

Abstract
Oxidative stress-related diseases underlie many if not all of the major leading causes of death in United States and the Western World. Thus, enormous interest from both academia and pharmaceutical industry has been placed on the development of agents which attenuate oxidative stress. With that in mind, great efforts have been placed in the development of inhibitors of NADPH oxidase (Nox), the major enzymatic source of reactive oxygen species and oxidative stress in many cells and tissue. The regulation of a catalytically active Nox enzyme involves numerous protein-protein interactions which, in turn, afford numerous targets for inhibition of its activity. In this review, we will provide an updated overview of the available Nox inhibitors, both peptidic and small molecules, and discuss the body of data related to their possible mechanisms of action and specificity towards each of the various isoforms of Nox. Indeed, there have been some very notable successes. However, despite great commitment by many in the field, the need for efficacious and well-characterized, isoform-specific Nox inhibitors, essential for the treatment of major diseases as well as for delineating the contribution of a given Nox in physiological redox signalling, continues to grow.
Keywords: NADPH oxidase, Nox, inhibitors, peptides, small molecules, therapeutics
INTRODUCTION
ROS and Disease
Oxidative stress is implicated as a common underpinning in hypertension [1], cancer [2], diabetes [3], ischemia reperfusion injury [4], neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease [5] to name a few. As these diseases underlie many if not all of the major leading causes of death in United States [6], and the Western World, enormous interest from both academia and pharmaceutical industry, has been placed on the development of agents which attenuate oxidative stress. This attenuation can be either via the agent’s role as antioxidants or as inhibitors of enzymatic sources implicated in altering the redox state within cells and tissues. Oxidative stress is a term describing a shift towards a pro-oxidative cell or tissue state whereby reactive oxygen (ROS) and nitrogen species (RNS) overwhelm antioxidant defense mechanisms. The latter serve to (a) scavenge excessive ROS and repair attendant damage by such excess; and/or (b) maintain reduced (GSH)/oxidized (GSSG) glutathione ratios responsible to preserve adequate reducing equivalents for overall cell function as well as key antioxidant and non-antioxidant enzymes. While multiple enzymatic sources are capable of generating ROS, a wide consensus in the literature accepts that the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (Nox) family are major “professional” producers of ROS and linked to the aforementioned and many other pathologies [1,2,5,7–17] An extensive discussion of the important roles of ROS from a variety of other subcellular sources can be found in more comprehensive reviews elsewhere [1,18,19].
Nox Enzymes
NADPH oxidases (Noxs) are considered “professional” ROS-producing enzymes as their primary, defined function is the generation of superoxide and/or hydrogen peroxide (H2O2) via the controlled transfer of electrons from NADPH to molecular oxygen by way of flavin adenine dinucleotide (FAD)-binding and NADPH-binding sites on the enzymes’ C-terminal tail. Importantly, the Nox family of functionally- and structurally-related enzyme systems is comprised of seven members; namely Nox1 through 5 and DUOX1 & 2. Interestingly, these isoforms differ in their tissue distribution, level of expression, nature of ROS produced, and control by distinct signaling modulators. Of these, Nox2, which is present in neutrophils and macrophages, was the first to be discovered [20,21] and is the most thoroughly characterized isoform. As the structure, localization and activation mechanisms for the Nox family members have been the subject of numerous in-depth reviews, [4,22] they will not be mentioned in detail here, except to give the reader a deeper perspective of the complexity of interactions required for a fully functional enzyme. This perspective will then inform the reader of the wide variety of strategic interventions that are plausible for Nox inhibition.
All Nox isoforms are characterized by a catalytic core, consisting of a bis-heme-containing transmembrane domain (6 – 7) and a C-terminal intracellular dehydrogenase tail that carries the required sites for FAD and NADPH binding. p22phox, a membrane-bound component present in Nox1-, Nox2, Nox3 and Nox4 complexes (but not in those of Nox5, Duox1 or Duox2), stabilizes its Nox counterpart and serves as a docking site for other regulatory subunits depending on the particular Nox system. [23]. These other regulatory subunits can act as organizers (targeting other subunits to the membrane) or as activators (directly modulating catalytic activity). The active Nox2 oxidase system comprises the Nox2 subunit and p22phox(membranal) as well as cytosolic p47phox (organizer), p67phox(activator), p40phox and the small Rho-family GTP-binding protein Rac2 and also Rac1 [24,25]. Similarly, the active Nox1 system is comprised of membrane-bound Nox1 and p22phox and in its generally accepted, canonical complex, of organizing subunit NoxO1 (homolog of p47phox), activating subunit NoxA1 (homologue of p67phox), and Rac1 [26]. Increasing evidence supports a non-canonical-Nox1 system which utilizes p47phox, in lieu of NoxO1, for activation [27] in vascular smooth muscle cells. As it is for Nox1, evidence supports the notion that the rodent Nox3 oxidase system requires NoxO1, NoxA1, and Rac1 besides its core membranal Nox3 and p22phox subunits [28] while human Nox3 has been reported to be activated by NoxO1 alone [29]. On the other hand, p47phox and p67phox apparently can, in certain settings, supplant the role of NoxO1 and NoxA1 in Nox3 oxidase albeit to lesser effect [30,31]. To our knowledge, Nox3 expression is restricted to the inner ear and some fetal tissues [28]. In contrast, the Nox4 isozyme includes the Nox4 subunit and p22phox, but the only other reported protein to our knowledge to modulate its function is Poldip2 [32]. Another differential feature of Nox4 is that is reportedly constitutively active and preferentially produces H2O2 over superoxide anion [33]. Incidentally, Nox4 has been proposed as an oxygen sensor [33]. Nox5 and DUOXs 1and 2 are distinct from Noxs 1 through 4 as they putatively do not require p22phox for membrane stabilization and are regulated by calcium binding to EF-hand motifs present in their N-terminal calmodulin homology domains [34–36]. Interestingly, Duox1 and Duox2 have an extra membrane-spanning domain with a peroxidase-like domain in their extracellular N-terminal region. Processing of Duox1 and 2 involving endoplasmic reticulum-to-Golgi transition, maturation, and translocation to the plasma membrane requires the presence of DUOXA1 and DUOXA2, respectively, to constitute a fully functional H2O2-generating enzyme [37].
As described above, the assembly and regulation of a catalytically active Nox system involves numerous protein-protein interactions [38–40]. Depicted in Figure 1 are common sites of interaction between the individual components of an active Nox enzyme. In the case of the Nox2 isozyme, a key interaction exists between a pro-line-rich domain (PRD) on p22phox and the bis-Src Homology 3 (SH3) domains of the organizer p47phox(Fig. 1: #1)[41,42]. p47phox also interacts with the SH3 domains from the activator p67phox through its C-terminal PRD region and through an additional surface, a helix-turn-helix motif, downstream from PRD (Fig. 1: #2)) [43–46]. As an organizer, p47phox binds to the C-terminus of Nox2 through its polybasic region (Fig. 1: #3) [47]. On the other hand, p67phox is able to interact with Nox2 C-terminus (Fig. 1: #4) [48,49] and with Rac (Fig. 1: #5) [50]. Intramolecular interactions within individual Nox components important for enzymatic activity have also been described (Fig. 1: #6). For example, in dormant phagocytes the SH3 domains of p47phox interact with its auto-inhibitory region (AIR) [42,51]. This intramolecular interaction blocks the SH3 domains’ interaction with other subunits. It is only upon serine phosphorylation by protein kinase C that these SH3 domains are unmasked and enzyme activation proceeds. In the case of the canonical Nox1 system, similar protein-protein interactions have been demonstrated for NoxO1 and p22phox. However, the interaction between NoxO1 and NoxA1 were shown to be significantly different from that between their homologous subunits in Nox2 in that the surface of interaction between both proteins seems to have lower affinity and to involve only the helix-turn-helix motif of NoxO1 interacting with the SH3 domain of NoxA1 [52]. This difference may offer insight into functional differences between the Nox1 and 2 isoforms. In addition, Yamamoto et al., demonstrated that interaction of NoxO1 and NoxA1 is further regulated by phosphorylation of T341 of NoxO1 that in turn increases Nox1 activity [53]. Intramolecular interactions within NoxO1 are also distinct from its counterpart, p47phox. That is, NoxO1’s auto-inhibitory potential at its C-terminal region is less pronounced, which may allow for the observed higher basal Nox1-derived superoxide anion generation [52]. Intramolecular interactions within the Nox anchoring subunits have also been described for Nox4 and Nox2 in which the polybasic region in the second intracellular loop, loop B, putatively binds to the DH-domain linking the heme-binding transmembrane domain with the FAD- and NADPH-binding domains. Thus, it is proposed that this intramolecular folding facilitates electron transfer and superoxide anion production [54]. In the case of Nox5, whose B-loop does not contain a polybasic region, the intramolecular interaction involves region in the EF-hands domain and a regulatory EF-hand-binding domain within the DH domain [55].
Fig. (1).

Sites of interaction among Nox subunits.
