasagittal sections; data are plotted as mean ± SEM, n = 3. Floxed indicates CASK floxed mice and GCKO indicates granule cell-specific CASK knockout mice
We next deleted CASK from the cerebellar granule cells by crossing the CASKfloxed mouse line to a Math5-Cre transgenic line. Math5-Cre is expressed in many different areas of brain including the hippocampus, the fourth layer of the cortex and the cerebellar cortex. In the cerebellum, cells expressing Math5-Cre give rise to the tightly packed cells in the granular layer [70]. We confirmed this distribution by crossing the Math5-Cre mouse line to an indicator mouse line that expresses tdTomato in a Cre-dependent manner [35]. Our experimental results show that Math5-Cre is expressed in granular cells as well as in a few cells in the molecular layer (Fig. 1h, middle panels). Granular cells are formed in the external granular layer during cerebellar genesis and subsequently migrate inwards. The CASK null granular cells are capable of this migration as indicated by their proper localization in the adult mouse cerebellum (Fig. 1h, lower row). Furthermore, we found that deleting CASK from the cerebellar granule cells did not alter the size of the cerebellum or layering of neurons compared to the CASKfloxed mice (Fig. 1i, j, k). No specific motor deficits were observed in any of these mice (data not shown). Our data therefore imply that although CASK may play an important role in cell survival in the postnatal brain, deleting it from specific neuronal subtypes is compatible with their survival. Furthermore, our data indicates that the cerebellar hypoplasia phenotype observed in CASK mutation subjects may not be directly related to the loss of CASK function in cerebellar neurons.
MICPCH pathology is not purely neuronal in origin
CASK knockout mice exhibit perinatal lethality and heightened apoptotic rates in the brain, indicating that CASK is essential for ex utero survival of brain cells [3]. However, our cerebellum-specific deletion experiments suggest that CASK may not be important for cell-autonomous survival. A large body of literature suggests that CASK may have a potential synaptic function (Reviewed in [26]), abnormality in which may lead to death of brain cells. Previously, attempt to generate brain specific CASK knockout mice was done using a transgenic mice expressing cre recombinase driven by nestin promoter to delete CASK from embryonic brain. The resultant mice exhibited perinatal lethality like the constitutive CASK knockout mice (personal communication with Prof. Susanne Schoch McGovern). However besides neurons, nestin is also expressed in non-neuronal brain cells and even in extra-neuronal tissue which made it difficult to interpret this data [65, 66]. We have therefore generated the pan-neuronal-specific CASK knockout mice by crossing CASKfloxed mice [3] with a synapsin1-Cre line which expresses Cre exclusively in post-mitotic neurons [54, 71]. In parallel, we also crossed the synapsin-Cre mice with the reporter mouse line that expresses tdTomato in a Cre-dependent manner. Our experiments confirmed the previous findings that Cre-mediated recombination in these mice occurs in nearly all neurons at an embryonic stage (as early as E12.5) ([71] and Additional file 1: Figure S2A,B). Since most CASK-associated patients are females harboring a heterozygous mutation in CASK, we initially examined female mice heterozygous for deletion of CASK pan-neuronally. Surprisingly, female mice heterozygous for neuron-specific CASK deletion exhibit no major phenotype and have normal-sized brains, including the cerebellum (Additional file 1: Figure S2C,D,E,F). This observation strongly suggests that CASK-associated MICPCH in humans is not specifically a neuronal pathology. Consistent with this assumption, complete deletion of CASK from all neurons did not produce the neonatal lethality observed in the constitutive CASK knockout mice [3] (Fig. 2a). The CASK neuronal-null mice are indistinguishable from their wild-type littermates until ~ 10 - 12 days after birth and maintain mobility until 21 days after birth, indicating that they have functional synapses (Additional file 2: Movie S1). After 10 days of birth, the CASK neuronal-null mice begin to exhibit a slower growth rate and by ~ 20 days of age, CASK neuronal-null mice are less than half the size of the control CASKfloxed mice or wild-type littermates (Fig. 2b). Brain weight is also significantly reduced in the CASK neuronal knockout pups, although the cerebellum appears normal compared to CASKfloxed mice and wildtype mice (Fig. 2c and Additional file 1: Figure S3). True microcephaly has been defined by some studies as an adult brain which is not only small but small for body weight [13, 23]. We therefore next measured the ratio of brain weight to body weight in CASK neuronal-null mice. The brain weight to body weight ratio is however significantly increased in the CASK neuronal-knockout mice compared to the control CASKfloxed mice or wildtype mice (Fig. 2d and data not shown), suggesting that the reduction in brain weight is secondary to overall growth retardation. Brain lamination is unaltered, and the neuronal arrangement in CASK neuronal knockout hippocampi and cerebella are comparable to control mice (Fig. 2e and Additional file 1: Figure S3). Neuronal CASK knockout mice express ~ 15 % of wild-type CASK in the brain (Fig. 2f, g), which may be due to CASK expression in other cell types including astrocytes [36] and oligodendrocytes [2]. Although CASK is deleted from most neurons at the embryonic stage, the levels of pre-synaptic and post-synaptic markers are unchanged, consistent with the previous study that demonstrated synapse formation is unaltered in CASK knockout mice [3]. The level of glial fibrillary acidic protein (GFAP) is also unaltered, indicating that although CASK is specifically deleted in neurons, there is no reactive gliosis and the ratio of neurons to astroglia may be unaltered (Fig. 2f, g). Beginning around 17-18 days of age, these mice suffer from progressively increasing bouts of epileptic seizures and spasms, which often prove fatal before 23 - 24 days of age (Additional file 3: Movie S2). Incidentally, epileptic spasms and epileptic disorders have been demonstrated in patients with CASK mutations [46, 51]. It has to be however considered that the CASK neuronal-null mice are produced on the CASKfloxed mice background, so glial cells are generating only ~ 33 % of CASK. The strong phenotype in the CASK neuronal-null mice therefore may indicate a combination of effect from CASK-null neurons and CASK-hypomorphic glial cells. Altogether, these data suggest that although loss of CASK function in neurons contributes to some components of the phenotypic spectrum of CASK-associated pathology (e.g. growth retardation and epilepsy), it does not contribute to microcephaly or cerebellar hypoplasia. Many CASK missense mutations in human males are associated with intellectual disabilities and growth retardation in the absence of microcephaly, clearly indicating that these phenotypes are not interdependent and may represent loss of different molecular functions of CASK [20].
Fig. 2.
