

Abstract
Ion-coupled secondary transport is utilized by multiple integral membrane proteins as a means of achieving the thermodynamically unfavorable translocation of solute molecules across the lipid bilayer. The chemical nature of these molecules is diverse and includes sugars, amino acids, neurotransmitters, and other ions. LeuT is a sodium-coupled, nonpolar amino acid symporter and eubacterial member of the solute carrier 6 (SLC6) family of Na+/Cl−-dependent neurotransmitter transporters. Eukaryotic counterparts encompass the clinically and pharmacologically significant transporters for γ-aminobutyric acid (GABA), glycine, serotonin (5-hydroxytryptamine, 5-HT), dopamine (DA), and norepinephrine (NE). Since the crystal structure of LeuT was first solved in 2005, subsequent crystallographic, binding, flux, and spectroscopic studies, complemented with homology modeling and molecular dynamic simulations, have allowed this protein to emerge as a remarkable mechanistic paradigm for both the SLC6 class as well as several other sequence-unrelated SLCs whose members possess astonishingly similar architectures. Despite yielding groundbreaking conceptual advances, this vast treasure trove of data has also been the source of contentious hypotheses. This chapter will present a historical scientific overview of SLC6s; recount how the initial and subsequent LeuT structures were solved, describing the insights they each provided; detail the accompanying functional techniques, emphasizing how they either supported or refuted the static crystallographic data; and assemble these individual findings into a mechanism of transport and inhibition.
1. INTRODUCTION
Communication across chemical synapses is the principal mode by which electrical signals are transmitted among neurons in the brain (Vanhatalo & Sohila, 1998). Subsequent to Ca2+-dependent release of neurotransmitters from the presynaptic neuron and activation of fast-acting ionotropic or slower-acting metabotropic postsynaptic receptors, these small molecules are cleared from the synaptic cleft primarily by presynaptic sodium-dependent neurotransmitter transporters. These integral membrane proteins rely on preexisting ion gradients to catalyze the thermodynamically unfavorable movement of their substrates across the phospholipid bilayer (Masson, Sagné, Hamon, & El Mestikawy, 1999).
Members of the SLC6 group, also known as neurotransmitter sodium symporters, depend on Na+/Cl− to transport an expansive spectrum of small molecules such as amino acids (glycine and GABA); osmolytes; and the monoamines (serotonin (5-hydroxytryptamine, 5-HT), dopamine (DA), and norepinephrine (NE); Kristensen et al., 2011). They are of particular clinical significance because their dysfunction has been implicated in multiple debilitating neurological and neuropsychiatric illnesses (Hahn & Blakely, 2007), and they are the target of numerous psychoactive agents (Kristensen et al., 2011).
Prior to cloning of any SLC6 members, seminal experiments detecting NE transport in the heart (Iversen, 1963) as well as nerve endings (Hetting & Axelrod, 1961); and unearthing the uptake’s sodium-dependence (Iversen & Kravitz, 1966), stereospecificity (Iversen, Jarrott, & Simmonds, 1971), saturability (Iversen, 1971), and inhibition by cocaine (Iversen, 1965) as well as tricyclic antidepressants (TCAs; Iversen, 1965) reinforced the concept that an integral membrane protein was responsible. These early studies were further enhanced by exploiting membrane vesicles enriched in transporter, prepared from brain synaptosomes (Kanner, 1978) and blood platelets (Rudnick, 1977), as well as partially purified transporter reconstituted into lipid vesicles (Radian & Kanner, 1985). Properties such as substrate/inhibitor specificity, ion selectivity, and substrate–ion stoichiometry were soon characterized.
Cloning of the rat GABA type 1 transporter (GAT1; Guastella et al., 1990) followed by the human norepinephrine transporter (NET; Pacholczyk, Blakely, & Amara, 1991) and others each represented breakthroughs in SLC6 research. At the most basic level, they showed that seemingly functionally diverse transporters all belonged to the same family. They enabled hydropathy analyses with the prediction of 12 transmembrane (TM) segments and intracellularly located amino- and carboxy-termini, a topology that was later verified by site-directed chemical labeling (Chen, Liu-Chen, & Rudnick, 1998). Notably, recombinant expression in heterologous systems permitted scientists to examine the consequences of mutating distinct amino acids on activity, trafficking, and regulation.
As more data amassed, the need for a three-dimensional template became evident. Although attempts to solubilize and purify representative eukaryotic SLC6 members from heterologously expressing cells and some native tissues were sufficient for functional assays, they fell far short of the milligram quantities demanded for crystallography. Comprehensive efforts began by targeting mammalian proteins, such as the rat serotonin transporter (SERT). However, this protein has proven problematic (Tate et al., 2003), being stable and active exclusively in the heterogeneous detergent digitonin (Fig. 1A), from which no membrane protein has ever been directly crystallized (Tate, 2010).
Figure 1.

Detergents used in structural and functional studies of some eukaryotic SLC6 members and the prokaryotic counterpart LeuT. (A) Digitonin. (B) n-Dodecyl-β-D-maltoside (DDM). (C) n-Decyl-β-D-maltoside. (D) n-Octyl-β-D-glucoside (β-OG). (E) n-Octyl-β-D-thioglucoside (C8SG). (F) Lauryl maltose neopentyl glycol (LMNPG). (G) 3-[(3-Cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO). Note that C8SG is somewhat larger than β-OG due to sulfur’s slightly longer atomic radius. Permission provided by Anatrace.
2. LeuT EXPRESSION, PURIFICATION, CRYSTALLIZATION, AND STRUCTURE DETERMINATION
2.1 Background
Structure determination of integral membrane proteins remains a daunting task. Before attempts at crystallization are even made, obtaining enough pure monodisperse protein with which to work remains a major bottleneck. Prerequisites include adequate expression levels, monodispersity, long-term stability, large hydrophilic surface area, and activity. Probably the most crucial variable is detergent. There is a diverse array now available whose properties vary by headgroup, charge, and alkyl tail length (Moraes, Evans, Sanchez-Weatherby, Newstead, & Stewart, 2014). Generally, nonionic shorter chain-length ones promote formation of crystals capable of diffracting to higher resolution, but they can also disrupt quaternary structure and/or induce aggregation. Thus, the goal is to identify the shortest chain-length detergent that preserves stability, function, and oligomeric state.
Transporters are particularly challenging because they are almost completely buried in the hydrophobic bilayer, are conformationally heterogeneous, and frequently have long, flexible termini and loops. One way of improving the odds of success is to evaluate monodispersity and stability of homologs from multiple organisms via fluorescence-detection size-exclusion chromatography (FSEC), a high-throughput method in which the target protein is fused to a fluorescent tag, expressed, and behavior monitored on a gel filtration column (Kawate & Gouaux, 2006). The fact that FSEC requires only nanograms of unpurified protein versus micrograms of purified protein greatly expedites the process. Even among thermophilic prokaryotes, whose proteins are inherently more stable than those of their eukaryotic cousins and generally express well in inexpensive hosts like Escherichia coli and/or Lactococcus lactis NZ9000 (Kunji, Slotboom, & Poolman, 2003), FSEC screening can make an enormous difference.
For the SLC6 family, a pivotal discovery was that bacterial counterparts exist (Nelson, 1998). The tryptophan transporter (TnaT) from Symbiobacterium thermophilum was the first to be functionally characterized (Androutsellis-Theotokis et al., 2003), suggesting that it might be the first homolog to unveil its atomic secrets. However, despite the fact that the protein expressed to high levels and could be purified to homogeneity via metal affinity chromatography, most of it was aggregated when expressed in standard E. coli strains and solubilized in the mild, nonionic detergent n-dodecyl-β-D-maltoside (DDM) (Fig. 1B). For many prokaryotic membrane proteins, expression in L. lactis or the E. coli “Walker” variants, C41(λDE3) and C43(λDE3) (Miroux & Walker, 1996) has proven more beneficial. After exhaustive trials, L. lactis and C41(λDE3) were found to be the best strains for overexpressing two other prokaryotic SLC6s, Tyt1 (Quick & Javitch, 2007) and LeuT (Yamashita, Singh, Kawate, Jin, & Gouaux, 2005), respectively, in fully functional monodisperse form.
