

Abstract
Background
A variety of NADPH oxidase (Nox) isoforms including Noxs 1, 2, 4 and 5 catalyze the formation of reactive oxygen species (ROS) in the vascular wall. The Nox2 isoform complex has arguably received the greatest attention in the progression of atherogenesis in animal models. Thus, in the current study we postulated that specific Nox2 oxidase inhibition could reverse or attenuate atherosclerosis in mice fed a high-fat diet.
Methods
We evaluated the effect of isoform-selective Nox2 assembly inhibitor on the progression and vascularization of atheromatous plaques. Apolipoprotein E-deficient mice (ApoE−/−) were fed a high fat diet for two months and treated over 15 days with Nox2ds-tat or control sequence (scrambled); 10 mg/ kg/day, i.p. Mice were sacrificed and superoxide production in arterial tissue was detected by cytochrome C reduction assay and dihydroethidium staining. Plaque development was evaluated and the angiogenic markers VEGF, HIF1-α and visfatin were quantified by real time qRT-PCR. MMP-9 protein release and gelatinolytic activity was determined as a marker for vascularization.
Results
Nox2ds-tat inhibited Nox-derived superoxide determined by cytochrome C in carotid arteries ( protein; P < 0.01) and caused a significant regression in atherosclerotic plaques in aorta (66 ± 6 μm2 vs 37 ± 1 μm2; scrmb vs. Nox2ds-tat; P < 0.001). Increased VEGF, HIF-1α, MMP-9 and visfatin expression in arterial tissue in response to high-fat diet were significantly attenuated by Nox2ds-tat which in turn impaired both MMP-9 protein expression and activity.
Conclusion
Given these results, it is quite evident that selective Nox inhibitors can reverse vascular pathology arising with atherosclerosis.
Keywords: Vascular Nox, Vulnerable plaque, Oxidative stress
1. Introduction
Chronic vascular inflammation, an important feature of atherosclerosis, propagates vasculopathy in large conduit vessels exhibiting atherosclerotic plaque formation [1]. This process is accelerated by the adhesion, migration and accumulation of lymphocytes and monocytes into the vessel wall [2]. In atherosclerosis, inflammatory leukocyte recruitment into the vessel wall is facilitated by the increasingly dense, wide and arborized vasa vasorum, whose development seems to originate via the recruitment of vascular progenitor cells in the vascular adventitia [3,4]. The concept that neovascularization from the vessel wall may play a key role in the pathophysiology of atherosclerosis was theorized over a century ago [5]. Experimental evidence associating angiogenesis in atherosclerotic plaque with a more unstable and progressive atherosclerotic disease has been highlighted by the fact that neointimal microvessel may increase delivery of cellular and soluble lesion components to the vessel wall [6]. The vasa vasorum constitutes a network of microvasculature that originates primarily in the adventitial layer of large arteries. The adventitia is where formation and regression of microvessels that penetrate and nourish the media and intima are controlled [7]. Considerable interest has been focused on the study of the formation of these microvessels and its role in atherosclerotic plaque. Recent evidence suggests a key role for reactive oxygen species (ROS) in these processes [8,9]. In most blood vessels the main source of ROS appears to be the NADPH oxidase (Nox) family of proteins (Noxs 1, 2, 4 and 5) variably expressed by vascular endothelial cells, smooth muscle cells and adventitial fibroblasts [10]. ROS are known players involved in the initiation and progression of cell proliferation and migration. Changes in cellular biochemistry, such as levels of inflammatory marker molecules and redox imbalance in reducing systems, have fundamental importance in the atherogenic process [11,12]. Antioxidants, like polyphenols [13], and agents such as apocynin [14] (albeit non-specific) that disrupt ROS production derived from NADPH oxidase, reverse vascular remodeling, improve endothelial function and reduce inflammation. Coronary artery disease (CAD) is associated with increased NADPH oxidase subunit expression, mainly p22phox and Nox2, related in part to higher monocyte/macrophage infiltration [8]. Indeed, a study by Barry-Lane et al. supports the latter, that is, deletion of essential Nox2 oxidase subunit p47phox prevents the progression of plaque formation in ApoE−/− mice [15]. In an attempt to gain maximal selectivity for some Nox isoforms, development of peptidic inhibitors targeted at disrupting assembly of Nox complexes has garnered significant interest [16]. Nox2ds-tat, a cell-permeant inhibitor targeting the assembly of Nox2, is a peptide that binds to the p47phox subunit and prevents its key interaction with the core membrane-integrated cytochrome b558 protein Nox2 [17]. Nox2ds-tat also blocked angiotensin II (AngII)-induced superoxide production in human resistance artery smooth muscle cells [18]. Many other studies have proven its efficacy to inhibit Nox2 in disrupted and intact cells as well as whole tissue and animal models [18–21]. Thus, we hypothesized that specific Nox2 inactivation would arrest atheroma plaque progression and instability and thus reverse the dangers of an unstable plaque. For that purpose, this study was designed to evaluate the ability of Nox2ds-tat peptide to disrupt the progression and vascularization of atheroma plaque in apolipo-protein E-deficient (ApoE−/−) mice and address the underlying mechanism, focusing on the association between inflammation, oxidative stress and accelerated atherosclerosis.
