

PEARLS
Atypical features that should alert neurologists to the possibility of alternative diagnoses include symmetric findings, disease duration greater than 2 years at presentation, young age at onset (less than 50), and pain.
While timely diagnosis of amyotrophic lateral sclerosis (ALS) is important, it is equally important that clinicians avoid premature closure by ruling out disease mimics, and following patients' clinical change over time.
OY-STERS
The presence of upper and lower motor neuron signs is not enough to diagnose ALS.
Misdiagnosis of ALS is not rare. Common ALS mimics include structural spinal pathology, hereditary spastic paraplegia, and multifocal motor neuropathy. Structural disease may be addressed surgically, and multifocal motor neuropathy is treatable.
Lack of disease progression is the most common reason for diagnostic reconsideration.
The clinical diagnosis of amyotrophic lateral sclerosis (ALS) is based on the presence of upper and lower motor neuron abnormalities spreading between body segments, along with the exclusion of rare disease mimics. No definitive diagnostic testing exists. Accurate, timely diagnosis of ALS is essential in order to provide appropriate patient counseling. While the revised El Escorial criteria provide standards for research purposes,1 trainees should be aware that many patients never meet criteria for definite ALS. ALS is a clinically heterogeneous disorder and presentations vary in regards to the distribution of weakness, rate of progression, and survival.2,3 Approximately 10% of patients with ALS will live more than 10 years.4 Patients carrying certain mutations in TARDBP and MART3 may have a longer survival.5,6 Despite this heterogeneity, a small number of patients carry an inaccurate diagnosis.
This case series describes patients in a US tertiary care center's multidisciplinary ALS clinic who were diagnosed by neuromuscular specialists and later determined to have been misdiagnosed.
CASE SERIES
Patients in this cross-sectional analysis were identified during ALS clinic visits between August 2011 and December 2013 at the University of Michigan. Each patient was determined not to have ALS by an ALS physician (B.C.C., S.A.G.). All patients had previously been diagnosed with ALS by a neuromuscular specialist at our institution. We performed a medical record review beginning with each patient's first neuromuscular consultation. The initial symptomatology, physician's reasoning for diagnostic reconsideration, and revised diagnoses were noted.
Thirteen of 332 patients initially diagnosed with ALS (3.9% prevalence) were later determined not to have ALS. The actual prevalence of misdiagnosis is likely higher given that multiple patients diagnosed with ALS are currently under investigation. The initial symptomatology, physician's reasoning for diagnostic reconsideration, and revised diagnoses are summarized in the table. Patients reported a median of 2 years of symptoms with an interquartile range (IQR) of 1–5.25 years (range 5 months–12 years). The median age at diagnosis was 61 years, with an IQR of 49–69.5 years (range 45–75 years). The median time from the initial visit to diagnostic reconsideration was 4 years (IQR 2–7 years, range 2 months–12 years).