Finally, protein-protein interactions between the membrane-spanning subunits have also been described (Fig. 1: #7). For example, Ambasta et al., using fluorescence resonance energy transfer techniques, demonstrated that p22phox forms complexes with Nox1, Nox2, and Nox4 disrupted by mutation of histidine 115 [56]. Differential interactions between Nox4 and p22phox have also been shown by mutation analysis [57]. Notwithstanding the need for specific Nox enzyme inhibitors for diseases in which oxidative stress is a causative factor, Nox isozymes have emerged as pivotal to homeostatic redox signaling. Thus, a better understanding of the aforementioned interactions is expected to provide key insight into modalities to “tweak” Nox activity in the positive or negative sense. Indeed, Noxs play an important role in signal transduction, i.e. participating in pathways including ERK1 and ERK2, NF-kB, JNK, and others leading to salutary and/or compensatory phenotypes [58,59]. Our growing appreciation of Noxs modulating fundamental physiological processes has shed important new light on the complex yet elemental significance of this oxidase family of enzymes [60].
History of Nox Inhibitors
Our understanding of the contribution of any given Nox isoform to a specific signaling pathway has been limited by the lack of isoform-selective inhibitors. The utility of inhibitors to gain insight into NADPH oxidase functionality was realized early in the discovery of the respiratory burst enzyme, now known as Nox2 oxidase [61,62]. Among the first Nox inhibitors to appear in the literature in the late 1980s through the 1990s were small molecule iodonium compounds [63,64], apocynin [65], AEBSF [66], S17834 [67], and the peptidic inhibitors PR-39 [68]. Soon thereafter, a first-in-class Nox inhibitor was designed to specifically inhibit Nox2 [69,70] and named gp91ds-tat, now named Nox2ds-tat, which will be discussed in more detail below. At the time of introduction of these early inhibitors to the field, other members of the Nox family had yet to be identified. Thus, it is not surprising that more thorough investigations of their mechanism of action and specificity since have in most cases revealed significant off-target effects.
Some of the characteristics of those early inhibitors have been reviewed previously [71–77]. The advent of the discovery of a 7-member family of NADPH oxidases ushered in an urgency to find selective inhibitors of each isozyme, be they peptidic or small molecule. It is the goal of this review to survey the literature and categorize each identified inhibitor by its effects on individual Nox enzymes and perhaps better appreciate the need for isoform specificity.
PEPTIDIC NOX INHIBITORS
The more information that is obtained in terms of the key regulatory regions for assembly and activation of the Nox protein and its specific subunits, the more rationally peptidic inhibitors can be designed. Below we describe a few promising peptidic inhibitors available to date with special emphasis on those which exemplify strategies for inhibition of activity and exploit defining characteristics of each of the target enzymes (Fig. 2). Importantly, some of these strategies include peptides replicating amino acid sequences within the Nox subunit itself, which are pivotal for the assembly of active enzyme (Fig. 2A). Others include peptides derived from one of the cytosolic subunits required for activity (Fig. 2B), and peptides mimicking regions within the Nox subunit and key to processes such as auto-inhibition (Fig. 2C). Fundamentally, these rationally-designed inhibitors take advantage of intrinsic regulatory interactions of the Nox.
Fig. (2).

Mechanism of action of peptidic inhibitors.
Inhibitors of Nox2
A wealth of information has been amassed over the years by the Pick, Jesaitis and Quinn laboratories on the structure and mechanisms associated with Nox2 activation. This is attributable largely to the development of peptidic inhibitors, many of which were identified using phage display[78,79] and peptide walking [80,81]. A wide spectrum of peptides capable of inhibiting Nox2 activity in vitro has been identified based on sequences from Nox2 or p47 phox proteins or from p22phox sequences and tested with respect to Nox2 activity [82]. A small subset of these peptides has been characterized in terms of their specificity and await testing of their effectiveness in vitro and in vivo. For detailed analysis of these peptides, please refer to outstanding reviews by El-Benna and coworkers [83,84].
PR-39
An endogenous proline-arginine (PR)-rich antibacterial peptide, PR-39 (RRR PRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP), was first described as an inhibitor of Nox2 based on the knowledge that the assembly of phagocyte NADPH oxidase required protein-protein interactions between SH3 domains of one and proline-rich domains (PRDs) of other Nox subunits. In fact, as shown by Shi et al. [68], PR-39 inhibited phagocytic Nox2 in whole cells and in cell-free preparations presumably by binding to SH3-domain in p47 phox and interfering with its binding to the PRDs within the C-terminus of p22phox. Indeed, PR-39 exerted cardioprotective effects following ischemia reperfusion which often involves activation of Nox [85]. However, further analysis demonstrated that this peptide could bind to other proteins besides p47phox, including other SH3-containing proteins, p130Cas [86] and PI3Kp85α [87]. Thus, careful interpretation of the results obtained using PR-39 is required and its use as a Nox2 inhibitor is recommended with consideration of potential off-target effects.
Nox2-Based Peptides
A thorough study by Dahan et al. [88] employing peptide walking analysis within the dehydrogenase region of Nox2 identified 10 clusters of inhibitory peptides that, in turn, helped to define ten functional domains in the C-terminal region of Nox2. Two of these domains, 288FWRSQQKVVITKVVT302 (cluster A) and 312MKKKG FKMEVGQYIF326 (cluster B), are found in the N-terminal region of Nox2 dehydrogenase domain. One, 348EDFFSIHIRIVGDWT362 (cluster C), in the ribityl chain-binding FAD subdomain. Four clusters overlapped portions of three NADPH-binding subdomains, that is peptides 393TASEDVFSYEVVMLV407 (cluster D) and 414TPFAS ILKSVWYKYC428 (cluster E) located to the pyrophos -phate-binding subdomain, while 432TNLKLKKIYFYWLCR446 (cluster F) and 528PNTRIGVFLCGPEAL542 (cluster H) contain residues required for the binding of ribose and nicotinamide moieties of NADPH, respectively. Cluster F1 (447DTHAFEWFA455) is contiguous to Cluster F, so it probably participates also in the binding of the ribose moiety. Peptide 468RNNAGFLSYNIYLTG482 (cluster G) corresponds to a region of unknown functional importance. Peptide 552SNSESGPRGVHFIFN566 (cluster I) was located in the C-terminal region of the enzyme and overlaps with a peptide previously described as binding to p47phox[47]. Interestingly, the pep-tides corresponding to FAD- and NADPH-binding regions did not exhibit the expected competitive kinetics regarding their respective substrates and were also able to inhibit enzymatic activity when added after complex assembly, suggesting a more complex mechanism of action. The validity of these peptides as isoform-specific Nox2 inhibitors remains to be determined since they primarily target regions involved in catalysis shared by all members of the Nox family.
Nox2ds-tat
Nox2ds-tat, designed in our laboratory, appears to distinguish itself from other peptide inhibitors as the only isoform-selective Nox inhibitor currently available. Nox2ds-tat was designed based on data from random-sequence peptide phage display showing that peptides corresponding to the B-loop of the Nox2 catalytic core, a short loop between helix 2 and 3, exhibited inhibitory activity in cell-free activity assays [79].

This together with the knowledge of a small 9-aa peptide derived from the HIV viral coat (HIV-tat) [89] capable of delivering conjugated proteins across membranes, gave rise to the design of an 18-mer chimeric peptide, Nox2ds-tat (originally named gp91ds-tat) [70] (sequence above; for mechanism of action see Fig. 2A). To date, Nox2ds-tat having been used in an array of experimental models to investigate the role of Nox2, appears to be the best characterized and most widely-used Nox2-selective inhibitor in the field. From the outset, this peptide was shown to be highly efficacious at inhibiting Nox2-derived ROS in vitro as well as in vivo [70,90,91] and from then on has been employed in a range of cell and animal models of disease. The ROS-generating stimuli that can be blocked by Nox2ds-tat include nutrient deprivation [92], hypoxia [93] atrial natriuretic peptide [94] angiopoietin-1 [95], interleukin-4 [96], shear stress [97], calcineurin inhibitors [98], endothelin-1 [99], TNF-α [100], and phenylephrine [101] to name a few. Nox2ds-tat also blocked angiotensin II (AngII)-induced superoxide production in human resistance artery smooth muscle cells [102] and collagen-induced Nox2 activity in platelets [103]. Furthermore, Nox2ds-tat has been applied to elucidate the involvement of Nox2 in various processes leading to function and dysfunction of various organ systems, e.g. hypertension [104], diabetes [14], retinopathy [93], Alzheimer’s disease [105] and aging [106]. Importantly, the specificity of Nox2ds-tat was rigorously tested utilizing heterologous cell-free systems expressing the classical Nox2 (p22phox, Nox2, p47phox, and p67phox), canonical Nox1 (p22phox, Nox1, NOXA1, and NOXO1), or Nox4 (p22phox and Nox4) oxidases, as well as the non-canonical Nox1 (p22phox, Nox1, p47phox, and NoxA1) system [107]. The findings demonstrated that Nox2ds-tat inhibits the canonical Nox2 system, but not Nox1 (canonical or non-canonical) or Nox4-derived ROS production. The IC50 calculated for Nox2 inhibition was 0.74µM, thus demonstrating that Nox2ds-tat is a moderately potent, efficacious, and selective inhibitor of Nox2. The mechanism of action of Nox2dstat seems to involve binding to p47phox as shown by enzyme-linked immunosorbent assay using biotinylated peptide [107].