2. Materials and methods
2.1. Ethical approval
All animals were cared for in accordance with the Guiding Principles in the Care and Use of Animals of the US National Institutes of Health (NIH). All procedures were approved by the Animal Research Committee of the National University of Cuyo (protocol approval #8/2012 CICUAL, School of Medical Science, Mendoza, Argentina).
2.2. Animal model
Male C57/BL6J ApoE−/− mice 8 weeks of age (20–22 g; The Jackson Laboratories, Bar Harbor, ME) were used for this study. Mice had unrestricted access to water and standard chow (GEPSA, Argentina) and were maintained on a 12 h light/dark cycle under pathogen-free conditions in the Genetically Modified Animal Lab, School of Medical Science, National University of Cuyo. Mice were randomly divided into four groups (n = 4–6 mice each) and fed a control diet (Control), high fat diet (HFD) (15.8% fat, 1.25% cholesterol; Gepsa, Argentina) for eight weeks, HFD + Nox2 inhibitor peptide (a chimeric 18-amino acid peptide ([H]-R-K-K-R-R-Q-R-R-R-C-S-T-R-I-R-R-Q-L- [NH2]) [18] that has been shown to inhibit NADPH oxidase activity in vivo and in vitro) and HFD plus control sequence (scrambled). Peptides (10 mg/kg/day) were injected i.p. over a 2 week period.
2.3. Biochemical determinations
After overnight fasting, blood samples for glucose, triglycerides, and cholesterol determinations (GT Lab, Buenos Aires, Argentina) were drawn from all animals, collected from cardiac puncture under anesthesia at the end of the experimental period.
2.4. Determination of tissue Nox-derived superoxide and vascular oxidant level
Tissue was calculated from the initial linear rate of SOD-inhibitable cytochrome C reduction quantified at 550 nm using the extinction coefficient of 21.1 mM−1 cm−1 as previously described [22]. Briely, isolated carotid arteries were homogenized in buffer containing 8 mM potassium, sodium phosphate buffer, pH 7.0, 131 mM NaCl, 340 mM sucrose, 5 mM MgCl2, 1 mM EGTA, and protease inhibitors (Roche); and centrifuged at 10,000 rpm for 15 min at 4 °C to remove unbroken cells, nuclei and debris. The assay of the supernatant was carried out using acetylated cytochrome C (0.2 mM, Sigma–Aldrich), in buffer containing catalase (300 U/ml) to prevent re-oxidation of reduced cytochrome C by H2O2. An identical set of samples was incubated in the presence of SOD (150 U/ml) for subtraction of the SOD-inhibitable signal. After 5 min baseline measurement, NADPH (180 μM) was added and production was measured at 550 nm using a UV–visible spectrophotometer (Mutiskan FC, Thermo Fisher). Superoxide production is expressed as nmoles protein using the extinction coefficient.
In situ vascular superoxide anion was approximated using dihydroethidium (DHE) as previously described [23]. Briefly, freshly isolated vascular segments (aortas and carotid arteries) from each treatment were embedded in tissue freezing medium and 6 μM cryosections were mounted on cover slips. DHE dissolved in PBS to final concentration of 5 μmol/l was added and incubated at 37 °C in dark for 30 min. Images were captured with a fluorescent microscope and three to six images were acquired from three sections per vessel ring for each experimental condition (n = 4 in each experimental group). Images were analyzed with ImageJ 1.37v software (NIH) and changes in total fluorescence intensity were calculated relative to control.
2.5. Quantification of atherosclerotic plaques
Plaque area was quantified by Oil-red-O staining of lipid deposits. Vessels were incubated for 45 min with Oil red O (0.5% in 60% isopropyl alcohol). Excess stain was removed with 60% isopropyl alcohol. Quantification of atherosclerosis was performed in the aortic arch region up to the descending abdominal aorta by computer-assisted image analysis. Subsequently images of en face preparations of the whole mounted aorta were taken and the percentage of plaques in relation to the entire aortic surface calculated as plaque score in percent of total area using ImageJ 1.37v software (NIH).
2.6. Quantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated with the Trizol (Invitrogen) method from carotid tissue pools (6 per group), 1 μg of total RNA was reverse-transcribed, using random primer hexamers (Biodynamics, SRL) and M-MLV reverse transcriptase (Promega), according to the manufacturer's instructions. Real-time PCR was performed with the cDNA samples, primers (Invitrogen) and EVA Green (GenBiTech, Argentina) by using a Rotor-Gene 6000 Series Software version 1.7 (Corbett). All samples were amplified in triplicate. The relative changes in the amount of transcripts in each sample were determined by normalizing with the actin RNA levels.