Table.
Initial symptomatology, physician's reasoning for diagnostic reconsideration, and revised diagnoses
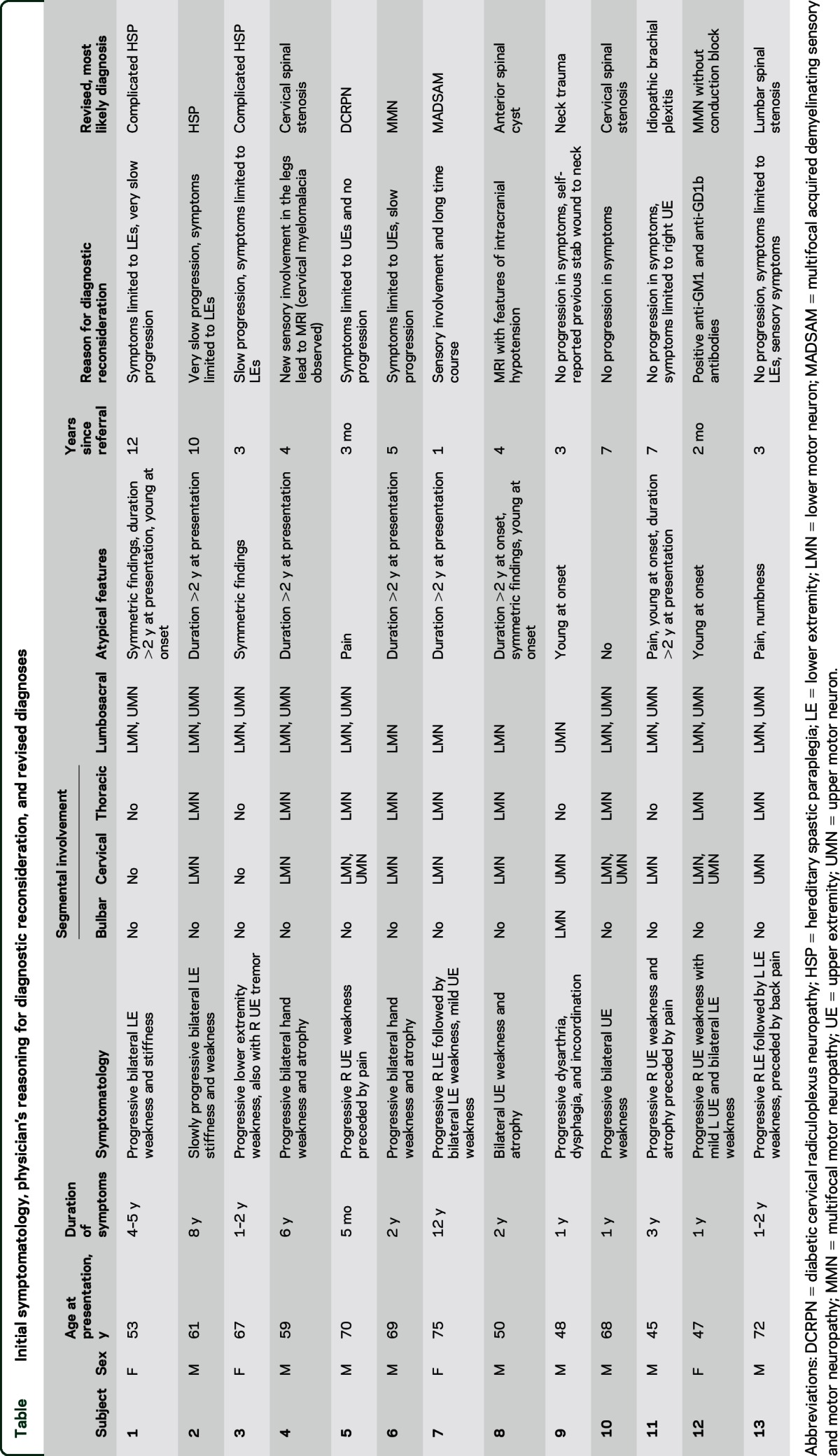
Segmental involvement at the time of presentation is indicated in the table. The atypical features included symmetric findings (3 patients), symptom duration of more than 2 years at diagnosis (7 patients), and age younger than 50 years at diagnosis (5 patients). Three patients reported pain as part of the initial clinical picture. Only one patient had no atypical features at the time of diagnosis. While it should be noted that age younger than 50 years is not entirely unusual for true ALS, it is vital that neurologists consider the appropriateness of the diagnosis in these younger individuals. Slow or no progression was the main reason for diagnostic reconsideration in 9 of the 13 patients. Other reasons included 2 patients with new or previous sensory deficits, one with signs of intracranial hypotension on initial MRI, and one with a history of immunoglobulin M (IgM) monoclonal gammopathy prompting GM-1 antibody testing.
The ALS physicians determined alternative diagnoses of spinal cord pathology (4 patients), hereditary spastic paraplegia (HSP) (3 patients), multifocal motor neuropathy (MMN) (2 patients), and other conditions (4 patients).
Structural spinal pathology.
Disease localizing to the spinal cord is a well-known ALS mimic, and 4 of our patients had structural spinal pathology identified. This included spinal stenosis in 3 cases and a symptomatic spinal cyst in another. Patients 4 and 10 were determined to have symptomatic cervical spondylosis, one after MRI of the spine was re-reviewed and another after the study was repeated years later. Patient 13 was found to have severe lumbar stenosis. In contrast to the initial EMG, a repeat EMG did not show denervation outside the lumbosacral segment. Patient 18 was later diagnosed with a large anterior spinal cyst found on magnetic resonance myelogram with intrathecal gadolinium. Additional testing was prompted by the finding of intracranial hypotension on initial MRI.
HSP phenotype.
All 3 patients had symmetric findings early in the disease course, often had a long duration of symptoms prior to diagnosis, and had symptoms predominantly in the legs. Only one patient has undergone genetic testing. Subject 1's genetic testing revealed a known nonsense mutation as well as a duplication of 2 exons in the SPG11 gene. Subject 2 had an initial EMG revealing lower motor neuron involvement, but repeat testing was normal. Given the normal EMG and spasticity limited to the lower extremities after 10 years without structural explanation, a diagnosis of HSP was made. Subject 3 had a thin corpus callosum in addition to a phenotype typical of HSP.
MMN.