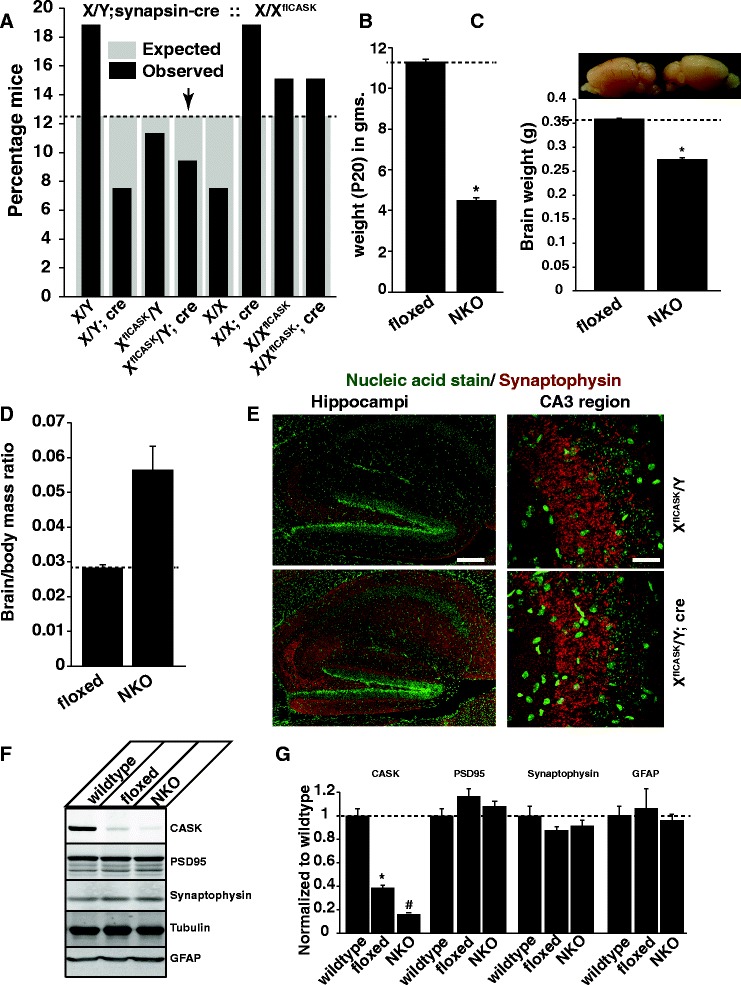
Survival and normal brain formation in CASK neuronal knockout mice. a Shows the expected (grey bar) and observed (black bar) percentage of mice with each genotype obtained from crossing the X/Y;synapsin-Cre males with XflCASK/X females; n = 62. Arrow indicates the neuronal CASK knockout group. b Weight of CASK floxed (floxed) and neuronal CASK knockout male mice (NKO) at 20 days of age. (* indicates p < 0.05; n = 5) (c) Representative brain halves and brain weight at 21 days of age from CASK (XflCASK/Y) (floxed) and neuronal CASK knockout (NKO) mice. (* indicates p < 0.05; n = 4). d Ratio of brain to body weight of CASK (XflCASK/Y) (floxed) and neuronal CASK knockout mice (NKO). (* indicates p < 0.05; n = 4). e Image showing hippocampus from 21 day old CASK floxed and CASK neuronal knockout mice. Green staining indicates nucleic acid and red staining indicates synaptophysin. Note that there is no significant change in the lamination and CA3 region synaptophysin staining in CASK neuronal knockout mice. f Representative Western blot showing endogenous levels of CASK, PSD95, synaptophysin and tubulin from 21 day old mice. g Western blot protein quantitation where data is normalized to tubulin and expressed relative to wild-type levels. Bar graphs are plotted as mean ± SEM. (* indicates p < 0.05; n = 3). Wild-type represents littermate mice with unperturbed CASK gene, floxed indicates CASK floxed mice and NKO indicates neuron-specific CASK knockout mice
CASK(+/-) mice mimic the human MICPCH phenotype
Since CASK-related neurological disorders occur due to deletion of a single CASK allele in females, we engineered a CASK(+/-) mouse line to model the neurodevelopmental defects. (Methods section and Additional file 1: Figure S4). Similar to the CASK(-/-) complete knockout mice, the brains of CASK(+/-) knockout mice were indistinguishable from their wild-type littermates at birth (Fig. 3a) [3]. However, pronounced microcephaly was observed beginning one week after birth, and the brains of adult CASK(+/-) females were ~ 25 % smaller in weight compared to sex-matched wild-type littermate controls (Fig. 3a, b). The hematoxylin and eosin (H&E) staining of brain sections showed no significant change in hippocampal size and layering of cells in the CASK(+/-) mice (Fig. 3c, g), but a marked decrease in cerebellar size (Fig. 3d, g) and number of cells in the cerebellum (Fig. 3e) was observed in the CASK(+/-) mice compared to the CASK(+/+) mice, suggesting disproportionate cerebellar hypoplasia. In contrast to lissencephaly [17], the CASK(+/-) cerebellum exhibited normal layering of cells (Fig. 3c, d, e). Examination of the cerebella of 5-day-old CASK(+/-) mice revealed that the thickness of the external granular layer (where granule cells are formed) remains unchanged (Additional file 1: Figure S5A,B). These data are consistent with our previous findings that lack of CASK increases cell loss by apoptosis rather than by producing any deficiency in the formation of cells [3]. Similar to CASK heterozygous deletion mutation patients, the CASK(+/-) mice displayed optic nerve hypoplasia (ONH); the optic nerve diameter was significantly reduced compared to sex-matched CASK(+/+) littermate controls (Fig. 3f, g). Although we observed a trend towards microphthalmia in CASK(+/-) mice, the optic globe size difference did not reach a statistical significance (Fig. 3g). ONH is frequently accompanied by malformation of the brain midline [5], but we did not observe such an anomaly in the CASK(+/-) mice (Additional file 1: Figure S6A), suggesting that ONH associated with CASK heterozygous deletion mutation has a distinct pathology. Similar to CASK patients, CASK(+/-) mice display scoliosis with a high degree of penetrance (~85 %) (Fig. 4a). CASK(+/-) mice also readily display a hind-limb clasping phenotype (Fig. 4b) that is indicative of neurological and motor dysfunction [19]. Atypical limb clasping phenotype involving four-limb clasping was also frequently observed. We did not find any anatomical aberrations in peripheral nerves such as myelination defects (data not shown) or neuromuscular junction abnormalities (Additional file 1: Figure S6B), which indicates that both hypotonia and the hind-limb clasping phenotype most likely originate from central nervous system impairment. CASK(+/-) mice also display increased diurnal activity relative to their sex-matched wild-type littermate controls (Fig. 4c) and are ataxic. CASK(+/-) mice fall off a rotarod much earlier (<2 min) than control mice (~6 min) due to ataxia in the first two days of training. Upon repeated training, however, these mice exhibit motor learning (Fig. 4d). Ataxia in mice can also be visualized by tracking movements within their home cage using a force plate. We observed that the CASK(+/-) mice traveled a longer distance to cover the same area compared to the CASK(+/+) mice (Fig. 4e, f), a parameter that is indicative of a higher ataxic index [22]. In sum, these data suggest that CASK(+/-) mice recapitulate multiple phenotypes observed in patients with MICPCH and therefore MICPCH represents heterozygous loss of CASK function (i.e. functional haploinsufficiency).
Fig. 3.