2.7. Western blotting
Western blot analysis of MMP-9 expression was performed on carotid tissue homogenates from 4 animals per group using rabbit-anti MMP-9 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), and HRP-conjugated secondary antibodies (1:10,000 Jackson laboratory, US). Proteins were visualized by performing luminol-enhanced chemiluminescence. Loading of equal amounts of proteins was confirmed by re-probing the membrane with an antibody against β-actin (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). Protein expression was quantified using ImageJ 1.37v software (NIH).
2.8. MMP-9 Activity by gelatin zymography
Soluble protein extracts were obtained by pulverizing pooled carotid arteries (n = 4) from each treatment in liquid nitrogen, placed in lysis buffer without protease inhibitors and centrifuged at 21,000 g for 20 min at 4 °C. Protein quantification was performed by the Bradford method and samples were diluted with a buffer containing 12.5% 0.5M Tris HCl pH 6.8, 10% glycerol, 4% SDS, and 0.05% bromophenol blue, without mercaptoethanol to avoid denaturation and to maintain biological activity. Gelatin zymography is mainly used for the detection of the gelatinases, MMP-2 and MMP-9, respectively. The assay is highly sensitive because levels of 10 pg of MMP-9 can be detected [24]. It was performed together with molecular weight markers using 8% SDS-polyacrylamide separating gels with 1% of gelatin (a suitable substrate for MMP-2 and MMP-9), 30% bis-acrylamide, 1.5M tris-HCL pH 8.8, 10% ammonium persulfate, 0.04% TEMED, 10% SDS. Electrophoresis was carried at 4 °C using a constant current of 20 mA during 3 h. Gels were washed twice in 2% Triton-x 100 for 15 min and three times with distilled water, incubated in buffer (Tris 0.25 M, CaCl2 25 mM, NaCl 1M, pH 7.6) for 48 h at 37 °C, stained with 0.1% Coomassie brilliant blue, 40% methanol and 10% acetic acid, for 1 h, under gentle shaking, and de-stained for 20 min with 25% ethanol/8% acetic acid solution. Gels were photographed and the enzyme gelatinolytic activity was determined by densitometry and analyzed with ImageJ Software. Collagenase was used as a positive control.
2.9. Data analyses
All values are expressed as means ± S.E.M. Comparisons across the four groups were analyzed using a one-way ANOVA, followed by a Bonferroni's post-hoc test to determine significant differences between two groups (Prism v4, GraphPad). Differences between groups with a P-value <0.05 was considered statistically significant.
3. Results
3.1. Biochemical parameters
Metabolic characteristics for each experimental group are outlined in Table 1. Glycemia and dyslipidemia were largely unaffected by treatment with Nox2 inhibitor.
Table 1.
Average plasma glucose and lipid levels in ApoE-KO mice fed with high fat diet and treated with Nox2ds-tat peptide or scrambled.
| Control Chow (baseline) | HFD | HFD + Scrambled | HFD + Nox2ds-tat | Pa | |
|---|---|---|---|---|---|
| Fasting Glucose (mmol/l) | 7.6 ± 0.7 | 8.0 ± 0.5 | 6.5 ± 0.7 | 7.4 ± 0.4 | 0.08 |
| Total Cholesterol (mg/dl) | 521 ± 20 | 908 ± 75a | 932 ± 138a | 1017 ± 20a | < 0.0001 |
| Triglycerides (mg/dl) | 135 ± 10 | 210 ± 25a | 209 ± 30a | 187 ± 16a | 0.0011 |
P vs Control Chow (baseline), n = 5.
3.2. Vascular superoxide anion measurement and NADPH oxidase gene expression
Nox-derived superoxide anion ( ) determined by cytochrome C in homogenized carotid tissue from ApoE−/− mice on a high fat diet, significantly increased compared to control diet, and chronic treatment with Nox2ds-tat, 10 mg/kg*d, significantly reduced this effect (Fig. 1A). We evaluated in carotid arteries the mRNA levels of core subunits Nox1, Nox2, and Nox4 and observed that Nox2ds-tat treatment induce a moderate decrease in the expression of Nox1 which did not reach significance, and did not cause any change in the expression of the other core vascular isoforms, i.e. Nox2 and Nox4 (Fig. 1B). However, Nox2ds-tat significantly inhibited mRNA expression of both p47phox and p22phox, compared with scrambled control. Next, we confirmed an increased vascular superoxide anion level in situ in aortic sections from HFD-treated ApoE−/− mice injected with control peptide, using redox-sensitive dihydroethidium (DHE). Fig. 2 shows the qualitative analysis of DHE staining. Findings in aortic tissue from HFD-ApoE−/− mice treated with Nox2ds-tat are consistent with a decreased oxyethidium signal compared with aortas from HFD-ApoE−/− mice treated with scrambled peptide. Similar results were found in carotid artery cryosections (data not shown).
Fig. 1.