2.2 Toward the first SLC6 crystal structure: The LeuT Na+/Leu-bound outward-occluded state
LeuT is from the hyperthermophilic, chemolithotrophic eubacterium Aquifex aeolicus (Deckert et al., 1998) and was one of seven bacterial homologs selected from a PSI-BLAST search against the rat glycine transporter 1 (GlyT1). It, along with one from Methanococcus jannachi, expressed extremely well in C41(λDE3) cells, but LeuT crystallized much more readily. Hanging-drop vapor diffusion trays were initially set up with protein that had been solubilized and NiNTA-purified in DDM, subjected to gel filtration chromatography to remove any aggregates and exchange to the shorter-chain detergent n-decyl-β-D-maltoside (DM; Fig. 1C), and then concentrated. Crystals grew, but they diffracted to only ~15 Å, suggesting that molecular packing was likely mediated by detergent rather than protein. Because DDM is extremely difficult to remove, having a low critical micelle concentration (CMC) of only 0.2 mM, and detergent exchange to DM was achieved on a size-exclusion column rather than an affinity matrix, it is conceivable that the LeuT-detergent micelle was actually mixed, consisting of both DDM and DM, but this possibility was never explored.
The adjustment that dramatically improved diffraction was exchange from DDM to the much smaller n-octyl-β-D-glucoside (β-OG; Fig. 1D). As with DM, LeuT eluted as a sharp Gaussian peak with little or no aggregate. Although the protein was somewhat less stable in β-OG, with visible precipitate appearing in 1–2 days at 4 °C, trays set up immediately yielded crystals within a few days at ~20 °C and in HEPES buffer with PEG 550 monomethylether as the precipitant. These crystals were best cryoprotected by gradually increasing the PEG concentration to 35% instead of adding glycerol, the latter of which actually deteriorated resolution. The final structure was solved with native and selenomethionine multiwavelength anomalous diffraction (Se-MAD) X-ray data (Hendrickson, Horton, & LeMaster, 1990) extending to 1.65 and 1.90 Å, respectively. Because of the high resolution and excellent starting phases, model building was trivial, greatly assisted by the automatic tracing algorithm ARP/wARP (Langer, Cohen, Lazmin, & Perrakis, 2008). Despite the fortuitous appearance of Leu and two putative ions sitting at the core, there was no functional evidence beforehand that this protein was even a transporter, let alone a sodium-dependent Leu symporter. Nevertheless, it was designated “LeuT,” and the name has remained ever since.
Compared with the electron density for Leu, which was unequivocal, that for the two Na+ ions was less so because, even at such high resolution, these latter peaks could have easily been modeled as water molecules or other cations. Therefore, parameters, including valence calculations (Nayal & Di Cera, 1996); coordinating distances/geometry; and refined atomic positions, temperature factors, and residual peaks in Fo–Fc maps were employed to validate their identity (Yamashita et al., 2005).
2.3 Initial revelations (Fig. 13B)
Figure 13.

Schematic of transport and inhibition based exclusively on crystal structures and steady-state kinetic data of LeuT except for the substrate/sodium-bound inward-occluded state, which is based on MhsT, and the apo outward-open state, for which no representative structure is yet available. Conformational changes associated with isomerization from (A) sodium-bound, substrate-free outward-open to (B) substrate/sodium-bound outward-occluded state to (C) sodium/substrate-bound inward-occluded (MhsT) to (D) apo inward-open to (E) sodium/substrate-free outward-open. Panel (F) and (G), respectively, depict structures of the competitively inhibited, stabilized outward-open Trp/Na+-bound and the noncompetitively inhibited, stabilized outward-occluded Leu/Na+/CMI-bound states. TM5 is not shown for clarity, although, as mentioned, it does play an important role in the translocation cycle. (See the color plate.)
The first LeuT structure afforded unprecedented glimpses into SLC6 architecture, substrate/Na+-binding sites, putative extracellular/intracellular gates, and a speculative transport mechanism. Although it confirmed the 12-TM topology (Fig. 2) as well as juxtaposition of EL2 and EL4 (Fig. 3), it unexpectedly revealed an internal structural repeat relating TMs 1–5 and 6–10 by an antiparallel pseudo twofold in the membrane plane (Fig. 2). LeuT has a central substrate/ion-binding site (S1 hereafter) and another cavity, the extracellular vestibule (EV), located approximately 11 Å above. Although the EV, specifically a site within known as S2, was subsequently shown to bind a diverse array of hydrophobic molecules, including β-OG (Quick et al., 2009), only water molecules were originally built into the weak, discontinuous electron density. Nonetheless, the presence of β-OG was later validated by selenium-containing n-heptyl seleno-βD-glucoside (β-SeHG), a β-OG analog with an anomalous signal (Wang, Elferich, & Gouaux, 2012).
Figure 2.

LeuT topology. Leu and two sodium ions (purple (gray in the print version) circles) are drawn halfway across the bilayer. Membrane boundaries are demarcated by two thick black horizontal lines. TM helices are depicted as cylinders, with TMs 1 and 6 unwound close to Leu and the two sodiums ions. Faint triangles illustrate the component helices of the 5+ 5 inverted repeat, each pair of which is shown in the same color.
Figure 3.

(A) LeuT in the membrane plane. Transmembrane helices (TM) and extracellular/intracellular loops are depicted as cylinders and coils, respectively. Heavy black lines demarcate the membrane boundaries. (B) Same view as (A) except tilted ~15° toward the reader and with TMs 10–12 removed to expose the substrates Leu (in yellow [carbon]/red [oxygen] spheres) and the two sodium ions (cyan spheres), all bound near the unwound sections of TMs 1 and 6. Adapted from Singh (2008).
The two central TMs, 1 and 6, are unwound halfway across the lipid bilayer, unmasking backbone carbonyl oxygens and amide nitrogens for Na+ and leucine coordination (Fig. 4A). The two sodiums, Na1 and Na2, bind in a completely dehydrated site, also formed by the unwound sections of TMs 1 and 6, in addition to residues in the middle of TMs 3 and 8. Coordination is provided by five or six precisely arranged oxygen ligands with defined geometry (octahedral for Na1 and trigonal bipyramidal for Na2) and within a defined distance (2.28 Å) of each of the dehydrated sodium ions. Only Na1 is in direct contact with Leu, specifically its carboxylate, suggesting it plays a vital role in forming the substrate-binding pocket. The universally conserved Y108 interacts with Leu’s carboxylate and stabilizes TM1 near the unwound region. In the monoamine transporters, whose respective substrates possess a primary amine but lack a carboxylate, G24 in LeuT is replaced with Asp whose side-chain carboxylate likely coordinates Na1 and hydrogen bonds with both Y’s hydroxyl as well as the primary amine (Fig. 4A). Although these suppositions are supported by human SERT (Celik et al., 2008) and DAT (Huang & Zhan, 2007) homology models, they have yet to be substantiated by the structure of a Na+/substrate-bound eukaryotic monoamine transporter. For example, the recent structure of a thermostabilized Drosophila melanogaster dopamine transporter (dDAT), incapable of transport, complexed with Na+, Cl−, and presumed inhibitor, demonstrates that Na1 indirectly interacts with Asp’s side-chain carboxylate through a water molecule, but it bares no information about how the substrate DA is coordinated.
Figure 4.

Close-up of S1. (A) Leu and sodium ions are primarily coordinated by backbone amide nitrogens (blue (gray in the print version)), backbone carbonyl oxygens (red (dark gray in the print version)), and side chain hydroxyls (red (dark gray in the print version)). Labels for the invariant tyrosine in TM3 and glycine in TM1, an aspartate in the monoamine transporters, are boxed. Hydrogen bonds are shown as dashed lines. (B) Overlay of LeuT substrates (sticks) Leu (gray), Ala (green (gray in the print version)), Gly (magenta (dark gray in the print version)), Met (blue (gray in the print version)), and L-4-F-Phe (orange (gray in the print version)). Atoms of bound Leu are shown as semi-transparent van der Waals spheres. Left and right images respectively reproduced from Singh (2008) and Singh, Piscitelli, Yamashita, and Gouaux (2008), with permission from Landes Bioscience and AAAS, respectively.
In LeuT’s outward-occluded state, S1 is solvent inaccessible from both sides of the membrane, with extracellular access obstructed to a lesser degree (Figs. 5 [right panel] and 7A). Near the extracellular gate, at the bottom of the EV, is a water-mediated salt bridge between D404 (TM10) and R30 (TM1), the latter of which is indirectly coupled to S1 via a hydrogen-bond network (Fig. 5 [left panel]). At the opposite end of the bilayer, comprising part of the cytoplasmic gate, is another salt bridge, an ionic latch between D369 (TM8) and R5 (N-terminal domain/TM1), the latter of which is partly stabilized by a hydrogen bond to Y268 (TMs 6–7). W8 (TM8) fits snugly into a hydrophobic pocket formed by TMs 1 and 6, further anchoring the N-terminus and R5 in their positions (Fig. 6).