Inhibitors of Nox1
NoxA1ds
In the past two years another isoform-specific inhibitor of Nox was developed, in this case the targeted enzyme being Nox1 [108]. NoxA1ds (NoxA1 docking sequence) is a peptide that mimics a putative activation domain of the human Nox1 activator subunit NOXA1 homologous to a reported p67phox activation domain that spans amino acids 199–210 and participates in the catalytic reduction of FAD [48].

With the knowledge that mutagenesis of a tyrosine for an alanine at residue 199 of NoxA1 in a region corresponding to the defined “activation domain” of its homolog p67phox reduced Nox1-derived superoxide production by >95% [109], it was postulated that the same substitution would render a peptide as an effective inhibitor of Nox1 activity. To confer specificity, amino acids flanking this putative activation domain (that were not conserved between NOXA1 and p67phox) were included. NoxA1ds potently inhibited Nox1-derived ROS production in a reconstituted, heterologous Nox1 cell-free system (IC50 = 19 nM), displaying no inhibitory effect on Nox2-, Nox4-, Nox5-, or xanthine oxidase activity [108]. Further, FRET, FRAP, and competitive binding analyses were consistent with NoxA1ds eliciting its effect by binding to Nox1 and thus blocking its interaction with NoxA1 (Fig. 2B). NoxA1ds significantly inhibited whole HT-29 carcinoma cell-derived ROS and hypoxia-induced human pulmonary artery endothelial cell ROS production and migration [108]. It seems likely, although it has not been tested, that the unmutated NoxA1 peptide could have an inhibitory effect on Nox1 activity. Recently, in an attempt to define the sites of interaction between Nox1 and NoxA1, Streeter et al. [110] used a NoxA1-AD peptide (LEPMDFLGKAKVV) which corresponds to the rat sequence overlapping NoxA1ds and showed interaction of NoxA1 with T429-phosphorylated Nox1 using computational modeling. In that study, NoxA1-AD peptide showed higher affinity to a phosphorylated form of a Nox1 peptide (KLKTQKIYF) at T429 compared to its non-phosphorylated counterpart.
Inhibitors of Nox4
Elegant studies by the Knaus and Lambeth laboratories supported key interactions in the Nox4 complex that could be exploited as targets [54,57]. A systematic screening of synthetic peptides, encompassing various regions on Nox4 protein including B-loop and C-terminus, as well as peptides mimicking p22phox regions thought to be essential for activity, yielded no inhibitor for Nox4. Thus, similar strategies to those used for peptidic inhibitors of Nox1 and 2 did not prove successful [111]. Those results could suggest that Nox4 exists in a tightly-assembled and active conformation which, unlike other Noxs, cannot be disrupted by conventional means.
Inhibitors of Nox5
In contrast to Nox1, 2, 3 and 4, Nox5 does not require other membrane-associated or cytosolic activator proteins for its activity [34,112] but its activation is mediated by an increase in cytosolic Ca2+ concentration acting on EF-hand motifs.
Pep1 and Pep3
In an attempt to investigate how the N-terminal region of Nox5 containing the EF domain, interacts with its C-terminal catalytic dehydrogenase domain (CDHD) leading to activation of the enzyme, Tirone et al. [55] were able to narrow the site of interaction to a region between two NADPH binding sites in the C-terminal portion of Nox. Interestingly, specific peptides (Pep1 and Pep3) targeting this regulatory calcium-binding domain of NOX5 were capable of blocking Ca2+-dependent superoxide generation in a dose-dependent manner (IC50 = 30 µM).

It is noteworthy that Pep2 and Pep4 interacted very weakly or not at all with the Nox5-EF domain, as evidenced by pull-down assays, despite the fact that they overlapped substantially with Pep1 or Pep3. In fact, Pep4 and Pep1 differ by only 5 amino acid residues, emphasizing the importance of the KDSIT stretch for Nox5-EF/CDHD interaction [55]. Although the inhibitory activity of Pep1 and 3 was only tested in Nox5 activity assays, it is not expected that they would block the activity of other Noxs due to (a) the absence of EF-hands; and (b) poor homology of aligned corresponding sequences for Nox 1, 2, 3, and 4 to that of Nox5. Still, direct testing of specificity remains to be performed. Purportedly, these peptides may represent a site in the catalytic domain of Nox5 that has an auto-inhibitory role [55] (Fig. 2C).
Melittin
Melittin is a 26 amino acid peptide (GIGAVLKVLTTGLP ALISWIKRKRQQ) derived from bee venom that has demonstrated potent Nox5 inhibition displaying an IC50 of 101 nM. The binding was shown to be Ca2+-dependent and it seems to act through direct binding with the EF-hand domains [112]. Melittin, however, is not specific to Nox5 but also binds to other Ca2+-dependent proteins such as calmodulin and troponin C [113].
Inhibitors of Nox3, DUOX1 and 2
To our knowledge, no peptidic inhibitors have yet been developed to specifically target Nox3 or DUOX1 and 2.
Afterthoughts
Despite the general potential for specific targeting and effectiveness of biological inhibitors, such as peptides, few have entered the clinical pipeline thus far, and none of those are Nox inhibitors [114,115]. The use of peptides as potent inhibitors of enzymatic activities raises concerns, mainly due to the preconceived notion that peptides lack therapeutic potential due to poor oral bioavailability, gut degradation, and toxicity. However, as Dahan & Pick [116] elegantly submit, those limitations can be minimized by strategic design and identification of appropriate sequences. Then, optimization of peptide ADME (absorption, distribution, metabolism, and excretion) properties and means of delivery could circumvent the limitations and improve therapeutic use [117]. Some examples of these attempts include peptide modifications that provide protection from protease degradation by stapling or partial substitution of L-amino acids with their D-isomers [118,119]. In terms of targeted peptide delivery, unpublished studies by our group targeting pulmonary diseases, indicate that aerosolization of NoxA1ds directly into the nasal passages and lungs, markedly reduces right ventricular hypertrophy in rodent models of pulmonary hypertension.
SMALL MOLECULE INHIBITORS
Selective, potent, and efficacious inhibition of Nox isozymes remains a major challenge in the discovery of small molecules with potential for progress towards therapeutic development. As our understanding of the Nox enzymes improves, we anticipate multiple methods of inhibition will become available, thereby opening the door to different classes of inhibitors, for example, catalytic core manipulation versus protein assembly inhibition. In contrast to peptidic inhibitors the strategy used to identify small molecule inhibitors is usually less borne out of rational targeting of protein-protein interactions than it is the high throughput screening of small molecules libraries. This requires a careful and thoughtful workflow of specific assays and counter screens to select the most specific hits [76,120]. Once a hit is identified, random modification of elements of the parent molecule may be pursued. Increased use of computational modeling facilitates the rational design of lead molecules.
Inhibitors of Nox2
The first group of compounds initially identified as Nox2 inhibitors using a high throughput screening strategy were the Vaso-pharm triazolo pyrimidine derivatives, VAS2870 [121] and VAS3947 [122]. Interestingly, VAS2870 inhibited superoxide production from cell-free Nox2 containing neutrophil system with an IC50 of 10.6 µM [121] and completely abolished oxLDL-induced Nox-derived ROS in human umbilical vein endothelial cells (HU-VECs) at 10 µM [123]. Moreover, the more soluble VAS3947 showed efficacy similar to that of VAS2870 in cell-based assays [122]. That said, the VAS compounds are now commonly regarded as pan-Nox inhibitors due to their ability to completely inhibit ROS production in multiple agonist-induced cell models with varied Nox expression [124,125]. Structurally, VAS compounds share a central nucleotide core and it is therefore predicted that these compounds interact at the level of nucleotide binding, i.e. NADPH or FAD. This would, in fact, explain their lack of isoform specificity and also their ability to inhibit the pre-assembled Nox2 [126]. Recent report have shed light on the negative aspects of using VAS2870 as a Nox inhibitor, demonstrating off-target effects through thiol alkylation [127] and inhibition of mitochondrial respiration and cytotoxicity [120]. More detailed binding and mutagenesis assays are required to fully elucidate the mode of inhibition of VAS compounds for Nox2.
Another group of compounds suggested to possess Nox2 inhibiting capabilities are the pyrazolopyrimidine compounds [128]. This group of molecules inhibited Nox activity in bovine aortic endothelial cell membrane fractions with an IC50 < 1 µM and inhibited extracellular superoxide formation by intact PMA-stimulated human neutrophils [126]. However, the authors also noted that these compounds failed to produce a decrease in Nox2-derived superoxide in a reconstituted neutrophil system. Consequently, the mechanism of reduced ROS generation was demonstrated via potent inhibition of protein kinase C (PKC)-βII, a key regulator of p47 phox phosphorylation. Thus, until further information becomes available, the use of pyrazolpyrimidine compounds as Nox2-targeted inhibitors should be discouraged.