Effect of Nox2ds-tat peptide on Nox-derived superoxide ( ) and NADPH oxidase components expression: (A) Nox-derived superoxide was measured by cytochrome C reduction assay in homogenates of carotid arteries from ApoE−/− mice fed a control diet or high fat diet (HFD) for 8 weeks and treated with Nox2ds peptide or scrambled peptide the final 2 weeks. Values are mean ± SEM (n = 4), **P < 0.001 vs control chow; §P < 0.01 vs scrambled (B) Quantitative real-time RT-PCR show a significant decrease in expression levels of p47phox and p22phox in carotid tissue from ApoE−/− mice treated with Nox2ds peptide compared with ApoE−/− mice treated with scrambled (*P = 0.02, **P = 0.01 vs scrambled). A slight but not significant decrease in Nox1 expression is also observed in ApoE−/−mice treated with Nox2ds peptide.
Fig. 2.

Effect of Nox2ds-tat peptide on superoxide production: Immunofluorescence images of DHE-stained fresh-frozen sections of ApoE−/− arteries. Animals were treated with Control diet or HFD plus peptides. Results are typical of staining of sections from four different aortas in each group. Digital scans of intimal regions of DHE stained aortas were quantified using ImageJ (NIH) software. Results shown are mean ± SEM. *P < 0.05 vs control; §P < 0.05 vs HFD + scrambled.
3.3. Assessment of atherosclerotic lesions in the aorta
Eight weeks of high fat diet caused an increase in atherosclerotic plaques within the aorta as assessed by Oil Red O staining (Fig. 3). Two weeks subsequent treatment with Nox2ds-tat significantly reduced lesion area in aortas from ApoE−/− mice in comparison with aortas scrambled-treated ApoE−/− mice. Similar results were found in carotid arteries cryosections (data not shown).
Fig. 3.

Effect of Nox2ds-tat peptide on atherosclerotic lesion in ApoE−/− mice Fed with High-fat Diet. Atheroma development in the whole aorta (aortic arch up to abdominal aorta) was quantified by computerized morphometry. ApoE−/− mice were fed control diet or HFD during 8 weeks and treated with Nox2ds peptide or scrambled during last 2 weeks. Arteries were stained with Oil-red O and results represent the area of lesion (μm2 × 103) relative to total aortic surface (n = 6 mice per group). P < 0.001 vs HFD+ Nox2ds peptide.
3.4. Effect of Nox2 inhibition on pro-angiogenic mediators
Shared angiogenic and oxidative mechanisms underlie the pathophysiology of atherosclerosis. Thus, we sought to investigate the mRNA expression of pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and hypoxia-inducible factor-1α (HIF-1α) in ApoE−/− mice treated with Nox2ds-tat. We found a significant increase in both VEGF and HIF-1α expression in carotid arteries from high fat fed-ApoE−/− mice (Table 2), and treatment with Nox2ds-tat significantly reduced the expression of both angiogenic mediators.
Table 2.
Fold differences in gene expression of pro-angiogenic markers.
| Markers | Fold increase
|
||
|---|---|---|---|
| Control chow | HFD + Scrambled | HFD + Nox2ds-tat | |
| VEGF | 1.0 ± 0.20 | 11.1 ± 0.1*** | 7.8 ± 0.2***,# |
| HIF-1a | 1.0 ± 0.04 | 2.74 ± 0.15** | 1.6 ± 0.1*,# |
| VISFATIN | 1.0 ± 0.01 | 3.66 ± 0.52** | 0.66 ± 0.02# |
| MMP-9 | 1.0 ± 0.03 | 1.69 ± 0.14** | 0.64 ± 0.04# |
mRNA expression was analyzed in carotic artery wall from ApoE-KO mice (n = 4) treated with control diet, or high fat diet (HFD) plus peptides, Scrambled or Nox2ds-tat. Gene expression level of the measured genes in control diet was set at 1. The corresponding ΔCt values are means ± SEM.
P< 0.01;
P < 0.001;
P < 0.0001 vs control diet;
P < 0.01 vs HFD + Scrambled.
Visfatin, an adipokine that possesses angiogenic properties, can induce VEGF and VEGF receptor 2 (VEGFR2) expression by activating both the PI3K/Akt and the ERK1/2 signaling cascades [25], and enhanced VEGF signaling results in increased expression of metalloproteinases (MMP) which promote many of the adverse structural changes associated with plaque vulnerability. Based on these findings, we decided to quantify visfatin and MMP-9 gene expression in our model. We found a significant reduction in mRNA expression of visfatin and MMP-9 expression in carotid artery from HFD treated-ApoE−/− mice treated with Nox2ds-tat (Table 2) suggesting that Nox2 inhibition reverses neovascularization induced by a high fat diet.
3.5. Effect of Nox2 inhibition on the stability of the atheroma
We set out to investigate the effect of Nox2 inhibition on plaque stability by analyzing MMP-9 expression, by Western blot and determining MMP-9 activity by gelatin zymography. Nox2ds-tat significantly inhibited HFD-simulated MMP-9 protein expression (Fig. 4A). MMP-9 activity in homogenates of carotid arteries was enhanced by a fat-rich diet and this effect was inhibited by Nox2ds-tat (Fig. 4B) suggesting that Nox2 inhibition could help to maintain the stability of atherosclerotic plaques by decreasing the factors involved in the breakdown of the atheromatous plaque.
Fig. 4.