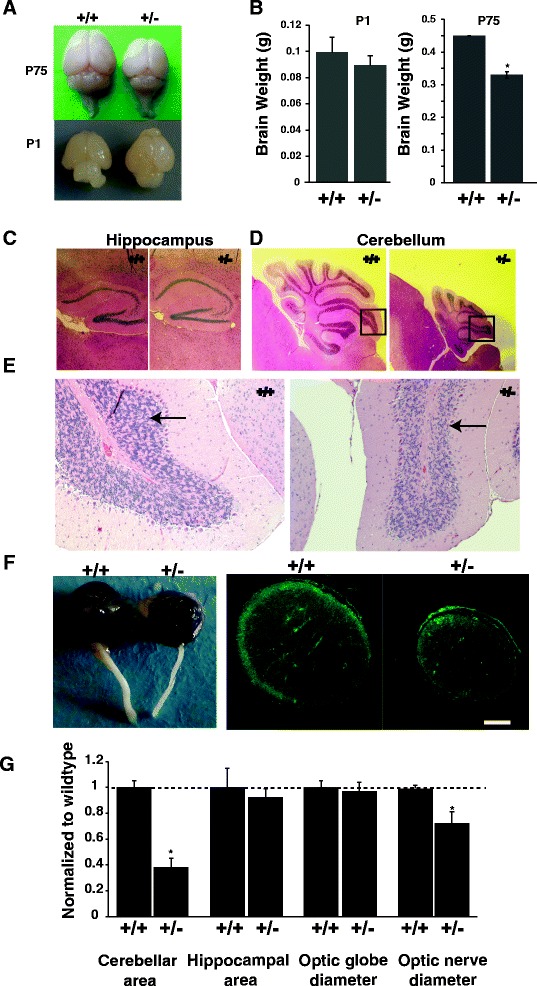
CASK (+/-) heterozygous mutant mice display postnatal microcephaly, cerebellar hypoplasia and optic nerve hypoplasia. a Representative brain images of the sex-matched CASK (+/+) and CASK (+/-) mutant littermates at postnatal day 1 and day 75 (P1 and P75), respectively. b Quantitation of brain weights from CASK (+/+) and CASK (+/-) mice at P1 and P75 (* indicates p < 0.05; n = 4). c Hematoxylin and eosin (H&E) stained sections of hippocampus derived from CASK (+/+) and CASK (+/-) mice at P75; note the hippocampi sizes are comparable. d H&E stained sections of the cerebellum at P75 showing pronounced cerebellar hypoplasia in CASK (+/-) mice relative to the CASK (+/+) control. e High magnification of the indicated square regions from panel D showing relatively fewer cells in the cerebellar folia of CASK (+/-) mice compared to the CASK (+/+) control. f Left panel shows representative images of the optic globe and optic nerve derived from three month old CASK (+/+) and CASK (+/-) mice . Right panel shows the optic nerve cross section stained with anti-tubulin antibody from CASK (+/+) and CASK (+/-) mice; note the decrease in optic nerve diameter . Scale bar = 100 μm. g Quantification of the cerebellar and hippocampal areas and the optic nerve and optic globe diameters obtained from CASK (+/+) and CASK (+/-) mice. Measurements were made using Image J software and normalized to the sex-matched wild-type littermate controls. Bar graphs are plotted as mean ± SEM; (* indicates p < 0.05; n = 4)
Fig. 4.
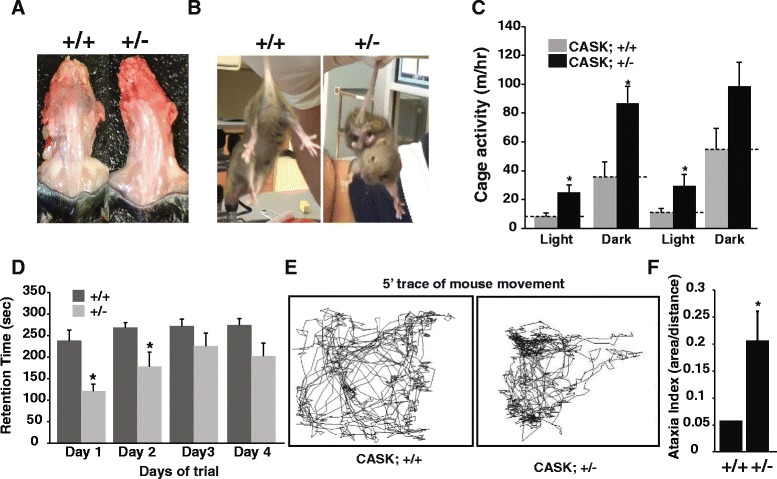
CASK (+/-) mice display musculoskeletal and locomotion defects. a Dissected torso from three month old CASK (+/+) and CASK (+/-) mice; CASK (+/-) mice display scoliosis. b CASK (+/-) mice display hind-limb clasping phenotype. c Activity of CASK (+/+) and CASK (+/-) mice measured in metabolic cages. Data plotted as mean ± SEM; (* indicates p < 0.05; n = 5) (d) CASK (+/-) mice were trained on a fixed speed rotorod and retention time noted. On each day, fresh sets of sex-and-age matched CASK (+/+) littermate mice were used as controls. Data are plotted as mean ± SEM; (* indicates p < 0.05; n = 5). e Representative 5-min trace of positions of CASK (+/+) and CASK (+/-) mice in home cage detected by a force plate. f Quantitation of locomotion observed on a force plate (area covered in 5 min/net distance traveled). Data are plotted as mean ± SEM; (* indicates p < 0.05; n = 5). All locomotion experiments were performed on mice between 25-30 days of age
CASK deletion affects cell number in a non-cell-autonomous manner
Although CASK(-/-) homozygous deletion has been shown to enhance the rate of apoptosis by nearly 3-fold in the brain [3], deleting CASK specifically from neuronal cells revealed that CASK is not essential for neuronal survival in a cell-autonomous manner (Figs. 1 & 2). We therefore next sought to determine if the microcephaly in CASK(+/-) mice was due to a reduction in cell number in a non-cell-autonomous manner. CASK is an X-linked gene and hence subject to random inactivation, therefore brains from female mice with a CASK(+/-) genotype are mosaic for CASK expression, with ~ 50 % of brain cells not expressing CASK. Cell death specifically of CASK-deleted brain cells in a CASK(+/-) mouse brain would result in the majority of surviving cells being CASK-positive (Additional file 1: Figure S7A,B,C). We tested this hypothesis by performing a quantitative immunoblotting experiment from CASK(+/+) and CASK(+/-) mouse brain homogenates. Total CASK levels were reduced by ~ 50 % in the brain of CASK(+/-) mice compared to the CASK(+/+) mice (Fig. 5a, b), but no significant change was observed in the level of a synaptic marker (synaptophysin) or a generic cell marker (ATP synthase β subunit), demonstrating that CASK null cells are equally viable in the brain of CASK(+/-) mice. These data suggest that the observed microcephaly in CASK(+/-) mice is not due to the cell-autonomous loss of CASK null cells.
Fig. 5.