Previously, the triterpenoid celastrol [129] originally referred to as an “antioxidant” was demonstrated to potently inhibit both Nox1-derived ROS (IC50 = 0.41 ± 0.20 µM) and Nox2-derived ROS (IC50 = 0.59 ± 0.34 µM) following PMA stimulation [130]. While the efficacy for both the canonical Nox1 and Nox2 systems was greater than the non-canonical Nox1 and 2 systems, the mode of inhibition is demonstrated as disrupting interaction of the proline rich region of p22phox and the tandem SH3 domains of either p47 phox or NoxO1. Structure-activity relationship (SAR) analogues are anticipated to generate a new class of competitive Nox inhibitors acting specifically at the level of p47phox-p22phox vs. NoxO1-p22phox interface. Whether this potential new class of compounds would discriminate between Nox1 vs Nox2 is still unknown.
A new group of Nox2 inhibitors identified to competitively inhibit the p47phox-p22phox binding interface are ebselen and its analogues. These compounds were identified utilizing a fluorescence polarization-based binding assay [131], as well as traditional ROS read-out assays. Importantly, this line of inquiry provides a rigorous methodology for the investigation of potential compounds impinging on protein-protein interactions. In the initial study, ebselen and some of its analogs were documented as potent Nox2 inhibitors which affected Nox1, 4, and 5 activities at a substantially lower potency depending upon the congener [131]. One derivative, JM-77b, had a selectivity for Nox2 (IC50= 0.4 µM) compared to Nox1 (IC50= 6.3 µM), Nox5 (IC50= 17 µM), and Nox4 (no significant inhibition) [131]. This class of compounds appears to hold significant promise for selective Nox2 inhibition. Mindful of ebselen’s reported glutathione peroxidase-like activity [132], congeners of ebselen found to be devoid of this activity should be of considerable interest.
Naloxone, a commonly-used antagonist of opioid receptors, was previously characterized as highly effective in preventing dopaminergic degeneration in different models of rodent Parkinson’s disease by inhibiting inflammation [133]. Importantly, it was shown that naloxone binds to Nox2 and blocks translocation of p47 phox to the plasma membrane, thus inhibiting ROS generation [134]. Naloxone’s IC50 was in the range of 2µM and inhibited the activity of the pre-assembled Nox2 enzyme [134]. However, no reports could be found disproving its binding or inhibition of other Nox subunits. Similarly, perhexiline, an approved prescription drug for angina, was recently demonstrated to inhibit neutrophilic Nox2 with an IC50 of 1.5–3.6 µM as well as in various vascular cells [135]. Importantly, perhexiline displayed no scavenging ability or xanthine oxidase inhibition. Its mechanism of action and its effect on other Nox isoforms requires further investigation.
Our group recently identified bridged tetrahydroquinolines as specific Nox2 inhibitors based on cellular assays employing heterologous Nox1,Nox2, Nox4, and Nox5 expressing systems [136]. In this report, compounds 11g and 11h showed selective inhibition of Nox2 in intact Cos-Nox2 cells stimulated with PMA (IC50 = 20 ± 1.9 and 32 ± 1.9 µM, respectively). Importantly, these compounds were unable to inhibit ROS production by Nox1-, Nox4-, and Nox5-expressing cells and exhibited no free radical scavenging or xanthine oxidase inhibitory activity. Although limited mechanistic information exists pertaining to their mode of inhibition within the current scientific literature, unpublished work employing modeling suggests a potential role as assembly inhibitors, acting on the p47phox-p22phox interface. That said, there is still considerable work to be done in the way of SAR to enhance potency, minimize potential off-target effects, and hence tissue and whole animal toxicity. Nevertheless, we contend that compounds 11g and 11h hold significant promise as prototype selective small molecule inhibitors of Nox2.
Inhibitors of Nox1
Given the promiscuous nature of the Nox1 oxidase as a canonical- or non-canonical-Nox1 system which utilizes p47phox, small molecule inhibitors intended for the canonical Nox2, p47phox-dependent enzyme could have effects on the non-canonical-Nox1 system (e.g. celastrol and ebselen derivatives) [75]. In that regard, a deeper understanding of the Nox1 activation process and potential distinct p47phox interactions with Nox1 versus Nox2 would be advantageous.
ML171 (2-acetylphenothiazine) belongs to a subset of phenothiazines and has an IC50 for Nox1 in the submicromolar range from cell-based assays, namely 0.129 µM in HT29 cells and 0.25 µM in a HEK293-Nox1 reconstituted cell system [137]. Moreover, the specificity of ML171 for other Noxs was reportedly >20-fold higher for Nox2, −3, and 4 as well as for xanthine oxidase compared to Nox1[137]. Importantly, the suggested mechanism of action surrounding ML171 was interaction with Nox1 catalytic subunit as only over-expression of Nox1, and not NoxA1 nor NoxO1, was able to restore the ML171-dependent inhibition of ROS generation in HEK293-Nox1-expressing cells [137]. However, by the same reasoning, ML171’s potential effect as an allosteric inhibitor of Nox1 at a region distinct from the catalytic site could also have been superseded by Nox1 overexpression.
A high-throughput screening (HTS) approach of small molecule libraries and subsequent optimization of lead compounds enabled the identification of dual Nox1/4 inhibitors by GenKyoTex (GKT) [138,139]. Among these compounds are GKT136901 and GKT137831, which belong to a pyrazolopyridine class of compounds, and are the first orally-active dual Nox1 and 4 inhibitors [140–142]. The first compound, GKT136901, was found to be a Nox inhibitor with a relatively high degree of potency for both Nox4 (inhibitory constant (Ki) = 165 ± 5 nM) and Nox1 (Ki =160 ± 10 nM). Similarly, the second compound, GKT137831, was shown to be a highly potent inhibitor of both human Nox4 (Ki =140 ± 40 nM) and human Nox1 (Ki=110 ± 30 nM). Indeed, these compounds are in phase II trials for the treatment of diabetic nephrophathy, which based on animal models, is thought to involve Nox4 but not Nox1[143]. One potential cause for concern is that Nox4 has been implicated broadly in cell differentiation and physiologic redox signaling and the drug may have deleterious effects in non-diseased organs. Additionally, GKT compounds were demonstrated to be a viable therapeutic for the treatment of idiopathic pulmonary fibrosis and liver fibrosis, both diseases with implicated Nox4 and/or Nox1 contributions [138,140]. GKT compounds are to date the best-characterized small molecule Nox inhibiting compounds, having been subjected to extensive in vivo screening analysis for off target effects, reportedly having favorable ADME profiles as well as being orally bioavailable [138,140]. It is expected that the mechanism of action of the GKT compounds could involve catalytic core modulation due to the lack of extensive protein assembly in Nox4 (see below). GKT compounds only exhibit 10–15-fold higher selectivity for Nox1/4 than Nox2. Therefore, consideration of the administration of high doses (>50mg/kg) in vivo [141] should be taken as these could obviate selectivity.
As alluded to previously, the inhibitors VAS2870 and VAS3947, considered pan-Nox inhibitors, are able to inhibit Nox-derived ROS production in multiple agonist-induced cell models.. Consequently, as with the GKT compounds, VAS compounds may interact at the level of nucleotide binding due to their lack of isoform specificity and also their ability to inhibit the pre-assembled Nox [126]. Further, ebselen and analogues are also predicted to inhibit Nox1-derived superoxide, especially when the non-cano-nical-Nox1 system is present or the agonist used favors p47phox-dependent Nox1 activation. In a similar vein, celastrol, being a recognized assembly inhibitor acting at the level of p47phox/NoxO1 and p22phox, potently inhibits Nox1-derived superoxide anion as discussed above.
Inhibitors of Nox3
The current biochemical literature surrounding the activation process of Nox3 remains limited. Moreover, the current dogma for cytosolic subunit preference and interchangeability of p47 phox for NoxO1 and p67phox for NoxA1 in Nox3 activation renders creation of protein assembly inhibitors a challenge. Thus, it is anticipated that the p47phox/NoxO1 class of assembly inhibitors e.g. celastrol and ebselen derivatives, could have an effect on Nox3. To date, no specific small molecule inhibitors to our knowledge exist for Nox3-dependent superoxide production despite its role in balance regulation, otoconia biosynthesis, and potentially hearing loss [28]. However, one report did identify a novel synthetic compound, 3-amino-3-(4-fluorophenyl)-1H–quinoline-2,4-dione (KR-22332) that significantly inhibited cisplatin-induced intracellular ROS generation in vitro and was protective against cisplatin-induced hearing loss in a rat model involving Nox3 ROS production [144]. Since no biochemical analysis or modeling was performed, it is not clear if these effects were elicited via catalytic inhibition or by other means. Furthermore, ML171 is also able to inhibit Nox3-dependent superoxide with an IC50 of 3 µM [137]; and further studies are required to determine the effects of other currently-available Nox inhibitors on the Nox3 system. Taken together, it is not outside the realm of possibilities that drugs that were designed or screened for other Noxs will exhibit an interesting profile of Nox3 inhibition.
Inhibitors of Nox4
To date, Nox4 transcriptional expression remains the most accepted mechanism for increased Nox4-derived ROS in cells and tissues [145,146]. Importantly, the interaction between Nox4 and p22phox appears to be tightly regulated as peptidic inhibitor strategies failed to affect Nox4 activity [111]. Despite this observation, a number of currently available inhibitors are reported to potently and effectively attenuate Nox4-derived ROS although often via unspecified mechanisms. As mentioned previously (for more detail, see Nox1 section) GKT compounds represent the first orally-available phase-II clinical trial candidates as dual Nox1/4 inhibitors for the treatment of diabetic nephropathy promoted in rodent models by Nox4 [147]. Emerging evidence, however, implicates Nox5 in human glomerulopathies [148]. These compounds also decrease angiogenesis and tumor volume, and improve diabetes-accelerated atherosclerosis, for which Nox1 vs. Nox4 are attributed, respectively [149,150].
