

Abstract
Key points
The real impact of physical exercise parameters, i.e. intensity, type of contraction and solicited energetic metabolism, on neuroprotection in the specific context of neurodegeneration remains poorly explored.
In this study behavioural, biochemical and cellular analyses were conducted to compare the effects of two different long‐term exercise protocols, high intensity swimming and low intensity running, on motor units of a type 3 spinal muscular atrophy (SMA)‐like mouse model.
Our data revealed a preferential SMA‐induced death of intermediate and fast motor neurons which was limited by the swimming protocol only, suggesting a close relationship between neuron‐specific protection and their activation levels by specific exercise.
The exercise‐induced neuroprotection was independent of SMN protein expression and associated with specific metabolic and behavioural adaptations with notably a swimming‐induced reduction of muscle fatigability.
Our results provide new insight into the motor units’ adaptations to different physical exercise parameters and will contribute to the design of new active physiotherapy protocols for patient care.
Abstract
Spinal muscular atrophy (SMA) is a group of autosomal recessive neurodegenerative diseases differing in their clinical outcome, characterized by the specific loss of spinal motor neurons, caused by insufficient level of expression of the protein survival of motor neuron (SMN). No cure is at present available for SMA. While physical exercise might represent a promising approach for alleviating SMA symptoms, the lack of data dealing with the effects of different exercise types on diseased motor units still precludes the use of active physiotherapy in SMA patients. In the present study, we have evaluated the efficiency of two long‐term physical exercise paradigms, based on either high intensity swimming or low intensity running, in alleviating SMA symptoms in a mild type 3 SMA‐like mouse model. We found that 10 months’ physical training induced significant benefits in terms of resistance to muscle damage, energetic metabolism, muscle fatigue and motor behaviour. Both exercise types significantly enhanced motor neuron survival, independently of SMN expression, leading to the maintenance of neuromuscular junctions and skeletal muscle phenotypes, particularly in the soleus, plantaris and tibialis of trained mice. Most importantly, both exercises significantly improved neuromuscular excitability properties. Further, all these training‐induced benefits were quantitatively and qualitatively related to the specific characteristics of each exercise, suggesting that the related neuroprotection is strongly dependent on the specific activation of some motor neuron subpopulations. Taken together, the present data show significant long‐term exercise benefits in type 3 SMA‐like mice providing important clues for designing rehabilitation programmes in patients.
Key points
The real impact of physical exercise parameters, i.e. intensity, type of contraction and solicited energetic metabolism, on neuroprotection in the specific context of neurodegeneration remains poorly explored.
In this study behavioural, biochemical and cellular analyses were conducted to compare the effects of two different long‐term exercise protocols, high intensity swimming and low intensity running, on motor units of a type 3 spinal muscular atrophy (SMA)‐like mouse model.
Our data revealed a preferential SMA‐induced death of intermediate and fast motor neurons which was limited by the swimming protocol only, suggesting a close relationship between neuron‐specific protection and their activation levels by specific exercise.
The exercise‐induced neuroprotection was independent of SMN protein expression and associated with specific metabolic and behavioural adaptations with notably a swimming‐induced reduction of muscle fatigability.
Our results provide new insight into the motor units’ adaptations to different physical exercise parameters and will contribute to the design of new active physiotherapy protocols for patient care.
Abbreviations
- ChAT
choline acetyltransferase
- Chodl
chondrolectin
- CK
creatine kinase
- CMAP
compound muscle action potential
- ERRβ
oestrogen‐related receptor β
- MyHC
myosin heavy chain
- NMJ
neuromuscular junction
- SMA
spinal muscular atrophy
- SMN
survival of motor neuron
- TBS
Tris‐buffered solution
Introduction
Spinal muscular atrophy (SMA), the leading genetic cause of death in childhood, is a neurodegenerative disorder characterized by spinal motor neuron degeneration, leading to progressive muscle weakness and atrophy (Russman et al. 1983). No cure for SMA is at present available. Patient care is usually provided through supportive ventilation, feeding assistance and passive limb and thoracic physiotherapy (Bladen et al. 2014). Deletion or mutation of the telomeric copy of the Survival‐of‐Motor Neuron gene SMN1 is responsible for SMA (Lefebvre et al. 1995). The inverted centromeric copy of the SMN1 gene, SMN2, produces a majority of transcripts lacking exon 7, resulting in the expression of an unstable survival of motor neuron (SMN) protein. SMN2 is thus unable to fully compensate for the SMN1 mutation (Lorson et al. 1998; Lorson & Androphy, 2000). Consequently, the number of SMN2 copies in the genome modulates the clinical heterogeneity of the disease. Three main types of SMA are commonly distinguished according to the age of onset and the severity of motor capacity impairments (Harding & Thomas, 1980), the severe type 1 SMA (Werdnig–Hoffman disease), the intermediate type 2 SMA, and the mild type 3 SMA (Kugelberg–Wellander disease).
Several studies have suggested that motor unit maturation was severely impaired in the severe type of SMA‐like mice, providing a tentative explanation for the motor neuron‐specific sensitivity to a loss of the ubiquitously expressed SMN protein (Biondi et al. 2008; Kariya et al. 2008; Kong et al. 2009). We have reported that a 5‐day training programme was sufficient to promote SMN expression in the spinal cord of severe type SMA‐like mice, resulting in the improvement of the motor unit postnatal maturation and motor neuron survival (Grondard et al. 2005; Biondi et al. 2008). Taken together, these data support the hypothesis of a link between neuronal activity, motor unit maturation, SMN expression and neuroprotection. However, no data are available in the case of less severe SMA, which is characterized by a late disease onset (Tsai et al. 2006), therefore raising questions about the potential role of motor unit maturation in the SMA‐induced neurodegeneration in adulthood on the one hand, and the potential exercise‐induced benefits in such a pathological context on the other.