Figure 5.

LeuT’s extracellular gate. Both S1 and S2 are labeled. On the left panel, note the water-mediated salt bridge between R30 and D404, the hydrogen-bond network linking R30 with S1, and the layering of F253 and Y108 on top of Leu. Residues involved in the gating conformational changes in SLC6 members are italicized, boldfaced, and boxed. Reproduced from Singh (2008) with permission from Landes Bioscience.
Figure 6.

LeuT’s intracellular gate. (Left panel) Note the direct salt bridge between D369 and R5. Labels for residues involved in the gating conformational changes in SLC6 members are italicized, boldfaced, and boxed. Reproduced from Singh (2008) with permission from Landes Bioscience.
The LeuT-fold was novel back in 2005, but, surprisingly, crystal structures of numerous transporters from diverse, sequence-unrelated families have since been found to exhibit the 5+5 inverted repeat. These comprise members of the amino acid–polyamine–organocation (APC) (AdiC, ApcT, and GadC); betaine/carnitine/choline (BCCT) (BetP and CaiT); nucleobase:cation symporter-1 (NCS1) (Mhp1); solute–sodium symporter (SSS or SLC5) (vSGLT) (Shi, 2013); and NRAMP (SLC11; ScaDMT) families (Ehrnstorfer, Geertsma, Pardon, Steyaert, & Dutzler, 2014).
2.4 Noncompetitive inhibitor-bound structures (Fig. 13G)
Akin to the outward-occluded structure, crystallizing subsequent cocomplexes with the noncompetitive tricyclic antidepressants (TCAs; Singh, Yamashita, & Gouaux, 2007; Zhou et al., 2007) and later, the selective serotonin reuptake inhibitors (SSRIs), sertraline (SRT) and fluoxetine (FLX; Zhou et al., 2009), was straightforward. It merely involved purifying LeuT as usual and then adding 10–40 mM clomipramine (CMI), imipramine (IMI), desipramine (DMI), SRT, or FLX to the protein for cocrystallization trials. Crystals grew within 1 week under comparable conditions and diffracted to 1.7–1.9 Å (Singh et al., 2007), 2.9 Å (Zhou et al., 2007), or 2.1–2.5 Å (Zhou et al., 2009). The cocomplexes were solved via difference Fourier analysis or molecular replacement and refined well. All LeuT–TCA/SSRI complexes assume the same outward-occluded conformation as the original LeuT structure with Leu and Na+ bound in S1 and a single antidepressant molecule bound in S2 (Figs. 7B and 8A), directly above the R30–D404 ion pair (Fig. 8B), displacing β-OG. The drug’s presence triggers an almost 180° flip in R30’s guanidium ring to expel two water molecules and form a direct salt bridge with D404 (Fig. 8B), a presumably stronger interaction than one mediated by water.
Figure 7.

Surface renderings of LeuT–Na+/Leu and inhibitor complexes with S1 and S2 labeled in (A). Na+ ions are purple (dark gray in the print version) spheres in all panels. Magnified sections with F253 and Y108 are indicated, colored orange (gray in the print version) in (A) and (B) and green (gray in the print version) in (C). (A) LeuT–Na+/Leu complex (outward-occluded state); Leu is colored by element, with carbon, oxygen, and nitrogen shown in green (light gray in the print version), red (gray in the print version), and blue (dark gray in the print version), respectively. (B) LeuT–Na+/Leu/CMI complex (noncompetitively inhibited stabilized outward-occluded state); CMI is colored by element, with carbon, nitrogen, and chlorine depicted in red (gray in the print version), blue (dark gray in the print version), and green (light gray in the print version), respectively. (C) Leu–Na+/Trp complex (competitively inhibited, trapped outward-open state); Trp atoms are colored by element, with carbon, oxygen, and nitrogen shown in orange (light gray in the print version), red (gray in the print version), and blue (dark gray in the print version), respectively.
Figure 8.

(A) The TCA clomipramine (CMI) binds to LeuT in S2 about 11 Å above S1 and just above the R30–D404 salt bridge. (B) Magnification of the CMI-binding site from LeuT–Na+/Leu/CMI complex (pink (gray in the print version)) overlaid onto that of the –Na+/Leu, depicting the flip of the guanidium group of R30 to form a direct salt bridge with D404 and the displacement of two water molecules. (C) Space-filling model of CMI (yellow (light gray in the print version)), R30 (blue (gray in the print version)) and F253 (gray), illustrating how CMI seems to stabilize R30 in its flipped position via a “layered” cation–π interaction with F253. (D) Dissociation of [3H]Leu from LeuT in the absence (filled circles) and presence (open circles) of 3 mM CMI. Reproduced from Singh (2008) with permission from Landes Bioscience.
2.5 Substrate- and competitive inhibitor-bound (Fig. 13F) structures
Relative to LeuT–Na+/Leu, crystallizing other substrate-bound complexes was more difficult because these other amino acids had to supplant the endogenously bound Leu. LeuT–Ala and –Trp were the easiest, simply requiring the inclusion of saturating Ala or Trp, respectively, to all buffers. The others were marginally more complicated, demanding the same high concentrations of Ala during solubilization and then gradual replacement with the desired amino acid during purification and dialysis. Because of Tyr’s extremely low solubility, the more soluble isosteric Tyr analog, L-4-fluorophenylalanine (L-4-F-Phe), was used instead. All cocrystals diffracted to 1.8–2.3 Å and the structures were solved via molecular replacement using outward-occluded LeuT–Na+/Leu as a phase probe. This strategy was successful for the substrate-bound complexes but failed for LeuT–Na+/Trp, signifying that a structural rearrangement had occurred. Thus, the search model was modified to delete parts of TM1, TM6, EL4a, and EL4b. Molecular replacement with this new probe revealed displaced density for the missing helices, indicating they had indeed moved. To corroborate this new conformation, experimental phases from a selenomethionine-substituted LeuT–Na+/Trp complex were obtained, and the resulting structure was indistinguishable from the refined molecular replacement solution (Singh et al., 2008).
What did the LeuT–Na+/substrate and –Na+/Trp structures uncover? As predicted, all of the substrate complexes adopt the same outward-occluded state, with Y108’s hydroxyl maintaining its critical interaction with the substrate carboxylate and L25’s amide nitrogen. The only variations are localized to F259 and I359 abutting the substrates’ R group. Depending on the size of this moiety, F259 and I359 pivot into or out from S1, with the greatest torsion in and out observed with the Gly and Tyr complexes, respectively (Fig. 4B). The Trp complex, on the other hand, adopts an outward-open conformation, in which TMs 1b, 2a, and 6a rotate approximately 9° outward, accompanied by substantial displacement of TM11 and ELs 2, 3, and 4a (Figs. 7C and 9 [left panel]), all of which open a solvent-accessible channel from the extracellular side to S1 (Fig. 7C). These movements can be traced to Trp’s broad rigid indole ring, which separates Y108 from the Trp carboxylate by ~5.1 Å to preclude formation of a critical hydrogen bond. It also increases the distance between Y108 and F253 by 3 Å (Fig. 9 [lower right panel]; Singh et al., 2008). Curiously, as model building and refinement progressed, density consistent with a second Trp molecule (Trp602) appeared in the more open permeation pathway, between R30 and D404, and this position, distinct from S2, might represent a low-affinity, transiently occupied site (Fig. 9 [upper right panel]).
Figure 9.

(Left panel) Cα superposition of the LeuT–Na+/Leu (gray cylinders) and –Na+/Trp (sand (light gray in the print version), red (dark gray in the print version), and magenta (dark gray in the print version) cylinders) complexes. Helices engaged in the domain shift (TMs 1b, 2a, and 6a) and EL4a are colored red (dark gray in the print version) and magenta (dark gray in the print version), respectively, as are their rotation axes. TM11 and ELs 2 and 3 are also involved but not highlighted. The bound Trps are shown as sticks, with the carbon atoms of 601 and 602 colored green (gray in the print version) and dark green (dark gray in the print version), respectively. (Lower right panel) Magnification of the Cα superposition depicting the hydrogen-bond network in S1 of LeuT–Na+/Trp. A double-headed arrow indicates disruption of the critical hydrogen-bond between Y108 and the Trp601 carboxylate. For clarity, view has been rotated approximately 90° toward the reader relative to that in the larger left panel. (Upper right panel) A second Trp molecule (602) is bound between R30 and D404, flanked by a π-helix in TM10, just below S2. Reproduced from Singh et al. (2008) with permission from AAAS.