Similarly, VAS3947, ML171 and celastrol exhibit Nox4 inhibition at higher concentrations than their intended Nox1 & 2 targets (see Table 1). Previously, triphenylmethane dyes, such as brilliant green and gentian violet, which have chemical characteristic resembling DPI, were shown to be potent and efficacious inhibitors of both Nox2 and Nox4 activity [151]. Therefore, from a structure-based approach, fulvene and its derivatives were generated as a new class of Nox2/4 inhibitors. Importantly, fulvene-5 at 5 µM equally inhibited Nox2 and Nox4 in vitro, and when applied in vivo, successfully blocked the growth of endothelial-mediated tumors in mice [152]. However, since the process of tumor angiogenesis is suggested to be a Nox4-driven mechanism, most likely it is the ability of fulvene-5 to inhibit Nox4 that confers its efficacy in vivo. This contention is buttressed by studies showing Nox4 shRNA produces similar effects [152]. That being said, fulvenes should be classified as putative inhibitors until which time a mechanism of action is identified and their possible role as scavengers can be ruled out. Finally, the recently-identified diarylheptanoid group of compounds obtained from edible plants yielded a compound named ACD084, which inhibited Nox4 with an IC50 of 3 µM without inhibition of either Nox2 or Nox5 [153]. Moreover, while other diaryl-heptanoids inhibited Nox4-derived ROS, they also were also capable of inhibiting Nox2. However, limited scavenging analysis was performed on the identified compounds and the authors reportedly did not investigate the effects on Nox1-derived superoxide. While mechanistic information is lacking for the mode on Nox4 inhibition by ACD084, it is predicted to interfere with nucleotide binding given its ability to inhibit ROS production from purified Nox4-dehydrogenase domain samples [153]. It remains to be determined whether this could also be true for the other Nox isoforms.
Table 1.
Isoform specificity of selected peptides and small molecule inhibitors of NADPH oxidase.
| Name | Structure | IC50s (µM) (* = Ki in µM) | Suggested Mecha- nism of Inhibition |
Refer- ences |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nox1 | Nox2 | Nox3 | Nox4 | Nox5 | Duox1 | Duox2 | XO | |||||
| Peptides | PR-39 | RRRPRPPYLPRPRPPPFFPPR LPPRIPPGFPPRFPPRFP |
↓ | Binding to SH3 domain of p47phox |
[68,85] | |||||||
| Nox2ds-tat | RKKRRQRRRCSTRIRRQL | ns | 0.74 | ns | ns | ns | p47phox-p22phox interface |
[69,70, 107] |
||||
| Cluster E | 414TPFASILKSVWYKYC428 | 8.4 | NADPH-binding site |
[88] | ||||||||
| Cluster I | 552SNSESGPRGVHFIFN566 | 5.38 | NK | [88] | ||||||||
| NoxA1ds | EPVDALGKAKV | 0.02 | ns | ns | ns | ns | Nox1-NoxA1 interface |
[108] | ||||
| Pep1 | DMKAIGLQMALDLLANKEKKDSITY | 30 | EF-hand-AID interface |
[55] | ||||||||
| Pep3 | RFLELHMYMTSALGKNDMKAIGL QMALDLLANKEKKDSIT |
30 | EF-hand-AID interface |
[55] | ||||||||
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ | 0.1 | EF-hand | [113] | ||||||||
| Small molecules | VAS2870 | 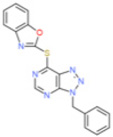 |
↓ | 10.6 (Neutrophils) 2 (HL-60) |
↓ | NK | [121–123] | |||||
| VAS3947 | 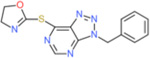 |
↓ | 1–2 (HL-60 cells) |
↓ | ns | NK | ||||||
| Fulvene-5 | 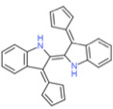 |
↓ | ↓ | NK | [152] | |||||||
| Naloxone | 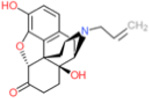 |
2 | p47phox binding | [134] | ||||||||
| Celastrol | 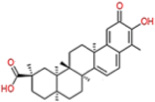 |
0.41 | 0.59 | 2.7 | 3.13 | ns | p47phox-p22phox interface |
[130] | ||||
| Ebselen | 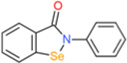 |
0.15 | 0.5 | ns | 0.7 | ns | p47phox and p67phox transloca- tion |
[131] | ||||
| GKT 136901 | 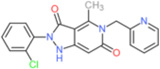 |
0.160* | 1.530* | 0.165* | 0.45 | >30* | NK | [138, 149] |
||||
| GKT137831 | 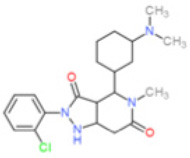 |
0.11 | 1.75 | 0.14 | 0.41 | >30* | NK | [139, 155] |
||||
| ML171 | 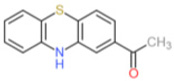 |
0.25 | 5.00 | 5.00 | 5.50 | NK | [137] | |||||
| ACD 084 | 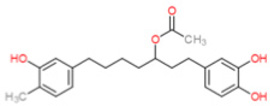 |
>5.00 | 3.08 | >5.00 | ? | NK | [153] | |||||
| 11g |  |
ns | 20 | ns | ns | ns | p47phox-p22phox interface |
[136] | ||||
| 11h | 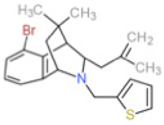 |
ns | 32 | ns | ns | ns | p47phox-p22phox interface |
[136] | ||||
↓= inhibits but no IC50 determined; ns= not significant; blank space=not determined; NK = Not known,
Ki
Inhibitors of Nox 5 & DUOXs 1 & 2
Recently, Nox5 has emerged as an attractive target for the development of inhibitors because of its potentially distinct and complicating role in humans. The inability to study this isozyme in rats and mice has greatly limited experimental inquiry. However, with enhanced procurement and study of human clinical samples, Nox5 is rapidly being viewed as an important ROS source in multiple disease settings. Its regulation by calcium binding and EF-hand domains also offers opportunities for new interventions. As with all Nox family members, Nox5 activity is inhibited by the nonselective flavoprotein inhibitor DPI and ROS generated by Nox5 can be partially suppressed with apocynin; however, this effect of apocynin could be a direct result of scavenging [154]. Ca2+-dependent Nox5 superoxide production has also been shown to be inhibited by GKT136901 in spermatozoa (Ki = ~450 nM; cells in which Nox1 or Nox4 are not detectable [155]), and by GKT137831 in cell free assay of Nox5-overexpressing cells [139]. Furthermore, celastrol was also shown to inhibit Nox5-derived superoxide in Nox5-expressing cells (IC50 of 3.13 µM) and purified membranes containing Nox5 (IC50 of 8.4 µM) [130]. That said, the associated inhibitory mechanisms are yet to be revealed. Incidentally, Nox5 contains structural domains not present in other members of the Nox family. We therefore anticipate that a target approach for its modulation is plausible; for example, melittin (see peptidic inhibitors section) through interactions with Nox5-EF-hands provides a potential site for small molecule Nox5 inhibition [112]. Similar to Nox5, DUOX1 and DUOX2 enzymes are regulated by calcium but require the presence of maturation subunits DUOXA1 or DUOXA2, respectively [37], and their role in disease and immunity is an emerging area. Accordingly, the modulation of DUOX1/2-derived ROS could prove to be an important area of research; however, to date, no known inhibitors of DUOX1/2 exist. Finally, DUOX1/2 enzymes also have EF-hand regions and, thus like Nox5, could be inhibited by EF-hand-binding small molecules or peptides such as melittin.
SUMMARY & LIMITATIONS
The current body of biochemical and medical literature augurs the need for efficacious Nox inhibitors and therapies. Importantly, we, like many others in the field, maintain that isoform selectivity is essential for disease treatment as well as its practical implications in delineating the contribution of a given Nox in physiological redox signaling. The demand for well-characterized, isoform-specific inhibitors including pharmacokinetic and pharmacodynamic profiles is yet to be adequately met. As shown in Table 1, it has becomes quite evident that data for even the most promising Nox inhibitors is, for the most part, incomplete as no compounds to our knowledge have been tested against the full array of Nox isozymes. Even the few compounds that have IC50s lower by 10–20-fold for a given Nox over other family members should be used mindfully in vivo where appropriate doses are more difficult to calibrate. Moreover, considerably more work is required to investigate mechanism of action. It is also noteworthy that Nox3, as well as Duox1 and Duox2 remain poorly characterized Nox targets. This is apparently a consequence of a greater intensity and interest in Noxs in mammalian cardiovascular physiology and disease to this point, where knowledge of these particular isoforms is limited or lacking. The established role of Nox3 and the Duoxs in important physiological functions, e.g. hearing and thyroid function, is also likely to be an impediment. In conclusion, the purpose and promise of both peptidic inhibitors and a handful of small molecules, each with its own advantages and limitations, has grown substantively in recent years and is only expected to burgeon further as interest in the Nox field widens. One promising step in the right direction would be employment of detailed information that peptide inhibitors provide in terms of protein-protein interactions and use of these as pharma-cophores for the design of stable and bioavailable small molecules [156].