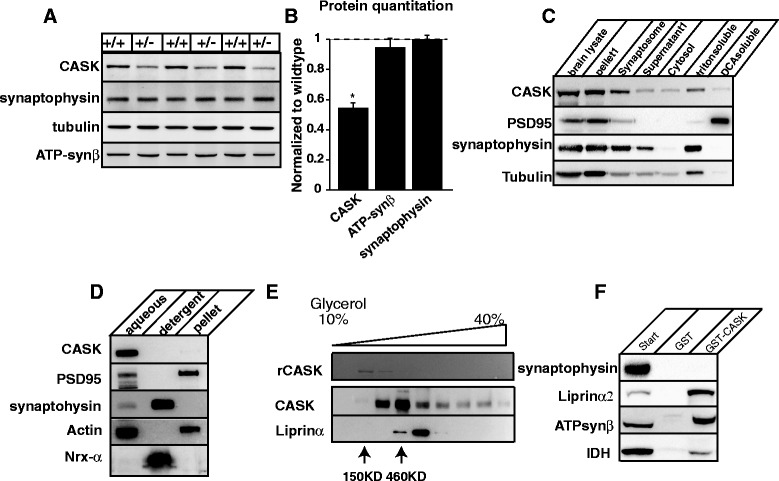
CASK is present in cytosol as a part of large protein complexes. a Western blot showing endogenous level of CASK, synaptophysin, ATP synthase β subunit and tubulin from CASK (+/+) and CASK (+/-) mutant mice (n = 3). b Western blot quantitation in CASK (+/-) mice relative to CASK (+/+) mice. Data is normalized to tubulin and expressed relative to wild-type levels. Bar graphs are plotted as mean ± SEM. (* indicates p < 0.05; n = 3). c Wild-type mice brains were initially fractionated to separate synaptosomal and cytosolic fractions. The synaptosomal fraction was solubilized initially in Triton X-100 and then in deoxycholic acid (DCA). Samples were blotted for the indicated antigen. d Wild-type mouse brain homogenate was solubilized in Triton X-114 at 4 °C, samples were warmed up to 37 °C and centrifuged to separate into an aqueous phase, a detergent phase and a pellet. Equal dilutions of each phase were blotted for the indicated proteins. e Aqueous phase (after Triton X-114 phase separation) from brain containing CASK was carefully layered on a 10-40 % continuous glycerol gradient. Following ultracentrifugation, samples were blotted for CASK and liprin-α3. Fig. also shows pure recombinant CASK. Arrows indicate the fractions where protein standards of 150 kDa and 460 kDa were observed. f GST-pulldown assay performed from rat brain homogenates using either full-length GST-CASK or GST (negative control) immobilized on glutathione resin. Synaptophysin (negative control); liprin α2 (positive control); ATPsynβ (ATP synthase β subunit); and IDH (isocitrate dehydrogenase)
CASK is an intracellular cytosolic protein and interacts with mitochondrial proteins
CASK is thought to be either completely or mainly membrane-anchored protein [21, 28]. Deletion of a signaling molecule at the membrane may explain a non-cell autonomous effect. We therefore decided to re-examine the cellular localization of CASK to confirm its presence at the membrane. We fractionated the wild-type mouse brain and looked for the solubility of CASK compared to PSD95, another MAGUK protein which is known to be membrane anchored [28, 63]. As expected, we found that PSD95 is completely membrane-anchored and is solubilized only with deoxycholic acid treatment, whereas a significant amount of CASK is also present in the cytosolic fraction isolated from the mouse brain, indicating that CASK is a soluble cytosolic protein (Fig. 5c). In this regard our finding is consistent with a previous report [28]. To further explore CASK’s cellular localization, we performed a Triton X-114 phase separation experiment. Triton X-114 solution is homogenous at 0 °C but separates into a detergent and aqueous phase at 20 °C, allowing for separation of hydrophilic cytosolic proteins from amphiphilic transmembrane proteins [6]. Since Triton X-114 is a mild non-ionic detergent, it is unlikely to disrupt strong interactions [6]. Interestingly, most of the CASK protein partitioned in the aqueous phase indicating that CASK is not tightly membrane-anchored and predominantly a cytosolic protein (Fig. 5d). In contrast to CASK, only a minor fraction of PSD95 is present in the aqueous phase (Fig. 5d). These results suggest that a large fraction of CASK protein may be cytosolic in the mouse brain, and in this aspect CASK differs substantially from other MAGUK proteins such as PSD95, which is tethered to the membrane to orchestrate cell-to-cell signaling.
In Drosophila, it is known that CASK interacts with different protein complexes in different cell types [44]. We therefore sought to determine if in the mammalian brain, CASK is also part of large protein complexes. We first expressed and purified full-length recombinant rat CASK (rCASK). CASK is a monomeric protein based on the gel exclusion chromatography experiments (data not shown). After glycerol gradient centrifugation, the rCASK (MW 120 kDa) protein was present in the second fraction along with a 150 kDa standard marker, as expected. Since rCASK migrates at ~ 120 kDa on SDS-PAGE, this confirms that rCASK is a monomer. In contrast, a large portion of the mouse brain cytosolic CASK sedimented in the fourth fraction along with a 460 kDa marker, indicating that when in cytosol, CASK is likely a part of a multi-protein complex (Fig. 5e). Surprisingly, small amounts of CASK were found to sediment in all fractions unlike the other well-known scaffolding protein e.g. liprin-α3 which displayed a well-defined sedimentation fraction indicating that the mouse brain CASK may be present in many complexes with molecular weights larger than 500 kDa (Fig. 5e).
Since CASK interacts with mitochondrial proteins in Drosophila [44], we next sought to determine if CASK also interacts with mitochondrial proteins in mammals. Using immobilized full-length recombinant CASK protein covalently attached to AminoLink™ agarose resin, we performed an affinity chromatography experiment using rat brain lysate and identified several CASK-interacting mitochondrial proteins from rat brain (unpublished data). To test the specificity of these interactions, we performed a pull-down assay with a GST (glutathione-S-transferase)-tagged CASK and found that CASK interacts with the mitochondrial ATP synthase β subunit and isocitrate dehydrogenase (IDH) (Fig. 5f).
CASK(+/-) heterozygous mice show aberrant metabolism
Since mitochondrial proteins co-precipitates with CASK in Drosophila [44] and mice, we hypothesized that CASK may play a role in regulating metabolism. To test this hypothesis, we characterized the consequences of CASK deletion on mitochondrial function and whole body metabolism. Our results showed that the one-month old CASK(+/-) mice weigh less (a small but statistically significant difference) than the sex-matched littermate controls (Fig. 6a), however there is no significant change in their lean body mass, fat weight or fluid weight, indicating an overall growth retardation (Fig. 6b, c, d). Surprisingly, we also found that CASK(+/-) mice expend significantly more energy at rest (Fig. 6e), which is likely attributable to ataxia and imbalance in these mice. It is known that an increase in muscle movement increases the respiratory exchange ratio (CO2 production/O2 uptake) [49], so we next examined the respiratory exchange ratio (RER). The RER of CASK(+/-) mice was indistinguishable from the CASK(+/+) littermate controls, despite an increased energy expenditure (Fig. 6 f).
Fig. 6.
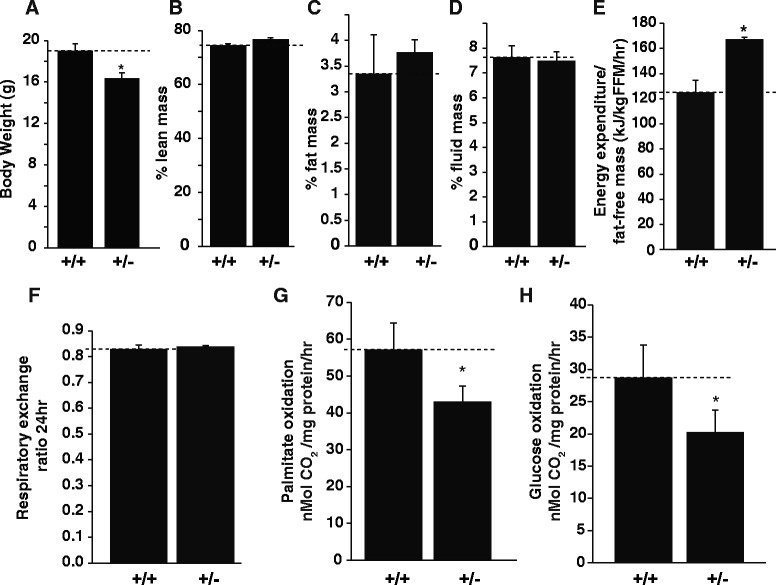
CASK (+/-) mutant mice exhibit growth retardation and skeletal muscle metabolic defects. Comparison of (a) body weight, (b) percentage lean mass, (c) percentage fat mass and (d) percentage fluid mass. All comparisons are presented as mean ± SEM, (* indicates p < 0.05; n = 5). e Energy expenditure/fat-free mass/hr in a 24-h period from CASK (+/-) mice compared to sex-matched CASK (+/+) littermate control. Data is presented as mean ± SEM (* indicates p < 0.05; n = 5). f Respiratory exchange ratio from CASK (+/-) mice compared to sex-matched CASK (+/+) littermate control. Data is presented as mean ± SEM. g Fatty acid oxidation rate and (h) glucose oxidation rate measured in skeletal muscle homogenates from CASK (+/+) and CASK (+/-) sex-matched littermate mice. Bar graphs are plotted as mean ± SEM. (* indicates p < 0.05; n = 5). All experiments were performed on one month old mice
Because some studies indicate that CASK plays a role in endocrine function such as insulin release [72], it is important to consider the fact that an overall measure of organism or organ metabolism may not be indicative of CASK’s direct involvement in cellular metabolism. We therefore examined the rate of metabolic fuel oxidation in skeletal muscle homogenates isolated from CASK(+/-) mice compared to the sex-matched CASK(+/+) littermate controls. Interestingly, we observed that the skeletal muscles from CASK(+/-) mice oxidize significantly lower amounts of fatty acids (Fig. 6g) and glucose (Fig. 6h) compared to the CASK(+/+) mice, suggesting that CASK is involved in the regulation of cellular substrate metabolism.