2.6 Outward-open, Na+-bound, substrate-free state (Fig. 13A)
Unlike the substrate- and inhibitor-bound complexes, crystallizing the substrate-free, sodium-bound outward-open state was more challenging because LeuT is not as stable in β-OG in the absence of ligands. Four components were critical for success: (1) use of a Y108F mutant to dramatically impair Leu binding by eliminating the stabilizing hydrogen bond with the Leu carboxylate (Piscitelli, Krishnamurthy, & Gouaux, 2010); (2) extensive washing of LeuT-Y108F membranes with sodium-free buffer supplemented with the sodium-specific chelator, 15-crown-5 (Christensen, Hill, & Izatt, 1971); (3) complexation with a conformation-specific Fab antibody fragment (2B12); and (4) detergent exchange to the slightly larger sulfur-containing n-octyl-β-D-thioglucoside (C8SG; Fig. 1E) rather than the smaller oxygen-containing β-OG. Although these crystals diffracted to only 3.1 Å, there was clearly no extra density in S1 except that for sodium, and LeuT had adopted an outward-open conformation (Fig. 10A), akin to the LeuT–Na+/Trp competitively inhibited complex, except for an additional 90° upward rotation of F253 (Krishnamurthy & Gouaux, 2012).
Figure 10.

Surface representation of the (A) Na+-bound/substrate-free, outward-open state and (B) apo inward-open conformation. Right panels magnify S1, with gating residues F253 and Y108 depicted as orange (gray in the print version) sticks. Panel A also includes the two sodium ions (purple (dark gray in the print version) spheres) partly behind the yellow (light gray in the print version) surface.
2.7 Inward-open “apo” state (Fig. 13D)
This structure was the most challenging and required an exhaustive search of the literature for clues. Two proved instrumental: First, molecular dynamic simulations with “LeuT-fold” transporters Mhp1 (Shimamura et al., 2010) and vSGLT1 (Watanabe et al., 2010), both of which share LeuT’s Na2 site (Shi, 2013), had implied that disrupting selected Na2-coordinating residues might promote intracellular substrate release. Second, mutation of the DAT intracellular gating residue Y335, analogous to Y268 in LeuT, had previously been shown to favor the DAT cytoplasmic-facing conformation (Kniazeff, Shi, Loland, Weinstein, & Gether, 2008). Based on these data, a new LeuT construct incorporating three mutations, two at Na2 (T354V and S355A) and one at the intracellular gate (Y268A), was generated.
The final modifications employed to crystallize the inward-open conformation included (1) use of this new mutant, LeuT-TSY; (2) utilization of another conformation-specific Fab (6A10); and (3) supplementation with the stabilizing lipid, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE; Fig. 11A). These steps were in addition to washing of membranes with sodium-free, 15-crown-5-supplemented buffer and exchange of LeuT-TSY into C8SG instead of β-OG. Like LeuT-Y108A-Fab crystals, these only diffracted to 3.2 Å. The structure was solved by molecular replacement, but the phase probe had to be heavily altered, using a combination of a previously solved Fab structure and a partial LeuT model. Initial maps definitively indicated that sizable rearrangements had occurred. First, TMs 1b and 6a flex to similar extents to partly close the extracellular pathway, while TM6b concomitantly bends away from the intracellular pathway by 17°. Second, TMs bracing TMs 1 and 6, namely TMs 2, 5, and 7, flex rather than tilt about their midpoints, facilitated by centrally located Gly or Pro residues. The bending of TM7 permits EL4 to “plug” the EV, mostly collapsing S2. The most striking movement is an almost 45° bend in TM1a away from the cytoplasmic side (Fig. 12) to open a solvent-accessible channel to S1 (Krishnamurthy & Gouaux, 2012; Fig. 10B). Such an enormous bend is startling, and, as the authors note, it is debatable if such a shift actually occurs in the membrane.
Figure 11.

Lipids used in LeuT structure/function experiments. (A) 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE). (B) 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). (C) 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC). Permission provided by Avanti Polar Lipids, Inc.
Figure 12.
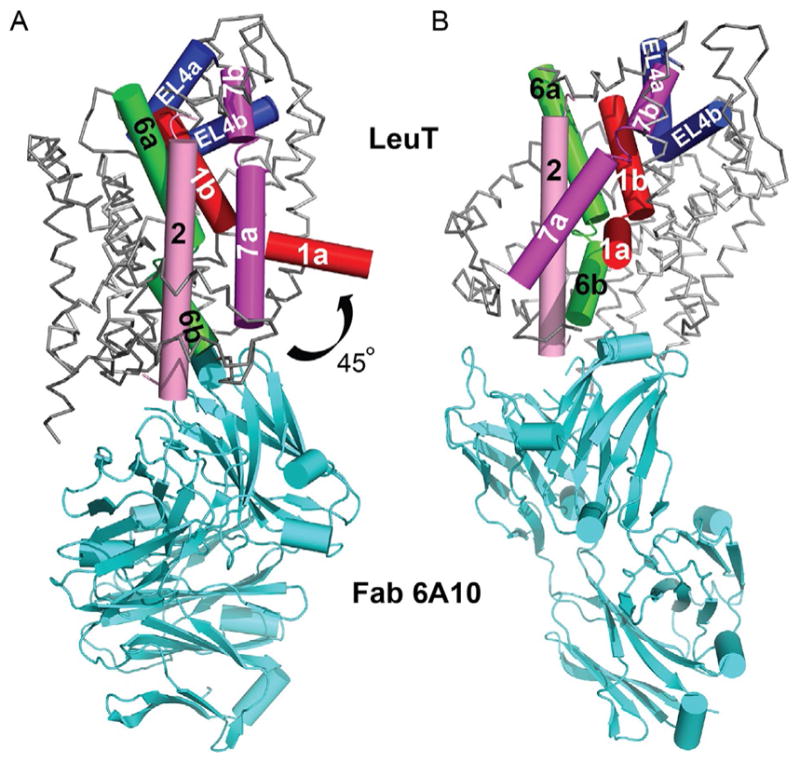
(A) Cα trace of the apo inward-open-Fab complex, with the “bundle” helices and EL4 depicted as cylinders. TMs 1, 2, 6, 7, and EL4 are colored red (dark gray in the print version), pink (gray in the print version), green (gray in the print version), magenta (dark gray in the print version), and blue (darkest gray in the print version), respectively. The 45° bend of TM1a into the predicted membrane is indicated. (B) Same image in (A) except rotated 90° toward the reader.
2.8 Inward-occluded Na+/substrate-bound state
Although structures of LeuT have been solved in multiple conformations along the translocation cycle, there are still two missing states—Na+/-substrate-bound inward-occluded (Fig. 13C) and Na+/substrate-free outward-open (Fig. 13E). Recently, however, the crystal structure of another bacterial SLC6 homolog from Bacillus halodurans (MhsT), a sodium-dependent aromatic amino acid transporter, was solved in the Na+/Trp-bound inward-occluded state in two separate lipidic environments (Fig. 13C; Malinauskaite et al., 2014). Unlike the LeuT apo inward-open structure, which was not crystallized in a membranous milieu. TM1a does not move nearly as far into the membrane and TM5 unwinds, enabling solvent access to Na2. Notably, a leucine at position 29 in LeuT is replaced with the larger tryptophan, as is the case with every other SLC6 member, and this Trp, in conjunction with EL4, completely collapses the EV, including S2.
3. FUNCTIONAL CHARACTERIZATION
Despite the undeniable power of crystallography, it alone is rarely a panacea for addressing every mechanistic question. Its full potential is realized only when combined with complementary biochemical, biophysical, and pharmacological techniques. Indeed, these methods were indispensable for delineating LeuT substrate specificity, elucidating TCA/Trp inhibition mechanisms, and characterizing solution conformational dynamics.