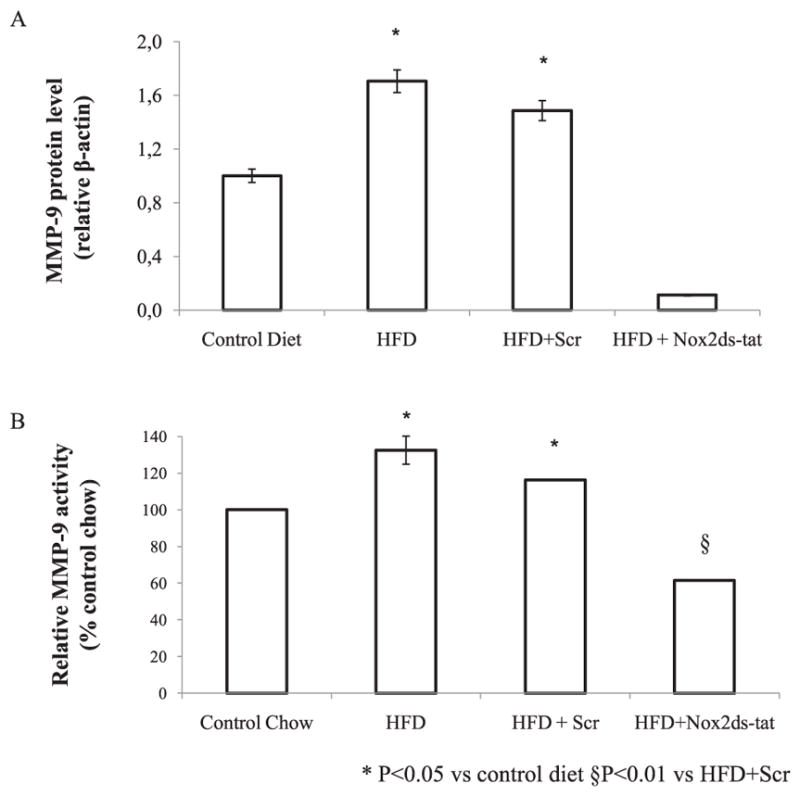
Effect of Nox2ds-tat peptide on protein expression and gelatinolytic activity of MMP-9. (A) Expression of MMP-9 determined by Western Blot in carotid arteries homogenates from ApoE−/ − ice (n = 4 each group) fed with control diet, HFD during 8 weeks and treated with Nox2ds peptide or scrambled during last 2 weeks. β-actin protein was used as control for relevant quantification. Results are means ± S.E.M, from two independent experiments (Density plot for n = 2 experiments) *P < 0.05 vs control diet. (B) MMP-9 activity was measured by gelatin zymography in carotid arteries homogenates from the same groups mentioned above. The enzyme gelatinolytic activity was determined by densitometry and analyzed with ImageJ Software. Collagenase was used as a positive control. Results, expressed as % relative control, are means ± S.E.M, from two independent experiments. *P < 0.05 vs control diet §P < 0.01 vs HFD + Scr.
4. Discussion
Increased cellular oxidative stress has been proposed to contribute to plaque progression leading to plaque instability. Previous studies in human indicate that Nox2 hereditary deficiency is associated with reduced atherosclerotic burden [26] and in this “human knockout” model of NADPH oxidase, arterial dilatation was also enhanced [27], thus providing evidence that this ROS-generating pathway is implicated in modulating arterial tone and in human atherosclerosis. Interestingly patients with p47phox hereditary deficiency had intermediate flow-mediated dilation and oxidative stress [28]. Changes in the NADPH oxidase system may contribute to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques [29], and new therapeutic strategies for cardiovascular disease based on prevention of ROS production, include the development of less toxic and more selective NADPH oxidase inhibitors, allowing clarification of the role of each NADPH oxidase isoform and its potential use in clinical practice [16]. In this study we sought to investigate whether specific inhibition of NADPH oxidase decreases the progression of atheromatous plaques and reduces the pro-angiogenic mediators that lead to increase plaque risk for rupture. Nox2ds-tat is an efficacious inhibitor of Nox2-NADPH oxidase and although as a peptide in its native state it has limited oral bioavailability, parenteral delivery methods are effective at reducing vascular pathologies associated with increased ROS production [30]. Furthermore, Nox2ds-tat is an isoform-specific inhibitor as evidenced by its lack of inhibition of ROS production in either the COS-Nox1 (canonical or hybrid), the COS-Nox4 oxidase systems or xanthine oxidase [31]. In our study, we demonstrated that the administration of Nox2ds-tat to ApoE−/− mice fed a Western type diet resulted in a significantly reduced in Nox-derived superoxide in the vascular wall, with associated downregulation of the expression of membrane-bound subunit p22phox and the cytosolic p47phox, a key interaction in the assembly of the active NADPH oxidase complex (the p47phox bis-SH3 domain and a proline-rich region present on the carboxy terminal of p22phox [32]). Although transcriptional regulation of Nox subunits by oxidants has not been an extensive focus of investigation, these data could suggest a feed forward transcriptional activation of Nox subunits by reactive oxygen species. Most importantly, the decrease in oxidative stress in the arterial wall led to a reduction of atheroma development in aorta and carotid arteries from high-fat fed ApoE−/− mice, suggesting an atheroprotective effect of this peptide. Sibley et al. reported reduced carotid but not coronary artery atherosclerosis in patients with chronic granulomatous disease (CGD) who have an immunodeficiency of the phagocyte NADPH oxidase and a concomitant reduction in reactive oxygen species [33]. CGD raises questions about the role of NADPH oxidase in the pathogenesis of clinically significant atherosclerosis. We focused on the effect of Nox2 inhibition on the progression (and regression) and vascularization of atheromatous plaques. The enlargement of the atheroma provokes intra-plaque hypoxia that triggers inflammatory cell infiltration, thus promoting local neo-vascularization. Interestingly, although intimal thickening is believed to be an early surrogate marker for atherosclerosis, pathological neovascularization is implicated in both early and late stages of the disease [34]. Angiogenesis is increased in lipid-rich plaque and neovascularization has been linked to the progression and vulnerability