Acknowledgments
The authors wish to acknowledge Dr. Gabor Csanyi for critical scientific review and Ms. Laura Pliske for editorial support during the preparation of this manuscript.
This work was supported by National Institutes of Health grants R01HL079207 and P01HL103455.
Biography

Patrick J. Pagano
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interests.
REFERENCES
- 1.Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal. 2014;20:164–182. doi: 10.1089/ars.2013.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison IP, Selemidis S. Understanding the biology of reactive oxygen species and their link to cancer: NADPH oxidases as novel pharmacological targets. Clin Exp Pharmacol Physiol. 2014;41:533–542. doi: 10.1111/1440-1681.12238. [DOI] [PubMed] [Google Scholar]
- 3.Gorin Y, Block K. Nox as a target for diabetic complications. Clin Sci Lond. 2013;125:361–382. doi: 10.1042/CS20130065. [DOI] [PubMed] [Google Scholar]
- 4.Kleikers PW, Wingler K, Hermans JJ, et al. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med Berl. 2012;90:1391–1340. doi: 10.1007/s00109-012-0963-3. [DOI] [PubMed] [Google Scholar]
- 5.Gao HM, Zhou H, Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci. 2012;33:295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61:1–117. [PubMed] [Google Scholar]
- 7.Bernard K, Hecker L, Luckhardt TR, Cheng G, Thannickal VJ. NADPH oxidases in lung health and disease. Antioxid Redox Signal. 2014;20:2838–2853. doi: 10.1089/ars.2013.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab. 2014;25:452–463. doi: 10.1016/j.tem.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. 2013;231:290–300. doi: 10.1002/path.4255. [DOI] [PubMed] [Google Scholar]
- 10.Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. 2014;20:2794–2814. doi: 10.1089/ars.2013.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landry WD, Cotter TG. ROS signalling, NADPH oxidases and cancer. Biochem Soc Trans. 2014;42:934–938. doi: 10.1042/BST20140060. [DOI] [PubMed] [Google Scholar]
- 13.Rodino-Janeiro BK, Paradela-Dobarro B, Castineiras-Landeira MI, et al. Current status of NADPH oxidase research in cardiovascular pharmacology. Vasc Health Risk Manag. 2013;9:401–428. doi: 10.2147/VHRM.S33053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sukumar P, Viswambharan H, Imrie H, et al. Nox2 NADPH Oxidase Has a Critical Role in Insulin Resistance-Related Endothelial Cell Dysfunction. Diabetes. 2013;62:2130–2144. doi: 10.2337/db12-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai H, Griendling KK, Harrison DG. The vascular NAD(P;H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 16.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 17.Nayernia Z, Jaquet V, Krause KH. New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal. 2014;20:2815–2837. doi: 10.1089/ars.2013.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brieger K, Schiavone S, Miller FJ, Jr, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly. 2012;142:13659. doi: 10.4414/smw.2012.13659. [DOI] [PubMed] [Google Scholar]
- 19.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 20.Babior BM, Curnutte JT, McMurrich BJ. The particulate superoxide-forming system from human neutrophils Properties of the system and further evidence supporting its participation in the respiratory burst. J Clin Invest. 1976;58:989–996. doi: 10.1172/JCI108553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patriarca P, Cramer R, Moncalvo S, Rossi F, Romeo D. Enzymatic basis of metabolic stimulation in leucocytes during phagocytosis: the role of activated NADPH oxidase. Arch Biochem Biophys. 1971;145:255–262. doi: 10.1016/0003-9861(71)90034-8. [DOI] [PubMed] [Google Scholar]
- 22.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 23.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes-prototype of the NOX electron transport chain systems. Biochim Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pick E. Role of the Rho GTPase Rac in the activation of the phagocyte NADPH oxidase: outsourcing a key task. Small GT Pases. 2014;5:e27952. doi: 10.4161/sgtp.27952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 28.Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 29.Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. J Biol Chem. 2004;279:34250–34255. doi: 10.1074/jbc.M400660200. [DOI] [PubMed] [Google Scholar]
- 30.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 31.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH Oxidase Nox3 Constitutively Produces Superoxide in a p22phox-dependent Manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 32.Lyle AN, Deshpande NN, Taniyama Y, et al. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nisimoto Y, Diebold BA, Constentino-Gomes D, Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014;53:5111–5120. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banfi B, Molnar G, Maturana A, et al. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 35.Bedard K, Jaquet V, Krause KH. NOX5: from basic biology to signaling and disease. Free Radic Biol Med. 2012;52:725–734. doi: 10.1016/j.freeradbiomed.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 36.Serrander L, Jaquet V, Bedard K, et al. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie. 2007;89:1159–1167. doi: 10.1016/j.biochi.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272. doi: 10.1074/jbc.C600095200. [DOI] [PubMed] [Google Scholar]
- 38.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 39.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. CardioVasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med. 2014;76C:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 41.Sumimoto H, Hata K, Mizuki K, et al. Assembly and activation of the phagocyte NADPH oxidase Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J Biol Chem. 1996;271:22152–22158. doi: 10.1074/jbc.271.36.22152. [DOI] [PubMed] [Google Scholar]
- 42.Meijles DN, Fan LM, Howlin BJ, Li JM. Molecular insights of p47phox phosphorylation dynamics in the regulation of NADPH oxidase activation and superoxide production. J Biol Chem. 2014;289:22759–22770. doi: 10.1074/jbc.M114.561159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finan P, Shimizu Y, Gout I, et al. An SH3 domain and proline-rich sequence mediate an interaction between two components of the phagocyte NADPH oxidase complex. J Biol Chem. 1994;269:13752–13755. [PubMed] [Google Scholar]
- 44.Hata K, Takeshige K, Sumimoto H. Roles for proline-rich regions of p47phox and p67phox in the phagocyte NADPH oxidase activation in vitro. Biochem Biophys Res Commun. 1997;241:226–231. doi: 10.1006/bbrc.1997.7807. [DOI] [PubMed] [Google Scholar]
- 45.Taylor RM, Lord CI, Riesselman MH, et al. Characterization of surface structure and p47phox SH3 domain-mediated conformational changes for human neutrophil flavocytochrome b. Biochemistry. 2007;46:14291–14304. doi: 10.1021/bi701626p. [DOI] [PubMed] [Google Scholar]
- 46.Lapouge K, Smith SJ, Groemping Y, Rittinger K. Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem. 2002;277:10121–10128. doi: 10.1074/jbc.M112065200. [DOI] [PubMed] [Google Scholar]
- 47.Nakanishi A, Imajoh-Ohmi S, Fujinawa T, Kikuchi H, Kanegasaki S. Direct evidence for interaction between COOH-terminal regions of cytochrome b558 subunits and cytosolic 47-kDa protein during activation of an O(2-)-generating system in neutrophils. J Biol Chem. 1992;267:19072–19074. [PubMed] [Google Scholar]
- 48.Han CH, Freeman JL, Lee T, Motalebi SA, Lambeth JD. Regulation of the neutrophil respiratory burst oxidase Identification of an activation domain in p67(phox) J Biol Chem. 1998;273:16663–16668. doi: 10.1074/jbc.273.27.16663. [DOI] [PubMed] [Google Scholar]
- 49.Dang PM, Cross AR, Quinn MT, Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558 II. Proc Natl Acad Sci USA. 2002;99:4262–4265. doi: 10.1073/pnas.072345299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diekmann D, Abo A, Johnston C, Segal AW, Hall A. Interaction of Rac with p67 phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265:531–532. doi: 10.1126/science.8036496. [DOI] [PubMed] [Google Scholar]
- 51.Ago T, Nunoi H, Ito T, Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phoxTriple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J Biol Chem. 1999;274:33644–33653. doi: 10.1074/jbc.274.47.33644. [DOI] [PubMed] [Google Scholar]
- 52.Dutta S, Rittinger K. Regulation of NOXO1 activity through reversible interactions with p22 and NOXA1. PLoS One. 2010;5:e10478. doi: 10.1371/journal.pone.0010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamamoto A, Takeya R, Matsumoto M, Nakayama KI, Sumimoto H. Phosphorylation of Noxo1 at threonine 341 regulates its interaction with Noxa1 and the superoxide-producing activity of Nox1. FEBS J. 2013;280:5145–5159. doi: 10.1111/febs.12489. [DOI] [PubMed] [Google Scholar]
- 54.Jackson HM, Kawahara T, Nisimoto Y, Smith SM, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. J Biol Chem. 2010;285:10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tirone F, Radu L, Craescu CT, Cox JA. Identification of the binding site for the regulatory calcium-binding domain in the catalytic domain of NOX5. Biochemistry. 2010;49:761–771. doi: 10.1021/bi901846y. [DOI] [PubMed] [Google Scholar]