3.1 Flux assays
Measuring ion-dependent radiolabeled substrate uptake in intact cells is often the first avenue traveled when investigating function. Surmising that LeuT could transport Leu based on this amino acid’s serendipitous presence in the original crystal structure, assays were initially attempted with [3H]Leu in LeuT-expressing cultures of the E. coli transposon knockout strain FB21219, which exhibits considerably reduced endogenous transport of branched amino acids (Adams et al., 1990). Unfortunately, the signal-to-noise was only twofold over untransformed knockouts. Therefore, DDM-purified LeuT was reconstituted into liposomes composed of E. coli total lipid extract and egg phosphatidylcholine, the predominant species of which is 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Fig. 11B), at a protein:lipid ratio of 1:400 (Yamashita et al., 2005) and later 1:100 (Singh et al., 2007, 2008). These assays were successful and demonstrated that [3H]Leu transport is electrogenic and Na+- but not Cl−-dependent, like other bacterial SLC6 members but unlike its eukaryotic cousins.
Nevertheless, sequence comparisons between LeuT and eukaryotic SLC6 members permitted identification of a putative Cl−-binding site in SERT (Forrest, Tavoulari, Zhang, Rudnick, & Honig, 2007), GAT1, GAT4, and DAT (Zomot et al., 2007) and demonstration that Cl−’s negative charge (E290 in LeuT) promotes Na+ binding and substrate binding/translocation. The proposed site was later confirmed with crystal structures of 1) LeuT-E290S complexed with Na+ and Cl− or Br−, the either anion coordinated by Y47, Q250, T254, and S290 (Kantcheva et al., 2013); 2) LeuBAT, a hybrid between LeuT and SERT/DAT/NET (Wang et al., 2013); and 3) the aforementioned thermostabilized dDAT mutant (Penmatsa, Wang, & Gouaux, 2013).
Successive flux assays illustrated that LeuT transports a wide array of aliphatic/aromatic amino acids, including Gly, Ala, Met, and Tyr, in addition to its namesake Leu, with catalytic efficiencies roughly corresponding to the inverse of substrate volume. Steady-state kinetics established that Ala is actually a much better substrate than Leu, with a catalytic rate constant (kcat) at least sixfold greater, and hence far superior for examining inhibition (Singh et al., 2007, 2008). These experiments indicated that although Trp can bind LeuT, it cannot be transported, and is instead a competitive inhibitor that stabilizes the outward-open conformation. Competitive inhibitory behavior was manifested by an obvious increase in Michaelis constant (Km) but no change in maximum velocity (Vmax) when concentrations of [3H]Ala and Trp were varied as substrate and inhibitor, respectively, graphically portrayed on an Eadie-Hofstee (EH) plot as nonparallel lines intersecting on the y-axis (Singh et al., 2008).
Since LeuT is a homolog of the clinically important SERT, DAT, NET, GAT1, and GlyTs, an immediate goal was to determine if any of the psychoactive agents that target these proteins can inhibit LeuT. A comprehensive screen with LeuT proteoliposomes revealed that the TCA CMI is the most potent, with IC50s of 250 and 5 μM, for inhibition of [3H]Leu and [3H]Ala uptake, respectively. Unlike the aliphatic and aromatic amino acids, none of the TCAs (CMI, IMI, or DMI) could displace bound [3H]Leu, provisionally excluding competitive inhibition as the mechanism. To conclusively define the mode of inhibition, steady-state kinetics with varying concentrations of [3H]Ala as the substrate and CMI as the inhibitor were conducted. Contrary to Trp inhibition kinetics, there was no change in Km but a noticeable reduction in Vmax, graphically illustrated by parallel lines on an EH plot, unambiguous indicators of noncompetitive inhibition (Singh et al., 2007). As mentioned above, all LeuT–TCA structures suggested that these drugs stabilize the occluded state. To provide functional support for this postulate, dissociation assays with bound [3H]Leu in the presence and absence of 3 mM CMI were performed. [3H]Leu instead of [3H]Ala was used for these experiments because the latter dissociates too quickly to accurately measure its off-rate. Data showed that CMI attenuates [3H]Leu release by almost 700-fold (Fig. 8D). Thus, the structure in combination with steady-state kinetic and dissociation data demonstrated that a noncompetitive inhibitor binding at S2 works by strengthening the R30/D404 salt bridge at the extracellular gate.
While it is tantalizing to surmise that the TCA/SSRI site observed in LeuT is present in NET and SERT, the actual targets in humans, almost all functional data on mammalian SERT, NET, and DAT indicate that these compounds inhibit monoamine transport competitively and bind at or near S1 (Kristensen et al., 2011). Moreover, alanine mutants of analogous residues within 5 Å of S2 in human SERT decreased CMI potency by only two- to fivefold (Singh, 2008; Zhou et al., 2007), far less than the values reported for S1 mutants and incompatible with molecular docking (Kristensen et al., 2011). The only region that seemed to have a more pronounced effect, at least for the SSRI sertraline, was termed the “halogen binding pocket” (Zhou et al., 2009), especially residue I111 in LeuT (I179, I155, and F168 in human SERT, NET, and DAT, respectively).
A recent structure of the thermostabilized dDAT variant complexed with Na+, Cl−, and the TCA nortriptyline (Penmatsa et al., 2013), along with several of LeuBAT, each complexed with Na+, Cl−, and a clinically relevant antidepressant (Wang et al., 2013), may seem to have resolved the issue, but there are serious caveats. The transporters are presumably trapped in an outward-open conformation, with all of the drugs bound in S1 except for the serotonin–norepinephrine reuptake inhibitor desvenlafaxine, which is bound in both S1 and S2. Although the authors contend that they have conclusively resolved the controversy over where these compounds bind and how they competitively inhibit substrate transport, their constructs cannot transport substrate and perhaps are perpetually frozen in the outward-open state. Furthermore, there is no evidence that these proteins can even bind substrate despite the fact that a simple displacement assay was already established to gauge relative antagonist potencies. Thus, it is impossible for the authors to directly correlate kinetic inhibition patterns with substrate/antagonist-binding sites and associated conformational changes, as was accomplished with both the noncompetitively inhibited LeuT–CMI and competitively inhibited LeuT–Trp complexes. Knowledge of both structure and kinetics is crucial for designing more selective pharmacological agents with fewer side effects.
Note that the ambiguity does not mitigate the significance of definitive drug binding in S2 of LeuT. First, the data advance a tangible, testable hypothesis for the general phenomenon of noncompetitive inhibition that may be applicable to eukaryotic SLC6 members. Second, they pinpoint a potential antagonist-binding site that may exist in eukaryotic cousins and be exploited in rational drug design efforts. Two possible examples of such inhibitors include the NET-specific χ-conotoxin MrIA (Paczkowski, Sharpe, Dutertre, & Lewis, 2007) and the GlyT2-specific N-arachidonylglycine (Edington et al., 2009). In addition, S2 cannot be excluded as the low-affinity allosteric site reported in SERT (Wennogle & Meyerson, 1982). Ibogaine, a noncompetitive inhibitor of SERT ( Jacobs, Zhang, Campbell, & Rudnick, 2007) and DAT (Bulling et al., 2012) does not fit into the “S2 category” because it stabilizes the inward-open conformation and binds to an unknown site, ostensibly accessible from the extracellular side.
3.2 Binding via scintillation proximity, equilibrium dialysis, and isothermal titration calorimetry
Another method of assessing transporter function is via binding of a cognate ligand. For LeuT, this can be any one of the amino acid substrates, the competitive inhibitor Trp, or the noncompetitive TCAs/SSRIs. Multiple means have been applied to evaluate LeuT function other than standard filter binding, including scintillation proximity assay (SPA), equilibrium dialysis (ED), and isothermal titration calorimetry (ITC). Probably the most convenient but also contentious is SPA. It utilizes fluoromicrospheres coated with a capture molecule (often copper chelate) to which a protein of interest (usually histidine-tagged) can adhere. The beads are filled with scintillant that emits light only when excited by a radiolabel bound either directly to the bead or to an attached target protein (Harder & Fotiadis, 2012; Quick & Javitch, 2007). Despite its advantages, one serious limitation is the unreliability of scintillation counting efficiency, potentially leading to errors when converting from cpm to moles substrate and thus, to substrate: protein molar binding stoichiometry. The “SPA” controversy has arisen from this very issue, which has led to divergence in published stoichiometry measurements, i.e., 2:1 versus 1:1 (Lim & Miller, 2012). That said, complementary binding methods such as ED have reinforced the disparity (see below).