CASK regulates mitochondrial respiration and oxidative metabolism
The human embryonic kidney 293 (HEK293) cells express CASK endogenously (Fig. 7a). To determine if CASK regulates mitochondrial function, we performed CASK knockdown experiments in HEK293 cells using two different shRNAs (out of 5 shRNAs tested; Additional file 1: Figure S8) that showed > 50 % reduction in CASK expression. Specifically, the shRNA 692 and shRNA 690 showed ~ 85 % and ~ 60 % CASK knockdown respectively in HEK293 cells compared to the vector transfected control cells (Fig. 7a, b and Additional file 1: Figure S9A,B). Knocking down CASK expression using shRNA 692 significantly reduced the number of HEK293 cells and increased lactate production (Fig. 7c, d). Total cell respiration was measured using a conventional Clark oxygen electrode [58], and was found to be significantly reduced in CASK knockdown HEK293 cells compared to the control cells (Fig. 7e). Moreover, expressing rat CASK in CASK knockdown HEK293 cells rescued the defect in respiration demonstrating the specificity of the phenotype (Fig. 7e). The decrease in respiration and cell numbers was also replicated using the shRNA 690 that showed > 50 % CASK knockdown, but not with other shRNAs (i.e shRNAs 691, 693 and 694) that showed < 50 % CASK knockdown indicating a dose dependency of this phenotype (Additional file 1: Figure S9). Taken together, these results suggest that mammalian CASK is a regulator of mitochondrial respiration.
Fig. 7.
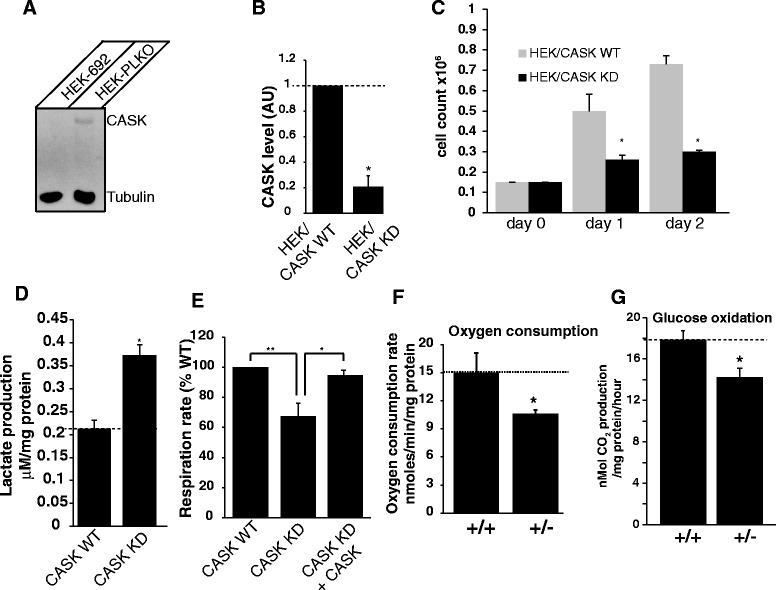
Reduction of CASK expression in cells and brain reduces oxidative metabolism. a Representative Western blot showing endogenous CASK expression in human embryonic kidney (HEK293-PLKO) cells as well as CASK knockdown in HEK293 cells using shRNA692 (HEK-692). b Quantitation of CASK knockdown (KD) in HEK293 cells using shRNA692 (* indicates p < 0.05; n = 3) (WT = wildtype). c Hemocytometer cell count in CASK knockdown (KD) HEK293 cells compared to wild-type control cells (WT). Bar graphs are plotted as mean ± SEM (n = 4; * indicates p < 0.05). d Lactate measurement from the culture media of wild-type control (WT) or CASK knockdown (KD) HEK293 cells. Bar graphs are plotted as mean ± SEM (* indicates p < 0.05; n = 3). e Total cellular respiration rate in CASK knockdown (KD) HEK293 cells and CASK knockdown HEK293 cells overexpressing recombinant rat CASK protein (KD + CASK) compared to the wild-type control cells (WT). Respiration is normalized to total protein levels (n = 4; * indicates p < 0.05, ** indicates p < 0.01). f Oxygen consumption rate measured in brain homogenates isolated from one month old CASK (+/+) and CASK (+/-) mice. Respiration rate is normalized to the total protein content (* indicates p < 0.05; n = 4). g Glucose oxidation rate measured in brain homogenates isolated from CASK (+/+) and CASK (+/-) sex-matched littermate mice (* indicates p < 0.05; n = 4)
Since the neonatal brain is highly susceptible to defects in metabolism, we next examined the effects of CASK deletion on oxidative metabolism in the brain. We adopted the same strategy as in muscles by using the brain homogenates to rule out any indirect effect/s of CASK deletion. Whole brain homogenates have been shown to exhibit a more tightly coupled respiration than isolated mitochondria [62]; and we also recently found that respiration can be measured more effectively from whole brain homogenates [11]. The rate of endogenous respiration in whole brain homogenates from CASK(+/-) mice was significantly reduced (~25 % decrease) compared to the CASK(+/+) brain homogenates (Fig. 7f), further suggesting that CASK is a regulator of mitochondrial respiration in the brain. Since glucose is the major source of fuel in the brain, we next asked whether glucose oxidation was altered in the brain of CASK(+/-) mice. We found a significant reduction (~20 % decrease) in the rate of glucose oxidation in CASK(+/-) mice brain compared to the CASK(+/+) brain (Fig. 7g), suggesting that CASK regulates oxidative metabolism in the brain.
Discussion
XLID affects males at a significantly higher rate than females because males have a single X-chromosome [50]. In females, X-linked mutations create a mosaic brain due to random X-inactivation, with ~ 50 % of brain cells expressing the mutant gene. This results in a relatively milder phenotype, as exemplified by the doublecortin mutations [1]. Most CASK deletion mutation patients are females, indicating that CASK expression is essential in the majority of cells for adequate brain function.
CASK heterozygous deletion mutation patient phenotypes have been classified as MICPCH or pontocerebellar hypoplasia. Here, we demonstrated that cerebellar hypoplasia associated with CASK mutations may not be a local neuronal pathology. One possible explanation for the disproportionate cerebellar hypoplasia could be the time of onset of this disorder; CASK heterozygous deletion mutations lead to aberrant postnatal brain growth and since cerebellar growth is largely postnatal, it may be affected disproportionately. In fact, it is known that although nutritional deprivation during postnatal brain development inhibits overall brain growth, it affects cerebellum disproportionately [57].