of atherosclerotic lesions [35]. HIF-1α is a key transcriptional regulator responding to hypoxia and activating genes, which promote angiogenesis, among them VEGF. Based on those data, we explored gene expression of different pro-angiogenic factors in carotid arteries homogenates. We found that high fat diet elevates the expression of VEGF and HIF-1α and that Nox2ds-tat treatment reduces this effect significantly. This is in accordance with previous report that showed that Nox2-containing NADPH oxidase deficiency (Nox2-deficient mice) protects against age-dependent impairment of neovascularization [36]. We also detected the expression of others genes associated with VEGF signaling, such as visfatin and metalloproteinases (MMP)-9. Both proteins displayed parallel overexpression in inflammatory cells, especially in the neovascularized plaque lesions. An intense expression of VEGF and MMP-9 in carotid plaques is related to plaque instability, high degree of stenosis and presence of symptomatic carotid occlusive disease [37]. On the other hand, Adya et al. demonstrate that the adipokine visfatin can induce endothelial cell (EC) proliferation and angiogenesis by stimulating the VEGF system. In particular, they showed that visfatin induces VEGF and VEGFR2 and enhanced VEGF signaling results in increased expression of MMP-2 and MMP-9 [25]. Visfatin plays important roles in angiogenesis, and was also shown to induce the expression of HIF1α and MMP production and activity [38,39]. This adipokine should be regarded as an inflammatory mediator, localized to foam cell macrophages within unstable atherosclerotic lesions, that potentially plays a role in plaque destabilization [40]. Interestingly, we found in our study that Nox2ds-tat administration significantly reduced the expression of visfatin and MMP-9 and specifically inhibited the gelatinolytic activity of MMP-9 which would be expected to result in decreased plaque vulnerability.
Inflammation and oxidative stress are closely associated with the activation of MMPs, which induces extracellular matrix degradation, resulting in the development and progression of atherosclerosis and plaque instability. Thus, MMPs are pivotal downstream components in the pathogenesis of atherosclerosis. Macrophages, which are abundant in ruptured atherosclerotic plaques, express multiple MMPs, weakening the plaque and making it ruptures prone [41]. Among MMPs, MMP-9 (92-kDa gelatinase B), which is a prevalent isozyme expressed by activated macrophages and foam cells, is found in human coronary atherosclerotic lesions [42]. Therefore, attenuation of macrophage infiltration may largely contribute to the decreased MMP activity.
Our findings are ostensibly in contrast with a recent experimental study in Nox2-deficient mice in which MMP-9 activity in the adventitia, but not media, was upregulated in aggravated abdominal aortic aneurysm (AAA) lesions, suggesting that a deficiency in Nox2 markedly perturbs proinflammatory cascades in macrophages present in AAA [43]. Thus, it is important to carefully monitor inflammatory phenotype when Nox2 intervention is conducted and this issue will also need to be investigated in humans.
In summary, we demonstrated for the first time to our knowledge that Nox2ds-tat, the inhibitor of Nox2 oxidase, exerts a significant protective effect against the progression of atherosclerosis by mechanisms including inhibition of oxidative stress and angiogenic mediator downregulation, resulting in regression of atheromatous plaques. This salutary effect appears to also decrease plaque neovascularization linked to the vulnerability of atherosclerotic lesions.
Acknowledgments
Funding
This work was supported by grant 06/J359 (to C Castro), 2011–2013 from Secretary of Science and Technology, National University of Cuyo, and PIP-CONICET 2012–2014; NIH grants R01HL079207, R01HL112914, and P01HL103455-04 to PJP.
AL (doctoral fellows) from National University of Cuyo and IQ (post-doctoral fellow) from CONICET.
The authors gratefully acknowledge the technical assistance of Mrs. María Teresa Lopez.
Abbreviation
- ApoE−/−
ApoE-deficient mice
- NADPH
nicotinamide adenine dinucleotide phosphate
- ROS
reactive oxygen species
- VEGF
vascular endothelial growth factor
- HIF
hypoxia inducible factor
- MMP-9
metalloproteinase 9
Footnotes
Conflict of interest
All authors disclose no conflict of interest including any financial, personal or other relationships with other people or organizations that could inappropriately influence, or be perceived to influence, their work.