What could be causing the discrepancy? One problem may stem from the fact that almost all crystallographic and binding experiments have been performed in two distinct detergents, β-OG versus DDM, the former of which binds in S2 (Wang et al., 2012) to conceivably displace a second substrate and inhibit transport (Quick et al., 2009). A second complication may focus on DDM concentration itself. SPA and ED conducted with LeuT at two different DDM concentrations (2 versus 6 mM) reported a substrate: protein binding stoichiometry of 1:1 in 6 mM (Piscitelli et al., 2010; Quick, Shi, Zehnpfennig, Weinstein, & Javitch, 2012) but 2:1 in 2 mM DDM (Quick et al., 2012). Even single mutations in S1 (F253A) and S2 (L400C, L400S, L400A), supposedly designed to obstruct substrate at these respective sites, have yielded conflicting outcomes. Moreover, whether these mutants actually obstruct the respective sites is also a source of debate (Piscitelli et al., 2010; Wang & Gouaux, 2012).
Although crystallography tends to negate the two-substrate theory, it is not irrefutable. First, despite the perception that LeuT purified in DDM and crystallized in bicelles composed of the detergent CHAPSO (Fig. 1G) and the lipid DMPC (Fig. 11C), revealed no substrate-like density in S2, DDM associates/dissociates extremely slowly (Quick et al., 2012) so it is possible that some DDM remained in the bicelles, invisible because of disorder (Lim & Miller, 2012). Second, although the inward-open LeuT conformation showed a collapsed S2, with presumably no room for a second substrate, there is some unspecified electron density present which has yet to be unequivocally identified (Krishamurthy & Gouaux, 2012). This finding directly contradicts the MhsT structure, which, with its bulkier Trp instead of Leu at position 29, possesses no mysterious density in this area (Malinauskaite et al., 2014).
3.3 SPA and LeuT-nanodiscs
The above binding methods were all performed in either DDM or the newly synthesized lauryl maltose neopentyl glycol (LMNPG; Fig. 1F), in which many membrane proteins, including LeuT, are more stable (Chae et al., 2010). Still, ligand binding should ideally take place with protein reconstituted into lipid bilayers. Proteoliposomes are an option and were employed in one investigation (Quick et al., 2012). Nanodiscs are another and have recently been tested in combination with SPA (Nasr & Singh, 2014). Equally crucial is the use of BioBeads to remove detergent, which were utilized in both proteoliposome and nanodisc preparations but not with bicelles.
SPA with both [3H]Leu and [3H]Ala showed that LeuT is almost twice as active in nanodiscs versus DDM, as would be expected for a membrane protein in a lipid bilayer, but the increase in Bmax values for both substrates is intriguing given the clash over the number of substrate-binding sites. It is plausible that the twofold rise is due to simultaneous binding of two substrates, but this interpretation will not be convincing until binding to the S1/S2 knockout mutant proteins is also gauged in the same hydrophobic nanodisc environment. Therefore, existence of dual substrate binding in LeuT remains uncertain at this juncture. Curiously, the protein-to-substrate molar binding stoichiometry in MhsT, which, as mentioned above, possesses the larger Trp instead of Leu in S2, like all other SLC6 members, is 1:1 (Malinauskaite et al., 2014) versus the 2:1 reported for LeuT. Thus, if the 2:1 stoichiometry is correct, it might hold true for LeuT exclusively and not the other members of the SLC6 family.
3.4 Spectroscopy
Two spectroscopic methods have been elegantly applied to LeuT, complementing static crystallographic experiments as well as bulk, steady-state binding/flux assays: pulsed electron paramagnetic resonance (EPR), along with double electron–electron resonance (DEER), and single-molecule Förster resonance energy transfer (smFRET). These techniques are fundamental for unraveling protein-mediated reaction mechanisms due to their intrinsic ability to track each step in real-time. Both require strategically placed cysteines on which to attach either nitroxide spin labels (EPR and DEER) or appropriate fluorophores (smFRET). These in turn report intervening distances and angles between two probes at various points along a reaction pathway, a reflection of associated conformational changes in response to the presence/absence of substrates, ions, and/or antagonists. LeuT is ideal for this work because it has no endogenous cysteines.
Four groundbreaking papers have capitalized on these spectroscopies, which have collectively validated, explained, and/or rebutted observations from bulk binding/transport and/or direct structural studies. One EPR study centered on the dynamics associated with Na+, substrate, and inhibitor binding (Claxton et al., 2010), the results of which are largely concordant with solved crystal structures. In the apo state (no Na+, substrate, or inhibitor), the nature of which remains impervious to crystallography, two populations emerged, one major (~26 Å) and one minor (~30–40 Å), interpreted as open and closed conformers, respectively. As anticipated, Na+ and Na+/Trp stabilized the outward-open, while Na+/Leu and Na+/Leu/CMI or Na+/Leu/β-OG stabilized the outward-occluded conformations. These results are nearly congruous with previously solved structures. Interestingly, distance distribution shits, coupled with accessibility measurements, suggested that the Na+-bound outward-open state is more conformationally heterogeneous than either the Na+/Leu-bound outward-occluded or Na+/Trp-bound outward-open states, which may clarify why LeuT–Na+ required Fab complexation to crystallize.
One smFRET study (Zhao et al., 2011) concentrated on ligand-induced gating dynamics and the relative activation energies required to transition from outward- to inward-open states. LeuT variants with cysteine substitutions on nonconserved, intracellular residues (H7C [distal N-terminus] and R86C [IL1] or T515C [TM12 C-terminal end]), were labeled with Cy3 or Cy5 maleimide, respectively. In the absence of Na+, two FRET states were detected, differing by ~13 Å between labeled pairs and thus inferred as outward- and inward-open conformations, consistent with EPR data. Increasing Na+ concentrations diminished the two FRET distributions, selectively stabilizing the inward-closed state by approximately sevenfold.
Aiming to discern conformational changes associated with the transport process, Leu was then added at a Na+ concentration sufficient for Leu binding, and the inward-closed state was stabilized by ~3.5-fold under these conditions. By contrast, when Ala was the substrate, rates between the two FRET states increased by approximately fourfold. Detailed transition state analysis indicates that whereas Ala substantially reduces, Leu raises the activation energy barrier between the outward- and inward-open states, providing a satisfying, time-resolved thermodynamic explication for Ala’s higher catalytic rate constant (Singh et al., 2008). The nature of the coupling cation and presence of inhibitors also impacted intracellular gating dynamics. Substituting Na+ with saturating concentrations of Li+, a cation with a ~0.3 Å smaller ionic radius (Shannon, 1976), permitted Ala binding to LeuT, but not transport, steady-state data buttressed by the fact that Ala/Li+ stabilizes the inward-closed state by twofold and thus is unable to enhance intracellular opening. As expected, the inhibitors CMI and β-OG, in the presence of Ala and Na+, completely blocked intracellular opening, stabilizing LeuT in a single high-FRET peak representing the inward-closed conformation. These time-resolved results nicely recapitulate the IC50 values reported previously (Singh et al., 2007) and again illustrate how time-resolved single-molecule spectroscopic data can furnish gratifying molecular explanations for bulk measurements.
Another smFRET study (Zhao et al., 2010) also focused on intracellular motions but this time further evaluated the degree of conformational coupling between both sides. It used the same three LeuT mutants delineated above and added two extracellular cysteine substitutions (K239C [EL3] and H480C [EL6]) as well as two more cytoplasmic ones (R185C [IL2] and K271C [IL3]). The second pair did not exhibit the Na+- or Na+/Leu-dependent changes of H7C/R86C or H7C/T515C, consistent with the view that IL2/IL3 does not move as much during intracellular gating. These mutations were made in otherwise WT LeuT, the results of which, predictably paralleled those of the 2011 smFRET study. To investigate the nature of coupling, the same series of mutants was also generated in LeuT-R5A, -Y268A, and R30A backgrounds, the former two of which disrupt the intracellular gate to favor the inward-open state (Kniazeff et al., 2008) and the latter of which putatively disturbs the extracellular gate by precluding formation of both the salt bridge with D404 and the cation–π interaction with F253. Interestingly, without Na+, the presence of R5A or Y268A markedly affected FRET distributions for both intracellular H7C/R86C as well as extracellular K239C/H490C, hinting that intracellular outward and extracellular inward movements occur concomitantly. By contrast, the presence of R30A affected only H7C/R86C.