Consistent with the notion that CASK is not involved in core neuronal functions [3], we demonstrate that neuronal-specific CASK deletion does not alter neuronal migration or survival. Although the neuron-specific CASK heterozygous knockout mice did not exhibit any major phenotype, the complete neuronal CASK knockout mice exhibited pronounced growth retardation and epilepsy, and both are symptoms of CASK-related syndromes. Since, the CASK neuronal-null mutants are made on a hypomorphic background, at present it is not possible to specifically point out the consequences of deleting CASK only from neurons. However, our data suggests that the disruption of neuronal CASK function does contribute to the overall CASK mutation-related phenotypes.
Phenotypes associated with CASK heterozygous deletion mutation in humans may represent a cumulative loss of CASK function in different brain cell types. Since CASK interacts with multiple synaptic proteins, it is presumed to possess a synaptic function [52]. Our recent proteomic screening experiments in Drosophila melanogaster confirms the synaptic protein interactions of CASK, but also revealed a hitherto unknown interaction of CASK with mitochondrial proteins [44]. In agreement with our previous findings, we demonstrate here that mammalian CASK also interacts with certain metabolic/mitochondrial proteins. Since a significant amount of CASK was found to be cytosolic in the mouse brain, we speculate that these interactions may be transient and taking place in the cytosol. Interestingly, the cytosolic interaction and phosphorylation of nuclear encoded mitochondrial proteins with other kinases have been shown to affect their import into mitochondria [53]. Reducing CASK expression in a cellular or animal model alters mitochondrial function, including oxygen consumption and fuel metabolism. Since the cell biological role of neuroglial cells and neurons are considerably different, specific deletion of CASK in each cell type is likely to produce different phenotypes. A metabolic function of CASK could also explain the phenotypic differences observed in multiple animal models of CASK deficiency, including the vulval phenotype in worms and a defective locomotion phenotype in flies, both of which are known to be associated with abnormal metabolism [12, 29]. In mammals, the high metabolic demand of the growing neonatal brain could explain the lethality and postnatal onset of microcephaly and cerebellar hypoplasia in cases of CASK deficiency. It is possible that the species-specific phenotypic differences represent a difference in the metabolic requirements of each specific nervous system rather than an evolutionary difference in the molecular function of CASK.
How does a cell-autonomous metabolic function of CASK translate into the reduction of cell numbers in a non-cell-autonomous manner? Although it may be assumed that a significant reduction in the mitochondrial function would be deleterious to the survival of neurons, the extent to which the energetic function is compromised may further determine whether neurons may survive abnormally or undergo cell death. In human mitochondrial encephalopathies, the symptoms typically do not manifest until two years of age, indicating adequate survival of neurons during development [15]. Importantly, the central nervous system pathology like stroke episodes in mitochondrial encephalopathies may also be related to an aberrant extracellular signaling e.g. nitric oxide mediated signaling [15]. The non-cell autonomous systemic manifestations of mitochondrial stress are gradually getting recognized [32]. It is possible that CASK deficiency modulates one or more metabolic pathway/s which are critical for the secretion of a factor(s) essential for cellular survival. Conversely, it is also possible that CASK deficiency leads to the release of toxic intermediates, resulting in the loss of cells in a non-cell autonomous manner. Mitochondrial function in the brain is not only critical to ATP production but also in regulating amino-acid metabolism, which governs neurotransmitter production. An altered milieu of different neurotransmitters in the brain may have profound implications on neurodevelopment. It can therefore be hypothesized that the postnatal microcephaly associated with CASK heterozygous deletion mutation may be caused by altered levels of one or more extracellular factors. Although much remains to be done to identify the specific players involved, the most exciting aspect of this study is the possibility that a metabolic intervention may potentially prevent some of the postnatal consequences associated with CASK deletion mutation.
Conclusions
Taken together our results suggest that lack of neuronal CASK function contributes to the phenotypic spectrum of CASK linked pathologies, however MICPCH is not purely neuronal in origin. Therefore, constitutive heterozygous CASK knockout females are better animal model to investigate MICPCH. Our data further indicates that although CASK is a cytosolic protein and may perform cell-autonomous function in regulating metabolism, CASK-linked microcephaly occurs in a non-cell autonomous manner which involves loss of both CASK positive and CASK null cells. Finally, the observed cerebellar hypoplasia is not a local neuronal pathology; disproportionate cerebellar hypoplasia may simply represent timing of the disorder which has a large postnatal component. It is known that nutritional deprivation during the postnatal brain growth spurt specifically affects the cerebellum. Therefore, our findings also provide a valid framework for the investigation of non-CASK related cerebellar hypoplasia.
Ethical statement
All procedures performed in studies involving animals were in accordance with the ethical standards of the VirginiaTech institutional animal care and use committee.
Acknowledgments
We gratefully acknowledge Prof. Thomas Südhof for kindly providing us with the mouse carrying the floxed CASK gene. We also thank Dr. Michael Fox for providing us with the Math5-Cre mice, Dr. Alexei Morozov for providing us with LSL-tdt-Tomato mice. We thank Dr. Leslie LaConte for careful reading of the manuscript. This work was partially supported by NIH award 1R01EY024712-01A1 to KM.
Additional files
Description of mouse lines used and effect of cre expression on floxed CASK gene. Figure S2. CASK deletion from neurons and characterization of female heterozygous mice with neuronspecific CASK deletion. Figure S3. CASK deletion from neurons does not affect cerebellum. Figure S4. Generation of CASK(+/-) heterozygous mutant mice. Figure S5. External granular layer of cerebellum is normal during development of CASK(+/-) heterozygous mutant mice. Figure S6. Characterization of CASK(+/-) heterozygous mutant mice. Figure S7. Schematic diagram depicting cell autonomous and non-cell autonomous reduction in cell numbersas possible cause of microcephaly. Figure S8. Table showing the nucleotide sequences of CASK shRNA used in the study. Figure S9. CASK knockdown using shRNA 690 reduces cellular respiration and proliferation. (PDF 73672 kb)
Additional file 2: Movie 1.
Demonstrates the movement and interaction of CASK neuronal knockout mice and wild-type littermate. The smaller mouse is the mutant mouse. (MOV 12948 kb)
Additional file 3: Movie 2.
Demonstrates recurrent severe seizures in neuronal CASK knockout mice at 21 days after birth. (MOV 69740 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS, MH and KM designed research; SS, RM, KM, JW, VC, HC, CL and HZ performed research; SS, RM, JW, HC, VC and KM analyzed data; and SS and KM wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Sarika Srivastava, Phone: 5405262047, Email: Sarika_Srivastava@vtc.vt.edu.
Konark Mukherjee, Phone: 5405262035, Email: konark@vtc.vt.edu.