References
- 1.Rosenfeld ME. Inflammation and atherosclerosis: direct versus indirect mechanisms. Curr Opin Pharmacol. 2013;13(2):154–160. doi: 10.1016/j.coph.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legein B, Temmerman L, Biessen EA, Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. 2013;70(20):3847–3869. doi: 10.1007/s00018-013-1289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watanabe M, Sangawa A, Sasaki Y, Yamashita M, Tanaka-Shintani M, Shintaku M, Ishikawa Y. Distribution of inflammatory cells in adventitia changed with advancing atherosclerosis of human coronary artery. J Atheroscler Thromb. 2007;14(6):325–331. doi: 10.5551/jat.e489. [DOI] [PubMed] [Google Scholar]
- 4.Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, Xu Q. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. 2004;113(9):1258–1265. doi: 10.1172/JCI19628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulligan-Kehoe MJ. The vasa vasorum in diseased and nondiseased arteries. Am J Physiol Heart Circ Physiol. 2010;298(2):H295–H305. doi: 10.1152/ajpheart.00884.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyle B, Caplice N. Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J Am Coll Cardiol. 2007;49(21):2073–2080. doi: 10.1016/j.jacc.2007.01.089. [DOI] [PubMed] [Google Scholar]
- 7.Ritman EL, Lerman A. The dynamic vasa vasorum. Cardiovasc Res. 2007;75(4):649–658. doi: 10.1016/j.cardiores.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guzik B, Sagan A, Ludew D, Mrowiecki W, Chwala M, Bujak-Gizycka B, Filip G, Grudzien G, Kapelak B, Zmudka K, Mrowiecki T, Sadowski J, Korbut R, Guzik TJ. Mechanisms of oxidative stress in human aortic aneurysms – Association with clinical risk factors for atherosclerosis and disease severity. Int J Cardiol. 2013 Oct 3;168(3):2389–2396. doi: 10.1016/j.ijcard.2013.01.278. http://dx.doi.org/10.1016/j.ijcard.2013.01.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zampetaki A, Dudek K, Mayr M. Oxidative Stress in atherosclerosis: The Role of microRNAs in Arterial Remodelling. Free Radic Biol Med. 2013 Sep;64:69–77. doi: 10.1016/j.freeradbiomed.2013.06.025. http://dx.doi.org/10.1016/j.freeradbiomed.2013.06.025. [DOI] [PubMed] [Google Scholar]
- 10.Violi F, Basili S, Nigro C, Pignatelli P. Role of NADPH oxidase in atherosclerosis. Future Cardiol. 2009;5(1):83–92. doi: 10.2217/14796678.5.1.83. [DOI] [PubMed] [Google Scholar]
- 11.Uno K, Nicholls SJ. Biomarkers of inflammation and oxidative stress in atherosclerosis. Biomark Med. 2010;4(3):361–373. doi: 10.2217/bmm.10.57. [DOI] [PubMed] [Google Scholar]
- 12.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14(1–2):70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kostyuk VA, Potapovich AI, Suhan TO, de Luca C, Korkina LG. Antioxidant and signal modulation properties of plant polyphenols in controlling vascular inflammation. Eur J Pharmacol. 2011;658(2–3):248–256. doi: 10.1016/j.ejphar.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Yue Q, Belcik T, Xie A, Inaba Y, McCarty OJ, Tormoen GW, Zhao Y, Ruggeri ZM, Kaufmann BA, Lindner JR. Molecular imaging of inflammation and platelet adhesion in advanced atherosclerosis effects of antioxidant therapy with NADPH oxidase inhibition. Circ Cardiovasc Imaging. 2013;6(1):74–82. doi: 10.1161/CIRCIMAGING.112.975193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J Clin Invest. 2001;108(10):1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodino-Janeiro BK, Paradela-Dobarro B, Castineiras-Landeira MI, Raposeiras-Roubin S, Gonzalez-Juanatey JR, Alvarez E. Current status of NADPH oxidase research in cardiovascular pharmacology. Vasc Health Risk Manag. 2013;9:401–428. doi: 10.2147/VHRM.S33053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini DJ, Egana L, Lopes LR, Jackson HM, Kelley EE, Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51(6):1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89(5):408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92(6):637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23(5):776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 21.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90(11):1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 22.Molshanski-Mor S, Mizrahi A, Ugolev Y, Dahan I, Berdichevsky Y, Pick E. Cell-free assays: the reductionist approach to the study of NADPH oxidase assembly, or “all you wanted to know about cell-free assays but did not dare to ask”. Methods Mol Biol. 2007;412:385–428. doi: 10.1007/978-1-59745-467-4_25. [DOI] [PubMed] [Google Scholar]
- 23.Cannizzo B, Quesada I, Militello R, Amaya C, Miatello R, Cruzado M, Castro C. Tempol attenuates atherosclerosis associated with metabolic syndrome via decreased vascular inf;ammation and NADPH-2 oxidase expression. Free Radic Res. 2014;48(5):526–533. doi: 10.3109/10715762.2014.889295. [DOI] [PubMed] [Google Scholar]