- 56.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 57.von Lohneysen K, Noack D, Jesaitis AJ, Dinauer MC, Knaus UG. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem. 2008;283:35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandes RP, Weissmann N, Schroder K. Redox-mediated signal transduction by cardiovascular Nox NADPH oxidases. J Mol Cell. Cardiol. 2014;73:70–79. doi: 10.1016/j.yjmcc.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. 2014;9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- 61.Cross AR. Inhibitors of the leukocyte superoxide generating oxidase: mechanisms of action and methods for their elucidation. Free Radic Biol Med. 1990;8:71–93. doi: 10.1016/0891-5849(90)90147-b. [DOI] [PubMed] [Google Scholar]
- 62.Jones OTG, Cross AR, Hancock JT, Henderson LM, O'Donnell VB. Inhibitors of NADPH oxidase as guides to its mechanism. Biochem Soc Trans. 1991;19:70–72. doi: 10.1042/bst0190070. [DOI] [PubMed] [Google Scholar]
- 63.Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophilsSpecific labelling of a component polypeptide of the oxidase. Biochem J. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doussiere J, Vignais PV. Diphenylene iodonium as an inhibitor of the NADPH oxidase complex of bovine neutrophilsFactors controlling the inhibitory potency of diphenylene iodonium in a cell-free system of oxidase activation. Eur J Biochem. 1992;208:61–71. doi: 10.1111/j.1432-1033.1992.tb17159.x. [DOI] [PubMed] [Google Scholar]
- 65.Hart BA, Simons JM, Knaan-Shanzer S, Bakker NP, Labadie RP. Antiarthritic activity of the newly developed neutrophil oxidative burst antagonist apocynin. Free Radic Biol Med. 1990;9:127–131. doi: 10.1016/0891-5849(90)90115-y. [DOI] [PubMed] [Google Scholar]
- 66.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 67.Cayatte AJ, Rupin A, Oliver-Krasinski J, et al. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets nadph oxidase. Arterioscler Thromb Vasc Biol. 2001;21:1577–1584. doi: 10.1161/hq1001.096723. [DOI] [PubMed] [Google Scholar]
- 68.Shi J, Ross CR, Leto TL, Blecha F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47 phox. Proc Natl Acad Sci USA. 1996;93:6014–6018. doi: 10.1073/pnas.93.12.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rey F, Cifuentes M, Quinn M, Pagano P. A competitive inhibitor of NADPH oxidase subunits p47 (phox) and gp91 (phox) attenuates blood pressure in angiotensin II-infused mice. FASEB J. 2000;14:A119–A119. [Google Scholar]
- 70.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P;H oxidase assembly attenuates vascular O(2-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 71.Aldieri E, Riganti C, Polimeni M, et al. Classical inhibitors of NOX NAD(P)H oxidases are not specific . Curr. Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 72.Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cifuentes-Pagano E, Csanyi G, Pagano PJ. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci. 2012;69:2315–2325. doi: 10.1007/s00018-012-1009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cifuentes-Pagano E, Meijles DN, Pagano PJ. The Quest for Selective Nox Inhibitors and Therapeutics: Challenges, Triumphs and Pitfalls. Antioxid Redox Signal. 2014;20:2741–2754. doi: 10.1089/ars.2013.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jaquet V, Scapozza L, Clark R, Krause KH, Lambeth JD. Small Molecule NOX Inhibitors: ROS-generating NADPH Oxidases as Therapeutic Targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 77.Streeter J, Thiel W, Brieger K, Miller FJ., Jr Opportunity Nox: The Future of NADPH Oxidases as Therapeutic Targets in Cardiovascular Disease. CardioVasc Ther. 2013;31:125–137. doi: 10.1111/j.1755-5922.2011.00310.x. [DOI] [PubMed] [Google Scholar]
- 78.Burritt JB, Quinn MT, Jutila MA, Bond CW, Jesaitis AJ. Topological mapping of neutrophil cytochrome b epitopes with phage-display libraries. J Biol Chem. 1995;270:16974–16980. doi: 10.1074/jbc.270.28.16974. [DOI] [PubMed] [Google Scholar]
- 79.DeLeo FR, Yu L, Burritt JB, et al. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc Natl Acad Sci USA. 1995;92:7110–7114. doi: 10.1073/pnas.92.15.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joseph G, Pick E. "Peptide walking" is a novel method for mapping functional domains in proteins Its application to the Rac1-dependent activation of NADPH oxidase. J Biol Chem. 1995;270:29079–29082. doi: 10.1074/jbc.270.49.29079. [DOI] [PubMed] [Google Scholar]
- 81.Morozov I, Lotan O, Joseph G, Gorzalczany Y, Pick E. Mapping of functional domains in p47(phox) involved in the activation of NADPH oxidase by "peptide walking". J Biol Chem. 1998;273:15435–15444. doi: 10.1074/jbc.273.25.15435. [DOI] [PubMed] [Google Scholar]
- 82.Dahan I, Issaeva I, Gorzalczany Y, Sigal N, Hirshberg M, Pick E. Mapping of functional domains in the p22(phox; subunit of flavocytochrome b(559) participating in the assembly of the NADPH oxidase complex by "peptide walking". J Biol Chem. 2002;277:8421–8432. doi: 10.1074/jbc.M109778200. [DOI] [PubMed] [Google Scholar]
- 83.El-Benna J, Dang PM, Perianin A. Peptide-based inhibitors of the phagocyte NADPH oxidase. Biochem Pharmacol. 2010;80:778–785. doi: 10.1016/j.bcp.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 84.El-Benna J, Dang PM, Perianin A. Towards specific NADPH oxidase inhibition by small synthetic peptides. Cell Mol Life Sci. 2012;69:2307–2314. doi: 10.1007/s00018-012-1008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikeda Y, Young LH, Scalia R, Ross CR, Lefer AM. PR-39, a proline/arginine-rich antimicrobial peptide, exerts cardioprotective effects in myocardial ischemia-reperfusion. Cardio Vasc Res. 2001;49:69–77. doi: 10.1016/s0008-6363(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 86.Chan YR, Gallo RL. PR-39, a syndecan-inducing antimicrobial peptide, binds and affects p130(Cas) J Biol Chem. 1998;273:28978–28985. doi: 10.1074/jbc.273.44.28978. [DOI] [PubMed] [Google Scholar]
- 87.Tanaka K, Fujimoto Y, Suzuki M, Suzuki Y, Ohtake T, Saito H, Kohgo Y. PI3-kinase p85alpha is a target molecule of proline-rich antimicrobial peptide to suppress proliferation of ras-transformed cells. Jpn J Cancer Res. 2001;92:959–967. doi: 10.1111/j.1349-7006.2001.tb01187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dahan I, Molshanski-Mor S, Pick E. Inhibition of NADPH oxidase activation by peptides mapping within the dehydrogenase region of Nox2-A "peptide walking" study. J Leukoc Biol. 2012;91:501–515. doi: 10.1189/jlb.1011507. [DOI] [PubMed] [Google Scholar]
- 89.Fawell S, Seery J, Daikh Y, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jacobson GM, Dourron HM, Liu J, et al. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 91.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23:776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 92.Lopes NHM, Vasudevan SS, Gregg D, et al. Rac-dependent monocyte chemoattractant protein-1 production is induced by nutrient deprivation. Circ Res. 2002;91:798–805. doi: 10.1161/01.res.0000040421.54108.81. [DOI] [PubMed] [Google Scholar]
- 93.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Furst R, Brueckl C, Kuebler WM, et al. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005;96:43–53. doi: 10.1161/01.RES.0000151983.01148.06. [DOI] [PubMed] [Google Scholar]
- 95.Harfouche R, Malak NA, Brandes RP, Karsan A, Irani K, Hussain SN. Roles of reactive oxygen species in angiopoietin-1/tie-2 receptor signaling. FASEB J. 2005;19:1728–1730. doi: 10.1096/fj.04-3621fje. [DOI] [PubMed] [Google Scholar]
- 96.Walch L, Massade L, Dufilho M, Brunet A, Rendu F. Pro-atherogenic effect of interleukin-4 in endothelial cells: modulation of oxidative stress, nitric oxide and monocyte chemoattractant protein-1 expression. Atherosclerosis. 2006;187:285–291. doi: 10.1016/j.atherosclerosis.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 97.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J Physiol. 2006;576:557–567. doi: 10.1113/jphysiol.2006.111070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krotz F, Keller M, Derflinger S, et al. Mycophenolate acid inhibits endothelial NAD(P;H oxidase activity and superoxide formation by a Rac1-dependent mechanism. Hypertension. 2007;49:201–208. doi: 10.1161/01.HYP.0000251162.14782.d4. [DOI] [PubMed] [Google Scholar]
- 99.Zeng Q, Zhou Q, Yao F, O'Rourke ST, Sun C. Endothelin-1 regulates cardiac L-type calcium channels via NAD(P)H oxidase-derived superoxide. J Pharmacol Exp Ther. 2008;326:732–738. doi: 10.1124/jpet.108.140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tarr JM, Ding N, Kaul K, Antonell A, Perez-Jurado LA, Chibber R. Cellular crosstalk between TNF-alpha, NADPH oxidase, PKCbeta2, and C2GNT in human leukocytes. Cell Signal. 2012;24:873–878. doi: 10.1016/j.cellsig.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 101.Hahn NE, Musters RJ, Fritz JM, et al. Early NADPH oxidase-2 activation is crucial in phenylephrine-induced hypertrophy of H9c2 cells. Cell Signal. 2014;26:1818–1824. doi: 10.1016/j.cellsig.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Touyz RM, Chen X, Tabet F, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P;H oxidase in smooth muscle cells from human resistance arteries Regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 103.Krotz F, Sohn HY, Gloe T, et al. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100:917–924. doi: 10.1182/blood.v100.3.917. [DOI] [PubMed] [Google Scholar]