References
- 1.Aigner L, Fluegel D, Dietrich J, Ploetz S, Winkler J. Isolated lissencephaly sequence and double-cortex syndrome in a German family with a novel doublecortin mutation. Neuropediatrics. 2000;31:195–198. doi: 10.1055/s-2000-7452. [DOI] [PubMed] [Google Scholar]
- 2.Anitei M, Ifrim M, Ewart MA, Cowan AE, Carson JH, Bansal R, Pfeiffer SE. A role for Sec8 in oligodendrocyte morphological differentiation. J Cell Sci. 2006;119:807–818. doi: 10.1242/jcs.02785. [DOI] [PubMed] [Google Scholar]
- 3.Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–98. doi: 10.1002/1526-968X(200011/12)28:3/4<93::AID-GENE10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 5.Birkebaek NH, Patel L, Wright NB, Grigg JR, Sinha S, Hall CM, Price DA, Lloyd IC, Clayton PE. Endocrine status in patients with optic nerve hypoplasia: relationship to midline central nervous system abnormalities and appearance of the hypothalamic-pituitary axis on magnetic resonance imaging. J Clin Endocrinol Metab. 2003;88:5281–5286. doi: 10.1210/jc.2003-030527. [DOI] [PubMed] [Google Scholar]
- 6.Bordier C. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- 7.Brusca JS, Radolf JD. Isolation of integral membrane proteins by phase partitioning with Triton X-114. Methods Enzymol. 1994;228:182–193. doi: 10.1016/0076-6879(94)28019-3. [DOI] [PubMed] [Google Scholar]
- 8.Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R, Zanni G, Petit F, Afenjar A, Goizet C, Barresi S, et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis. 2012;7:18. 10.1186/1750-1172-7-18. [DOI] [PMC free article] [PubMed]
- 9.Cacace AT, Pinheiro JM. The mitochondrial connection in auditory neuropathy. Audiol Neurootol. 2011;16:398–413. doi: 10.1159/000323276. [DOI] [PubMed] [Google Scholar]
- 10.Castro-Gago M, Blanco-Barca MO, Gomez-Lado C, Eiris-Punal J, Campos-Gonzalez Y, Arenas-Barbero J. Respiratory chain complex I deficiency in an infant with Ohtahara syndrome. Brain Dev. 2009;31:322–325. doi: 10.1016/j.braindev.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Chavan V, Willis J, Walker SK, Clark HR, Liu X, Fox MA, Srivastava S, Mukherjee K. Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS One. 2015;10:e0125185. doi: 10.1371/journal.pone.0125185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng Z, Tsuda M, Kishita Y, Sato Y, Aigaki T. Impaired energy metabolism in a Drosophila model of mitochondrial aconitase deficiency. Biochem Biophys Res Commun. 2013;433:145–150. doi: 10.1016/j.bbrc.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 13.Dobbing J. Vulnerable periods of brain development. In: lipids, malnutrition & the developing brain. Ciba Found Symp. 1971: 9-29. PMID: 4949882. [PubMed]
- 14.Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116:4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson EL, Horvitz HR. Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics. 1985;110:17–72. doi: 10.1093/genetics/110.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldowitz D, Cushing RC, Laywell E, D'Arcangelo G, Sheldon M, Sweet HO, Davisson M, Steindler D, Curran T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of reelin. J Neurosci. 1997;17:8767–8777. doi: 10.1523/JNEUROSCI.17-22-08767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-Z. [DOI] [PubMed] [Google Scholar]
- 19.Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010: doi10.3791/1787. [DOI] [PMC free article] [PubMed]
- 20.Hackett A, Tarpey PS, Licata A, Cox J, Whibley A, Boyle J, Rogers C, Grigg J, Partington M, Stevenson RE, et al. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet. 2010;18:544–552. doi: 10.1038/ejhg.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hata Y, Butz S, Sudhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho A, Morishita W, Atasoy D, Liu X, Tabuchi K, Hammer RE, Malenka RC, Sudhof TC. Genetic analysis of Mint/X11 proteins: essential presynaptic functions of a neuronal adaptor protein family. J Neurosci. 2006;26:13089–13101. doi: 10.1523/JNEUROSCI.2855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong ST, Mah W. A critical role of GIT1 in vertebrate and invertebrate brain development. Exp Neurobiol. 2015;24:8–16. doi: 10.5607/en.2015.24.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980;96:435–454. doi: 10.1093/genetics/96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoskins R, Hajnal AF, Harp SA, Kim SK. The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development. 1996;122:97–111. doi: 10.1242/dev.122.1.97. [DOI] [PubMed] [Google Scholar]
- 26.Hsueh YP. The role of the MAGUK protein CASK in neural development and synaptic function. Curr Med Chem. 2006;13:1915–1927. doi: 10.2174/092986706777585040. [DOI] [PubMed] [Google Scholar]
- 27.Hsueh YP, Wang TF, Yang FC, Sheng M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature. 2000;404:298–302. doi: 10.1038/35005118. [DOI] [PubMed] [Google Scholar]
- 28.Hsueh YP, Yang FC, Kharazia V, Naisbitt S, Cohen AR, Weinberg RJ, Sheng M. Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J Cell Biol. 1998;142:139–151. doi: 10.1083/jcb.142.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia K, Albert PS, Riddle DL. DAF-9, a cytochrome P450 regulating C. elegans larval development and adult longevity. Development. 2002;129:221–231. doi: 10.1242/dev.129.1.221. [DOI] [PubMed] [Google Scholar]
- 30.Kang HC, Lee YM, Kim HD. Mitochondrial disease and epilepsy. Brain Dev.2013: Doi 10.1016/j.braindev.2013.01.006. [DOI] [PubMed]
- 31.LaConte L, Mukherjee K. Structural constraints and functional divergences in CASK evolution. Biochem Soc Trans. 2013;41:1017–1022. doi: 10.1042/BST20130061. [DOI] [PubMed] [Google Scholar]
- 32.Lee MS. Effect of mitochondrial stress on systemic metabolism. Ann N Y Acad Sci. 2015;1350:61–65. doi: 10.1111/nyas.12822. [DOI] [PubMed] [Google Scholar]
- 33.Lewandoski M, Wassarman KM, Martin GR. Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr Biol. 1997;7:148–151. doi: 10.1016/S0960-9822(06)00059-5. [DOI] [PubMed] [Google Scholar]
- 34.Lincke CR, van den Bogert C, Nijtmans LG, Wanders RJ, Tamminga P, Barth PG. Cerebellar hypoplasia in respiratory chain dysfunction. Neuropediatrics. 1996;27:216–218. doi: 10.1055/s-2007-973792. [DOI] [PubMed] [Google Scholar]
- 35.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marquez-Rosado L, Singh D, Rincon-Arano H, Solan JL, Lampe PD. CASK (LIN2) interacts with Cx43 in wounded skin and their coexpression affects cell migration. J Cell Sci. 2012;125:695–702. doi: 10.1242/jcs.084400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matos MF, Mukherjee K, Chen X, Rizo J, Sudhof TC. Evidence for SNARE zippering during Ca2 + -triggered exocytosis in PC12 cells. Neuropharmacology. 2003;45:777–786. doi: 10.1016/S0028-3908(03)00318-6. [DOI] [PubMed] [Google Scholar]
- 38.Mattman A, Sirrs S, Mezei MM, Salvarinova-Zivkovic R, Lillquist Y. Mitochondrial disease clinical manifestations: an overview. BC Med J. 2011;53:183–187. [Google Scholar]
- 39.McMillan RP, Wu Y, Voelker K, Fundaro G, Kavanaugh J, Stevens JR, Shabrokh E, Ali M, Harvey M, Anderson AS, et al. Selective overexpression of Toll-like receptor-4 in skeletal muscle impairs metabolic adaptation to high-fat feeding. Am J Physiol Regul Integr Comp Physiol. 2015;309:R304–313. doi: 10.1152/ajpregu.00139.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, Major P, Meijer IA, Lemyre E, Cossette P, et al. The genetic landscape of infantile spasms. Hum Mol Genet. 2014;23:4846–4858. doi: 10.1093/hmg/ddu199. [DOI] [PubMed] [Google Scholar]