- 24.Snoek-van Beurden PA, Von den Hoff JW. Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. BioTechniques. 2005;38(1):73–83. doi: 10.2144/05381RV01. [DOI] [PubMed] [Google Scholar]
- 25.Adya R, Tan BK, Punn A, Chen J, Randeva HS. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008;78(2):356–365. doi: 10.1093/cvr/cvm111. [DOI] [PubMed] [Google Scholar]
- 26.Violi F, Pignatelli P, Pignata C, Plebani A, Rossi P, Sanguigni V, Carnevale R, Soresina A, Finocchi A, Cirillo E, et al. Reduced atherosclerotic burden in subjects with genetically determined low oxidative stress. Arterioscler Thromb Vasc Biol. 2013;33(2):406–412. doi: 10.1161/ATVBAHA.112.300438. [DOI] [PubMed] [Google Scholar]
- 27.Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, et al. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120(16):1616–1622. doi: 10.1161/CIRCULATIONAHA.109.877191. [DOI] [PubMed] [Google Scholar]
- 28.Loffredo L, Carnevale R, Sanguigni V, Plebani A, Rossi P, Pignata C, De Mattia D, Finocchi A, Martire B, Pietrogrande MC, et al. Does NADPH oxidase deficiency cause artery dilatation in humans? Antioxidants Redox Signal. 2013;18(12):1491–1496. doi: 10.1089/ars.2012.4987. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Chamseddine AH, Carrell S, Miller FJ., Jr Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2014;2:642–650. doi: 10.1016/j.redox.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cifuentes-Pagano E, Csanyi G, Pagano PJ. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci. 2012;69(14):2315–2325. doi: 10.1007/s00018-012-1009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Csanyi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, Pagano PJ. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol. 2012;32(12):2966–2973. doi: 10.1161/ATVBAHA.112.300031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibley CT, Estwick T, Zavodni A, Huang CY, Kwan AC, Soule BP, Long Priel DA, Remaley AT, Rudman Spergel AK, Turkbey EB, et al. Assessment of atherosclerosis in chronic granulomatous disease. Circulation. 2014;130(23):2031–2039. doi: 10.1161/CIRCULATIONAHA.113.006824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaipersad AS, Lip GY, Silverman S, Shantsila E. The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol. 2014;63(1):1–11. doi: 10.1016/j.jacc.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 35.Hutter R, Speidl WS, Valdiviezo C, Sauter B, Corti R, Fuster V, Badimon JJ. Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1alpha activation: a novel aspect of angiogenesis in atherosclerosis. J Cardiovasc Transl Res. 2013;6(4):558–569. doi: 10.1007/s12265-013-9469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turgeon J, Haddad P, Dussault S, Groleau J, Maingrette F, Perez G, Rivard A. Protection against vascular aging in Nox2-deficient mice: impact on endothelial progenitor cells and reparative neovascularization. Atherosclerosis. 2012;223(1):122–129. doi: 10.1016/j.atherosclerosis.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Papalambros E, Sigala F, Georgopoulos S, Panou N, Kavatzas N, Agapitos M, Bastounis E. Vascular endothelial growth factor and matrix metalloproteinase 9 expression in human carotid atherosclerotic plaques: relationship with plaque destabilization via neovascularization. Cerebrovasc Dis. 2004;18(2):160–165. doi: 10.1159/000079736. [DOI] [PubMed] [Google Scholar]
- 38.Park JW, Kim WH, Shin SH, Kim JY, Yun MR, Park KJ, Park HY. Visfatin exerts angiogenic effects on human umbilical vein endothelial cells through the mTOR signaling pathway. Biochimica Biophysica Acta. 2011;1813(5):763–771. doi: 10.1016/j.bbamcr.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Sun Z, Lei H, Zhang Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013;24(5):433–442. doi: 10.1016/j.cytogfr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, Damas JK, Tunheim SH, Ueland T, Smith C, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation. 2007;115(8):972–980. doi: 10.1161/CIRCULATIONAHA.106.665893. [DOI] [PubMed] [Google Scholar]
- 41.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005;3(1):63–68. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 42.Kwon SH, Ju SA, Kang JH, Kim CS, Yoo HM, Yu R. Chemokine Lkn-1/ CCL15 enhances matrix metalloproteinase-9 release from human macrophages and macrophage-derived foam cells. Nutr Res Pract. 2008;2(2):134–137. doi: 10.4162/nrp.2008.2.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kigawa Y, Miyazaki T, Lei XF, Nakamachi T, Oguchi T, Kim-Kaneyama JR, Taniyama M, Tsunawaki S, Shioda S, Miyazaki A. NADPH oxidase deficiency exacerbates angiotensin II-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2014;34(11):2413–2420. doi: 10.1161/ATVBAHA.114.303086. [DOI] [PubMed] [Google Scholar]