- 104.Ebrahimian T, Li MW, Lemarie CA, et al. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: regulation of oxidative stress. Hypertension. 2011;57:245–254. doi: 10.1161/HYPERTENSIONAHA.110.159889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.He Y, Cui J, Lee JC, et al. Prolonged exposure of cortical neurons to oligomeric amyloid-beta impairs NMDA receptor function via NADPH oxidase-mediated ROS production: protective effect of green tea (−)-epigallocatechin-3-gallate. ASN Neuro. 2011;3:e00050. doi: 10.1042/AN20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 107.Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, et al. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51:1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ranayhossaini DJ, Rodriguez AI, Sahoo S, et al. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J Biol Chem. 2013;288:36437–36450. doi: 10.1074/jbc.M113.521344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maehara Y, Miyano K, Yuzawa S, Akimoto R, Takeya R, Sumimoto H. A conserved region between the TPR and activation domains of p67phox participates in activation of the phagocyte NADPH oxidase. J Biol Chem. 2010;285:31435–31445. doi: 10.1074/jbc.M110.161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Streeter J, Schickling BM, Jiang S, et al. Phosphorylation of Nox1 Regulates Association with NoxA1 Activation Domain. Circ Res. 2014;115:911–918. doi: 10.1161/CIRCRESAHA.115.304267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Csanyi G, Pagano PJ. Strategies Aimed at Nox4 Oxidase Inhibition Employing Peptides from Nox4 B-Loop and C-Terminus and p22 (phox) N-Terminus: An Elusive Target. Int J Hypertens. 2013;2013:842827. doi: 10.1155/2013/842827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Banfi B, Tirone F, Durussel I, et al. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) J Biol Chem. 2004;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 113.Comte M, Maulet Y, Cox JA. Ca2+-dependent high-affinity complex formation between calmodulin and melittin. Biochem J. 1983;209:269–272. doi: 10.1042/bj2090269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 115.Sun L. Peptide-based drug development. Mod Chem Appl. 2013;1:e103. [Google Scholar]
- 116.Dahan I, Pick E. Strategies for identifying synthetic peptides to act as inhibitors of NADPH oxidases, or "all that you did and did not want to know about Nox inhibitory peptides". Cell Mol Life Sci. 2012;69:2283–2205. doi: 10.1007/s00018-012-1007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Di L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2014 doi: 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc Natl Acad Sci USA. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Walensky LD, Kung AL, Escher I, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zielonka J, Cheng G, Zielonka M, et al. High-throughput assays for superoxide and hydrogen peroxide: design of a screening workflow to identify inhibitors of NADPH oxidases. J Biol Chem. 2014;289:16176–16189. doi: 10.1074/jbc.M114.548693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.ten Freyhaus H, Huntgeburth M, Wingler K, et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. CardioVasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 122.Wind S, Beuerlein K, Eucker T, et al. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stielow C, Catar RA, Muller G, et al. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun. 2006;344:200–205. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 124.Altenhöfer S, Kleikers PM, Radermacher K, et al. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wingler K, Altenhoefer SA, Kleikers PW, Radermacher KA, Kleinschnitz C, Schmidt HH. VAS2870 is a pan-NADPH oxidase inhibitor. Cell. Mol Life Sci. 2012;69:3159–3160. doi: 10.1007/s00018-012-1107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gatto GJ, Jr, Ao Z, Kearse MG, et al. NADPH oxidase-dependent and -independent mechanisms of reported inhibitors of reactive oxygen generation. J Enzyme Inhib Med Chem. 2013;28:95–104. doi: 10.3109/14756366.2011.636360. [DOI] [PubMed] [Google Scholar]
- 127.Sun QA, Hess DT, Wang B, Miyagi M, Stamler JS. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870) Free Radic Biol Med. 2012;52:1897–1902. doi: 10.1016/j.freeradbiomed.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Seno K, Nishi K, Matsuo Y, Fujishita T. Pyrazolo[1, 5-A]pyrimidine derivative and NAD(P)H oxidase inhibitor containing the same. US 2006/0089362. Osaka, Japan: Shionogi & Co, Ltd; 2006. [Google Scholar]
- 129.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsycho Pharmacol Biol Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 130.Jaquet V, Marcoux J, Forest E, et al. NOX NADPH oxidase isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol. 2011;164:507–520. doi: 10.1111/j.1476-5381.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smith SM, Min J, Ganesh T, et al. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem Biol. 2012;19:752–763. doi: 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nakamura Y, Feng Q, Kumagai T, et al. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant Implication for inflammation-associated carcinogenesis. J Biol Chem. 2002;277:2687–2694. doi: 10.1074/jbc.M109641200. [DOI] [PubMed] [Google Scholar]
- 133.Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. Inhibition by naloxone stereoisomers of beta-amyloid peptide (1–42;-induced superoxide production in microglia and degeneration of cortical and mesencephalic neurons. J Pharmacol Exp Ther. 2002;302:1212–1219. doi: 10.1124/jpet.102.035956. [DOI] [PubMed] [Google Scholar]
- 134.Wang Q, Zhou H, Gao H, et al. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012;9:32. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kennedy JA, Beck-Oldach K, McFadden-Lewis K, et al. Effect of the anti-anginal agent, perhexiline, on neutrophil, valvular and vascular superoxide formation. Eur J Pharmacol. 2006;531:13–19. doi: 10.1016/j.ejphar.2005.11.058. [DOI] [PubMed] [Google Scholar]
- 136.Cifuentes-Pagano E, Saha J, Csanyi G, et al. Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors Med Chem Comm. 2013;4:1085–1092. doi: 10.1039/C3MD00061C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gianni D, Taulet N, Zhang H, et al. A novel and specific NADPH oxidase-1 (Nox1; small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5:981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Laleu B, Gaggini F, Orchard M, et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 139.Gaggini F, Laleu B, Orchard M, et al. Design, synthesis biological activity of original pyrazolo-pyrido-diazepine, -pyrazine, -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg.Med.Chem. 2011;19:6989–6999. doi: 10.1016/j.bmc.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 140.Aoyama T, Paik YH, Watanabe S, et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gray SP, Di Marco E, Okabe J, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 142.Jiang JX, Chen X, Serizawa N, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jha JC, Gray SP, Barit D, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol. 2014;25:1237–1254. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shin YS, Song SJ, Kang SU, et al. A novel synthetic compound, 3-amino-3-(4-fluoro-phenyl)-1H–quinoline-2,4-dione, inhibits cisplatin-induced hearing loss by the suppression of reactive oxygen species: In vitro and in vivo study. NeuroScience. 2013;232:1–12. doi: 10.1016/j.neuroscience.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 145.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Serrander L, Cartier L, Bedard K, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Thallas-Bonke V, Jha JC, Gray SP, et al. Nox-4 deletion reduces oxidative stress and injury by PKC-alpha-associated mechanisms in diabetic nephropathy. Physiol Rep. 2014;2:e12192. doi: 10.14814/phy2.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Holterman CE, Thibodeau JF, Kennedy CR. NADPH oxidase 5 and renal disease. Curr Opin Nephrol Hypertens. 2015;24:81–87. doi: 10.1097/MNH.0000000000000081. [DOI] [PubMed] [Google Scholar]
- 149.Garrido-Urbani S, Jemelin S, Deffert C, et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS One. 2011;6:e14665. doi: 10.1371/journal.pone.0014665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vendrov AE, Madamanchi NR, Niu XL, et al. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem. 2010;285:26545–26557. doi: 10.1074/jbc.M110.143917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Perry BN, Govindarajan B, Bhandarkar SS, et al. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J Invest Dermatol. 2006;126:2316–2322. doi: 10.1038/sj.jid.5700413. [DOI] [PubMed] [Google Scholar]
- 152.Bhandarkar SS, Jaconi M, Fried LE, et al. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J Clin Invest. 2009;119:2359–2365. doi: 10.1172/JCI33877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kofler PA, Pircher H, von Grafenstein S, et al. Characterisation of nox4 inhibitors from edible plants. Planta Med. 2013;79:244–252. doi: 10.1055/s-0032-1328129. [DOI] [PubMed] [Google Scholar]
- 154.Heumuller S, Wind S, Barbosa-Sicard E, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 155.Musset B, Clark RA, DeCoursey TE, et al. NOX5 in human spermatozoa: expression, function, and regulation. J Biol Chem. 2012;287:9376–9388. doi: 10.1074/jbc.M111.314955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Scognamiglio PL, Di Natale C, Perretta G, Marasco D. From peptides to small molecules: an intriguing but intricated way to new drugs. Curr Med Chem. 2013;20:3803–3817. doi: 10.2174/09298673113209990184. [DOI] [PubMed] [Google Scholar]