- 41.Moog U, Bierhals T, Brand K, Bautsch J, Biskup S, Brune T, Denecke J, de Die-Smulders CE, Evers C, Hempel M, et al. Phenotypic and molecular insights into CASK-related disorders in males. Orphanet J Rare Dis. 2015;10:44. doi: 10.1186/s13023-015-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moog U, Kutsche K, Kortum F, Chilian B, Bierhals T, Apeshiotis N, Balg S, Chassaing N, Coubes C, Das S, et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet. 2011;48:741–751. doi: 10.1136/jmedgenet-2011-100218. [DOI] [PubMed] [Google Scholar]
- 43.Mukherjee K, Sharma M, Jahn R, Wahl MC, Sudhof TC. Evolution of CASK into a Mg2 + -sensitive kinase. Sci Signal. 2010;3:ra33. doi: 10.1126/scisignal.2000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukherjee K, Slawson JB, Christmann BL, Griffith LC. Neuron-specific protein interactions of Drosophila CASK-beta are revealed by mass spectrometry. Front Mol Neurosci. 2014;7:58. doi: 10.3389/fnmol.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J, Christian SL, Ullmann R, Kuechler A, Haas CA, et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet. 2008;40:1065–1067. doi: 10.1038/ng.194. [DOI] [PubMed] [Google Scholar]
- 46.Nakajiri T, Kobayashi K, Okamoto N, Oka M, Miya F, Kosaki K, Yoshinaga H. Late-onset epileptic spasms in a female patient with a CASK mutation. Brain Dev. 2015;37:919–923. doi: 10.1016/j.braindev.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Matsumoto N, Saitsu H, Jinnou H, Ohki S, et al. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 2014;36:272–273. doi: 10.1016/j.braindev.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Piluso G, D'Amico F, Saccone V, Bismuto E, Rotundo IL, Di Domenico M, Aurino S, Schwartz CE, Neri G, Nigro V. A missense mutation in CASK causes FG syndrome in an Italian family. Am J Hum Genet. 2009;84:162–177. doi: 10.1016/j.ajhg.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos-Jimenez A, Hernandez-Torres RP, Torres-Duran PV, Romero-Gonzalez J, Mascher D, Posadas-Romero C, Juarez-Oropeza MA. The respiratory exchange ratio is associated with fitness indicators both in trained and untrained men: a possible application for people with reduced exercise tolerance. Clin Med Circ Respir Pulm Med. 2008;2:1–9. doi: 10.4137/ccrpm.s449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raymond FL. X linked mental retardation: a clinical guide. J Med Genet. 2006;43:193–200. doi: 10.1136/jmg.2005.033043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saitsu H, Kato M, Osaka H, Moriyama N, Horita H, Nishiyama K, Yoneda Y, Kondo Y, Tsurusaki Y, Doi H, et al. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia. 2012;53:1441–1449. doi: 10.1111/j.1528-1167.2012.03548.x. [DOI] [PubMed] [Google Scholar]
- 52.Samuels BA, Hsueh YP, Shu T, Liang H, Tseng HC, Hong CJ, Su SC, Volker J, Neve RL, Yue DT, et al. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56:823–837. doi: 10.1016/j.neuron.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schonfisch B, Guiard B, Sickmann A, Pfanner N, et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell. 2011;144:227–239. doi: 10.1016/j.cell.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 54.Schoch S, Cibelli G, Thiel G. Neuron-specific gene expression of synapsin I. Major role of a negative regulatory mechanism. J Biol Chem. 1996;271:3317–3323. doi: 10.1074/jbc.271.6.3317. [DOI] [PubMed] [Google Scholar]
- 55.Sitarz KS, Chinnery PF, Yu-Wai-Man P. Disorders of the optic nerve in mitochondrial cytopathies: new ideas on pathogenesis and therapeutic targets. Curr Neurol Neurosci Rep. 2012;12:308–317. doi: 10.1007/s11910-012-0260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slawson JB, Kuklin EA, Ejima A, Mukherjee K, Ostrovsky L, Griffith LC. Central regulation of locomotor behavior of Drosophila melanogaster depends on a CASK isoform containing CaMK-like and L27 domains. Genetics. 2011;187:171–184. doi: 10.1534/genetics.110.123406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smart JL, Dobbing J, Adlard BP, Lynch A, Sands J. Vulnerability of developing brain: relative effects of growth restriction during the fetal and suckling periods on behavior and brain composition of adult rats. J Nutr. 1973;103:1327–1338. doi: 10.1093/jn/103.9.1327. [DOI] [PubMed] [Google Scholar]
- 58.Srivastava S, Barrett JN, Moraes CT. PGC-1alpha/beta upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Hum Mol Genet. 2007;16:993–1005. doi: 10.1093/hmg/ddm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stevenson D, Laverty HG, Wenwieser S, Douglas M, Wilson JB. Mapping and expression analysis of the human CASK gene. Mamm Genome. 2000;11:934–937. doi: 10.1007/s003350010170. [DOI] [PubMed] [Google Scholar]
- 61.Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, van den Brand MA, Hendriks-Franssen MG, Gilissen C, Veltman JA, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet. 2013;22:656–667. doi: 10.1093/hmg/dds473. [DOI] [PubMed] [Google Scholar]
- 62.Tialowska B, Kaczor J, Stempak W, Grybos D, Popinigis J. Tightly coupled respiration in rat brain homogenates. Acta Biochim Pol. 1991;38:165–167. [PubMed] [Google Scholar]
- 63.Topinka JR, Bredt DS. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron. 1998;20:125–134. doi: 10.1016/S0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 64.Valayannopoulos V, Michot C, Rodriguez D, Hubert L, Saillour Y, Labrune P, de Laveaucoupet J, Brunelle F, Amiel J, Lyonnet Set al (2012) Mutations of TSEN and CASK genes are prevalent in pontocerebellar hypoplasias type 2 and 4. Brain. 2012;135: e199; author reply e200 Doi 10.1093/brain/awr108. [DOI] [PubMed]
- 65.Wagner N, Wagner KD, Scholz H, Kirschner KM, Schedl A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am J Physiol Regul Integr Comp Physiol. 2006;291:R779–787. doi: 10.1152/ajpregu.00219.2006. [DOI] [PubMed] [Google Scholar]
- 66.Walker AS, Goings GE, Kim Y, Miller RJ, Chenn A, Szele FG. Nestin reporter transgene labels multiple central nervous system precursor cells. Neural Plast. 2010;2010:894374. doi: 10.1155/2010/894374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Li R, Du D, Zhang C, Yuan H, Zeng R, Chen Z. Proteomic analysis reveals novel molecules involved in insulin signaling pathway. J Proteome Res. 2006;5:846–855. doi: 10.1021/pr050391m. [DOI] [PubMed] [Google Scholar]
- 68.Wei JL, Fu ZX, Fang M, Zhou QY, Zhao QN, Guo JB, Lu WD, Wang H. High expression of CASK correlates with progression and poor prognosis of colorectal cancer. Tumour Biol. 2014;35:9185–9194. doi: 10.1007/s13277-014-2179-3. [DOI] [PubMed] [Google Scholar]
- 69.Williams AN, Gray RG, Poulton K, Ramani P, Whitehouse WP. A case of Ohtahara syndrome with cytochrome oxidase deficiency. Dev Med Child Neurol. 1998;40:568–570. doi: 10.1111/j.1469-8749.1998.tb15416.x. [DOI] [PubMed] [Google Scholar]
- 70.Yang Z, Ding K, Pan L, Deng M, Gan L. Math5 determines the competence state of retinal ganglion cell progenitors. Dev Biol. 2003;264:240–254. doi: 10.1016/j.ydbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 71.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu ZQ, Wang D, Xiang D, Yuan YX, Wang Y. Calcium/calmodulin-dependent serine protein kinase is involved in exendin-4-induced insulin secretion in INS-1 cells. Metabolism. 2014;63:120–126. doi: 10.1016/j.metabol.2013.09.009. [DOI] [PubMed] [Google Scholar]