This pioneering smFRET work thus argues that conformational transitions between the two sides are communicated through a linked series of small local, flexible rearrangements instead of one concerted tilt of a rigid bundle and is also partly suggestive of large movements in TM1a. Nevertheless, there was no indication that TM1a moves as much as 45° into the membrane. Indeed, evidence from a subsequent series of EPR experiments supports this contention (Kazmier et al., 2014). Although the broad EPR line distributions were consonant with LeuT’s innately dynamic character, and TM1a of the Y268A mutant did move far into the presumed lipid bilayer, it did not exhibit such a large amplitude change in the wild-type protein, as implied by the structure of the inward-open, highly mutagenized Fab-complexed LeuT. The fact that R5A also mimicked the anomalous behavior of Y268A reinforces the notion that tampering with intracellular gating residues skews conformation and is likely an artifact. An additional complication with the inward-open structure is the fact that the 6A10 Fab abuts the intracellular face and protrudes into the intracellular-facing cavity (Fig. 12). Its presence on the same side as the one that moves in the inward-open conformation may prevent TM1a from resting there and possibly force it to flip up as much as it does. It is also conceivable that the drastic movement is a synergistic combination of the Y268A mutant and Fab. Regardless, such sizable bending is incompatible with solution EPR data of WT LeuT.
4. TRANSPORT MECHANISM UNVEILED FROM STRUCTURE, FUNCTION, AND COMPUTATIONAL BIOLOGY
The most prevalent theory to describe mechanism in secondary transporters is one in which substrates and ions bind to unique sites on the protein and an ensuing isomerization permits alternating access to either the extracellular or cytoplasmic side. This thought was first drafted in outline form by Peter Mitchell over 50 years ago with his vision of a “translocase,” a moving “barrier” that was alternately accessible from either side of the lipid bilayer (Mitchell, 1957). Some variations and details were added to this basic two-state concept over the next few years, reformulated and dubbed “gate-type non-carrier” by Patlak (1957), “two-shape allosteric” by Vidaver (1966), and “alternating access” by Jardetsky (1966).
The atomic framework for these nebulous ideas has emerged from dozens of crystal structures from many different SLC families. One model is based on the intriguing outward-occluded state of the original LeuT structure, which signifies that, in addition to outward- and inward-open configurations of the paradigmatic alternating access model, there is at least one more state where both gates are closed. The unwound helices of TMs 1 and 6 first portended that they might bend like “fingers” during the transport cycle. Movement of EL4 was implied by the lid-like structure it forms over EV, and movement of EL2 was suggested by its proximal position on top of EL4. The fact that access to the substrate-binding site from the extracellular side is blocked by only a few residues indicated that only modest rotations in key helices such as TMs 1b, 6a, 3, and 8, along with more dramatic shifts in EL2 and EL4, would be required for the transporter to open to the outside. By contrast, the 25 Å of ordered protein structure between the intracellular gate and S1 hinted that this entire region would have to rearrange considerably in order for the transporter to open to the inside. Indeed, LeuT’s Na+-bound/substrate-free outward-open, competitively inhibited outward-open, and apo inward-open as well as MhsT’s Na+/Trp inward-occluded states have largely confirmed such movements. Specifically, concomitant rotations of TMs 2, 5, and 7 along with considerable rearrangements of ELs 2, 3, and 4, complemented with spectroscopic data, all bolster these conclusions. However, the pronounced movement of TM1a into the bilayer is not supported by solution smFRET and EPR data and stands in stark contrast to the simple rotation of the structurally related transporters Mhp1 and BetP.
A second model is based on the 5+5 architecture and pseudosymmetry astutely noticed in the inverted repeats, which were “swapped” to create a model of the inward-occluded state (Forrest et al., 2008). This “rocking bundle” proposal has two elements, a “scaffold” comprised of TMs 3–5 and 8–10 and a “bundle” comprised of TMs 1–2 and 6–7, the latter of which rocks back and forth relative to the “scaffold” to alternately expose S1 to the external and internal milieu. Accessibility measurements in SERT as well as the inward-facing vSGLT, Mhp1, and several BetP structures mostly support the premise. Although the “rocking bundle” hypothesis emphasizes tilting of a rigid bundle and cannot inherently predict EL and IL movements, it does not exclude the possibility of internal rearrangements and/or bending of individual helices envisioned by the first model (Forrest & Rudnick, 2009).
A third model also posits that the occluded state is an essential intermediate but that binding of a second substrate molecule in S2 acts as an allosteric trigger for release of S1-bound substrate and sodium to the intracellular medium (Shi, Quick, Zhao, Weinstein, & Javitch, 2008). S2-bound substrate is hypothesized to act as a “symport effector,” whereas hydrophobic molecules that bind in S2 such as β-OG, TCAs, and SSRIs inhibit transport by serving as “symport uncouplers.” This unorthodox concept, derived from SPA data and “steered” molecular dynamics, deviates significantly from the traditional alternating access mechanism and has evolved into a contentious issue (Lim & Miller, 2012). Nevertheless, the idea may be applicable just to LeuT, being the only SLC6 member with published evidence for a 2:1 molar substrate:protein binding stoichiometry. Notably, LeuT is the only homolog with Leu at position 29 in S2, whereas all others possess the larger Trp, which in MhsT completely collapses the EV, including S2.
5. SUMMARY
Biophysical studies on LeuT have catapulted our knowledge of both SLC6 members and structurally related SLCs, unraveling molecular details about transport mechanism, inhibition, substrate/ion-binding sites, and conformational dynamics. More importantly, they have provided an experimental platform on which the clinically significant eukaryotic SLC6 members can eventually be pursued via complementary crystallographic, steady-state kinetic, and real-time spectroscopic methods.
Acknowledgments
The authors thank Gary Rudnick for insightful comments and Suraj Adhikary for assistance with figures. Research in the corresponding author’s laboratory was/is funded by Yale University (Goodman-Gilman Scholar Award), the Alfred P. Sloan Foundation, the US-Israel Binational Science Foundation (BSF 2013198), and the NIH (R00MH083050, R21MH098180, and R01MH100688).
References
- Adams MD, Wagner LM, Graddis TJ, Landick R, Antonucci TK, Gibson AL, et al. Nucleotide sequence and genetic characterization reveal six essential genes for the LIV-I and LS transport systems of Escherichia coli. The Journal of Biological Chemistry. 1990;265:11436–11443. [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Goldberg NR, Ueda K, Beppu T, Beckman ML, Das S, et al. Characterization of a functional bacterial homologue of sodium-dependent neurotransmitter transporters. The Journal of Biological Chemistry. 2003;278:12703–12709. doi: 10.1074/jbc.M206563200. [DOI] [PubMed] [Google Scholar]
- Bulling S, Schicker K, Zhang YW, Steinkeller T, Stockner T, Gruber CW, et al. The mechanistic basis for noncompetitive ibogaine inhibition of serotonin and dopamine transporters. The Journal of Biological Chemistry. 2012;287:18524–18534. doi: 10.1074/jbc.M112.343681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, et al. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. Journal of the American Chemical Society. 2008;130:3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, Goren MA, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization, and crystallization of membrane proteins. Nature Methods. 2010;7:1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Liu-Chen S, Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. The Journal of Biological Chemistry. 1998;273:12675–12681. doi: 10.1074/jbc.273.20.12675. [DOI] [PubMed] [Google Scholar]
- Christensen JJ, Hill JO, Izatt RM. Ion binding by synthetic macrocyclic compounds. Science. 1971;174:459–467. doi: 10.1126/science.174.4008.459. [DOI] [PubMed] [Google Scholar]
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, et al. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nature Structural & Molecular Biology. 2010;17:822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- Edington AR, McKinzie AA, Reynolds AJ, Kassiou M, Ryan RM, Vanderberg RJ. Extracellular loops 2 and 4 of GLYT2 are required for N-arachidonylglycine inhibition of glycine transport. The Journal of Biological Chemistry. 2009;284:36424–36430. doi: 10.1074/jbc.M109.017509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnstorfer IA, Geertsma ER, Pardon E, Steyaert J, Dutzler R. Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nature Structural & Molecular Biology. 2014;21:990–996. doi: 10.1038/nsmb.2904. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Rudnick G. The rocking bundle: A mechanism for ion-coupled solute flux by symmetrical transporters. Physiology. 2009;24:377–386. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B. Identification of a chloride ion binding site in Na+/Cl−-dependent neurotransmitter transporters. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12761–12766. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, et al. Mechanism for alternating access in neurotransmitter transporters. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, et al. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- Hahn MK, Blakely RD. The functional impact of SLC6 transporter genetic variation. Annual Review of Pharmacology and Toxicology. 2007;47:401–441. doi: 10.1146/annurev.pharmtox.47.120505.105242. [DOI] [PubMed] [Google Scholar]