Two patients were diagnosed with MMN. Patient 6 underwent a repeat EMG revealing possible but not definitive conduction block. IV immunoglobulin was trialed, with a therapeutic response. In patient 12, the concern for MMN was high despite a lack of conduction block on EMG/nerve conduction studies, because of a history of IgM monoclonal gammopathy. Anti-GM1 and anti-GD1b antibodies were subsequently found. Rituximab therapy resulted in clinical improvement.
Other conditions.
Patient 5 was diagnosed with diabetic cervical radiculoplexus neuropathy (DCRPN). He initially presented with right neck and shoulder pain followed by right hand weakness and atrophy. The diagnosis of DCRPN was made based on the clinical presentation coupled with no progression over the subsequent 2 years. Similarly, patient 7 was diagnosed with multifocal acquired demyelinating sensory and motor neuropathy after a repeat EMG showed conduction block and sensory involvement. Patient 9 later reported a previous penetrating neck injury that had caused hypoglossal nerve injury and unilateral tongue fasciculations. Patient 11 was diagnosed with brachial neuritis after lack of progression prompted additional history.
DISCUSSION
We identified 13 patients who were diagnosed with ALS and later determined to have an alternative diagnosis. Previous studies have investigated misdiagnosis of ALS. Thirty-two out of 437 (7.3%) patients in the Irish ALS Register7 and 46 out of 552 (8%) patients in the Scottish Motor Neuron Disease Register8 were misdiagnosed. These 2 cohorts, as well as an Italian registry cohort,4 highlight the surprising frequency of ALS misdiagnosis. Our study, however, is the first to investigate a population diagnosed and followed by tertiary neuromuscular specialists. Among these patients, ALS misdiagnosis is not rare.
There are significant differences between our findings and the populations described in Europe. In the Irish cohort, the 2 most common ALS mimics were MMN (22%) or Kennedy disease (13%) and suspicion for HSP was not frequent. While the Irish ALS patients had been diagnosed by general neurologists, all patients in our cohort had seen a neuromuscular specialist. It is possible these specialists recognized MMN and Kennedy disease, but not HSP. In the Scottish register, the most common alternative diagnoses were cervical spondylosis and cerebrovascular disease. The diagnosis of ALS was made by a wide array of physicians including generalists. In the Italian Registry, the 2 most common ALS mimics were cervical spondylosis and peripheral neuropathy.4 While most studies, including ours, reveal that cervical disease and MMN are frequent ALS mimics, our cohort is the first to demonstrate that complicated HSP may be a frequent mimic.
A notable similarity between these patients and previous cohorts is that all identified lack of progression as the most common reason to consider an alternative diagnosis. This point is key for current residents and fellows: the importance of longitudinal evaluation of patients diagnosed with ALS by well-trained experts cannot be overemphasized. When considering our cohort, it is noteworthy that only one patient had bulbar symptoms. Patients with bulbar involvement appear unlikely to be misdiagnosed with ALS. Further, patients who never develop bulbar symptoms despite long follow-up may prompt reconsideration.
Our patients whose ALS diagnosis was replaced had a number of striking features. Compared to previous cohorts, suspected HSP was frequent in misdiagnosed patients. Almost all of the patients had an atypical feature at diagnosis. One of the universally taught and recognized features of ALS is progression of symptoms and signs over time. However, these patients had minimal or no disease progression. These observations may be of use to trainees, both with regards to initial diagnosis of patients with suspected ALS and in following patients longitudinally. While prompt diagnosis of ALS is in the patient's best interest, so is continually rethinking his or her clinical presentation and evolution.
AUTHOR CONTRIBUTIONS
Dr. Jacobson was involved in study design, chart abstraction, and wrote the manuscript. Drs. Callaghan and Goutman were involved in study design, identified patients for inclusion, and contributed to the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Brooks BR, Miller RG, Swash M, Munsat TL, Diseases WFoNRGoMN. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 2.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 2014;10:661–670. [DOI] [PubMed] [Google Scholar]
- 3.Goutman SA, Feldman EL. Clinical trials of therapies for amyotrophic lateral sclerosis. JAMA Neurol 2015;72:743–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pupillo E, Messina P, Logroscino G, Beghi E; SLALOM Group. Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol 2014;75:287–297. [DOI] [PubMed] [Google Scholar]
- 5.Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 2014;17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corcia P, Valdmanis P, Millecamps S, et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012;78:1519–1526. [DOI] [PubMed] [Google Scholar]
- 7.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol 2000;57:109–139. [DOI] [PubMed] [Google Scholar]
- 8.Davenport RJ, Swingler RJ, Chancellor AM, Warlow CP. Avoiding false positive diagnoses of motor neuron disease: lessons from the Scottish Motor Neuron Disease Register. J Neurol Neurosurg Psychiatry 1996;60:147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]