- Harder D, Fotiadis D. Measuring substrate binding and affinity of membrane transport proteins using the scintillation proximity assay. Nature Protocols. 2012;7:1569–1578. doi: 10.1038/nprot.2012.090. [DOI] [PubMed] [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. The EMBO Journal. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetting G, Axelrod J. Fate of tritiated noradrenalin at the sympathetic nerve-endings. Nature. 1961;192:172–173. doi: 10.1038/192172a0. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhan CG. How dopamine transporter interacts with dopamine: Insights from molecular modeling and simulation. Biophysical Journal. 2007;93:3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL. The uptake of noradrenalin by the isolated perfused heart. British Journal of Pharmacology and Chemotherapy. 1963;21:523–537. doi: 10.1111/j.1476-5381.1963.tb02020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL. Inhibition of noradrenaline uptake by drugs. The Journal of Pharmacy and Pharmacology. 1965;17:62–64. doi: 10.1111/j.2042-7158.1965.tb07572.x. [DOI] [PubMed] [Google Scholar]
- Iversen LL. Role of transmitter uptake mechanisms in synaptic neurotransmission. British Journal of Pharmacology. 1971;41:571–591. doi: 10.1111/j.1476-5381.1971.tb07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL, Jarrott B, Simmonds MA. Differences in the uptake, storage and metabolism of (+)- and (−)-noradrenaline. British Journal of Pharmacology. 1971;43:845–855. doi: 10.1111/j.1476-5381.1971.tb07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen LL, Kravitz EA. Sodium dependence of transmitter uptake at adrenergic nerve terminals. Molecular Pharmacology. 1966;2:360–362. [PubMed] [Google Scholar]
- Jacobs MT, Zhang YW, Campbell SD, Rudnick G. Ibogaine, a non-competitive inhibitor of serotonin transport, acts by stabilizing the cytoplasmic-facing of the transporter. The Journal of Biological Chemistry. 2007;282:29441–29447. doi: 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- Jardetsky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Kanner BI. Active transport of gamma-aminobutyric acid by membrane vesicles isolated from rat brain. Biochemistry. 1978;17:1207–1211. doi: 10.1021/bi00600a011. [DOI] [PubMed] [Google Scholar]
- Kantcheva AK, Quick M, Shi L, Winther AM, Stolzenberg S, Weinstein H, et al. Chloride binding site of neurotransmitter transporters. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8489–8494. doi: 10.1073/pnas.1221279110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Kazmier K, Sharma S, Quick M, Islam SM, Roux B, Weinstein H, et al. Conformational dynamics of ligand-dependent alternating access in LeuT. Nature Structural & Molecular Biology. 2014;21:472–479. doi: 10.1038/nsmb.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Shi L, Loland CJ, Weinstein H, Gether U. An intracellular interaction network regulates conformational transitions in the dopamine transporter. The Journal of Biological Chemistry. 2008;283:17691–17701. doi: 10.1074/jbc.M800475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacy Review. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Kunji ER, Slotboom DJ, Poolman B. Lactococcus lactis as host for overproduction of functional membrane proteins. Biochimica et Biophysica Acta. 2003;1610:97–108. doi: 10.1016/s0005-2736(02)00712-5. [DOI] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lazmin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Miller C. It takes two to transport, or is it one? Nature Structural & Molecular Biology. 2012;19:129–130. doi: 10.1038/nsmb.2239. [DOI] [PubMed] [Google Scholar]
- Malinauskaite L, Quick M, Reinhard L, Lyons JA, Yano H, Javitch JA, et al. A mechanism for intracellular release of Na+ by neurotransmitter/sodium symporters. Nature Structural & Molecular Biology. 2014;21:1006–1012. doi: 10.1038/nsmb.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson J, Sagné C, Hamon M, El Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacy Review. 1999;51:439–464. [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. Journal of Molecular Biology. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Mitchell P. A general theory of membrane transport from studies of bacteria. Nature. 1957;180:134–136. doi: 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- Moraes I, Evans G, Sanchez-Weatherby J, Newstead S, Stewart PDS. Membrane protein structure determination—The next generation. Biochimica et Biophysica Acta. 2014;1838:78–97. doi: 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr ML, Singh SK. Radioligand binding to nanodisc-reconstituted membrane transporters assessed by the scintillation proximity assay. Biochemistry. 2014;53:4–6. doi: 10.1021/bi401412e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. Journal of Molecular Biology. 1996;256:228–243. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- Nelson N. The family of Na+/Cl− neurotransmitter transporters. Journal of Neurochemistry. 1998;71:1785–1803. doi: 10.1046/j.1471-4159.1998.71051785.x. [DOI] [PubMed] [Google Scholar]
- Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- Paczkowski FA, Sharpe IA, Dutertre S, Lewis RJ. chi-Conopeptide and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. The Journal of Biological Chemistry. 2007;282:17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- Patlak CS. Contributions to the theory of active transport: II. The gate type non-carrier mechanism and generalizations concerning tracer flow, efficiency, and measurement of energy expenditure. The Bulletin of Mathematical Biophysics. 1957;19:209–235. [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli CL, Krishnamurthy H, Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468:1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Javitch JA. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3603–3608. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch JA. Experimental conditions can obscure the second high-affinity site in LeuT. Nature Structural & Molecular Biology. 2012;19:207–211. doi: 10.1038/nsmb.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor bound conformation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radian R, Kanner BI. Reconstitution and purification of the sodium- and chloride-coupled gamma-aminobutyric acid transporter from rat brain. The Journal of Biological Chemistry. 1985;260:11859–11865. [PubMed] [Google Scholar]
- Rudnick G. Active transport of 5-hydroxytryptamine by plasma membrane vesicles isolated from human blood platelets. The Journal of Biological Chemistry. 1977;252:2170–2174. [PubMed] [Google Scholar]
- Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenics. Acta Crystallographica. 1976;A32:751–767. [Google Scholar]
- Shi Y. Common folds and transport mechanisms of secondary active transporters. Annual Review of Biophysics. 2013;42:51–72. doi: 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter—Inward release of Na+ and substrate is triggered by substrate in a binding site. Molecular Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Weyand S, Beckstein O, Rutherford NG, Hadden JM, Sharples D, et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328:470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK. LeuT: A prokaryotic stepping stone on the way to a eukaryotic neurotransmitter transporter structure. Channels (Austin, Tex) 2008;2:380–389. doi: 10.4161/chan.2.5.6904. [DOI] [PubMed] [Google Scholar]
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor stabilizes LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Tate CG. Practical considerations of membrane protein instability during purification and crystallization. Methods in Molecular Biology. 2010;601:187–203. doi: 10.1007/978-1-60761-344-2_12. [DOI] [PubMed] [Google Scholar]
- Tate CG, Haase J, Baker C, Boorsma M, Magnani F, Vallis Y, et al. Comparison of seven different heterologous protein expression systems for the production of the serotonin transporter. Biochimica et Biophysica Acta. 2003;1610:141–153. doi: 10.1016/s0005-2736(02)00719-8. [DOI] [PubMed] [Google Scholar]
- Vanhatalo S, Sohila S. The concept of chemical neurotransmission—Variations on a theme. Annals of Medicine. 1998;20:151–158. doi: 10.3109/07853899808999398. [DOI] [PubMed] [Google Scholar]
- Vidaver GA. Inhibition of parallel flux and augmentation of counter flux by transport models not involving a mobile carrier. Journal of Theoretical Biology. 1966;10:301–306. doi: 10.1016/0022-5193(66)90128-7. [DOI] [PubMed] [Google Scholar]
- Wang H, Elferich L, Gouaux E. Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nature Structural & Molecular Biology. 2012;19:212–219. doi: 10.1038/nsmb.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmasta A, Ressler R, Gouaux E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503:141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Gouaux E. Substrate binds in the S1 site of the F253A mutant of LeuT, a neurotransmitter sodium symporter homologue. EMBO Reports. 2012;13:861–866. doi: 10.1038/embor.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Choe S, Chaptal V, Rosenberg JM, Wright EM, Grabe M, et al. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature. 2010;468:988–991. doi: 10.1038/nature09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennogle LP, Meyerson LR. Serotonin modulates the dissociation of [3H] imipramine from human platelet recognition sites. European Journal of Pharmacology. 1982;86:303–307. doi: 10.1016/0014-2999(82)90333-8. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, et al. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–113. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, et al. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reup-take. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressants specificity of serotonin transporter suggested by three LeuT-SSRI complex structures. Nature Structural & Molecular Biology. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI. Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature. 2007;449:726–730. doi: 10.1038/nature06133. [DOI] [PubMed] [Google Scholar]