Moreover, the potential benefits induced by long‐term physical training in the case of SMA‐induced late‐onset and progressive neurodegeneration have never been investigated. Several investigations describing the effects of exercise in mouse models corresponding to different adulthood neurodegenerative disorders such as Parkinson's disease (Tuon et al. 2012; Goes et al. 2014), Alzheimer's disease (Liu et al. 2013; Xu et al. 2013), and amyotrophic lateral sclerosis (Kirkinezos et al. 2003; Veldink et al. 2003; Liebetanz et al. 2004; Deforges et al. 2009) have globally indicated beneficial effects of physical exercise, but these are related to either the intrinsic parameters of the exercise or the nature of neuronal alterations. Indeed, physical exercise consists in different practices involving various motor, metabolic and physiological solicitations, including concentric vs. eccentric contractions, anaerobic vs. oxidative pathways, voluntary vs. forced and long‐term vs. acute exercises. However the impact of these different exercise parameters has never been thoroughly investigated on diseased motor units, which limits the application of active physiotherapy for neurodegenerative disorders.
In the present study, we investigated the effects of two different exercises in adult type 3 SMA‐like mice (Tsai et al. 2006) over 10 consecutive months, i.e. a high intensity and amplitude swimming‐based training and a low intensity and amplitude running‐based training (Grondard et al. 2008). We report compelling evidence of specific exercise‐induced benefits for diseased adult motor units, at both the structural and the functional level.
Methods
Ethical approval
Animal handling and experimentation were performed in line with approved Institutional Animal Care and Use Committee protocols at the University of Paris Descartes and followed the national authority (Ministère de la Recherche et de la Technologie, France) guidelines for the detention, use and the ethical treatment of laboratory animals based on European Union Directive 2010/63/EU.
Type 3 SMA‐like mouse model
The knockout‐transgenic type 3 SMA‐like mice (FVB/NRj‐Smn Δ7/Δ7, huSMN2 +/+) derived from mice obtained from the Institute of Molecular Biology (Hsieh‐Li et al. 2000) (Academia Sinica, Taipei, Taiwan) have been purified on the FVB/NRj genetic background (Janvier Labs, Le Genest‐Saint‐Isle, France) by backcross for more than 10 generations and were designated as ‘SMA’ (n = 68). The control mice (control; n = 68) were heterozygous knock‐out for murin Smn, expressed homozygous human SMN2 transgene (FVB/NRj‐Smn Δ/Δ7, huSMN2 +/+). Only males were selected for experimentation to standardize the analyses, and all the experiments were performed in a blind systematic manner to minimize bias. From two to four animals were housed in each cage, with food and water ad libitum.
Physical training protocols and behavioural evaluation
Two‐month‐old mice were submitted either to running‐based training on a speed‐regulated treadmill at 13 m min−1, or to swimming‐based training in an adjustable‐flow swimming pool at 5 l min−1 (Charbonnier and Soude, 2006, Patent FR 06 53772). Both exercises were performed for 20 min per day and 5 days a week for 10 consecutive months. Mice were initially subjected to a preliminary 2‐week training period during which the speed of treadmill and the intensity of water flow were increased every day by 5% increment from 50% to the final speed. Sedentary control and SMA (n = 28) mice were placed in the pool without flow (14 SMA and 14 control mice) and floated at the water surface or on the treadmill without speed (14 SMA and 14 control mice). We formed four groups of trained mice, one running group of controls and one of SMA (Run control and Run SMA; n = 12 for each) and one swimming group of controls and one of SMA (Swim control and Swim SMA; n = 12 for each).
All groups of control and SMA mice were tested at 2, 3, 6, 9 and 12 months of age for motor capabilities through four different tests, i.e. a grip test for muscle resistance; grip strength for maximum muscle force; open‐field for exploratory abilities; and a fatigue test for the measure of time before exhaustion in the swimming condition. Both a 48 h actimeter for spontaneous activity and a fatigue test for the measure of time before exhaustion in the running condition were applied to all groups of mice at 12 months of age only.
For the grip test, the time during which the animals were able to sustain their weight holding onto a metal rail suspended at 50 cm from a table was recorded with a maximum time at 720 s. Only the maximum value of five successive trials was recorded with at least a 10 min resting period between each try.
For the grip strength test, the strength was measured using a grip tensiometer (Bioseb, Vitrolles, France) according to the TREAT‐NMD guidelines. Forelimb and four‐limb traction strength tests were separately performed using a single thin metal rod and a thin metal grid, respectively, interfaced with a tensiometer. Mice gripped the metal rod with forelimbs or the metal grid with four limbs and the manipulator pulled it progressively by the tail along the instrument axis (following the manufacturer's protocol). Only the maximum value of five successive tries was retained as maximum force for each mouse, and the measurement was given in grams. The hindlimb grip strength was calculated by subtracting forelimb from four‐limb grip measurements.
For the open‐field test, the mice were tested individually in a wooden box measuring 50 cm × 50 cm × 20 cm and divided into 25 equal squares of 10 cm. Squares adjacent to walls were referred to as the periphery and the nine remaining squares were referred to as the centre. The open field was washed after each session. A mouse was placed in a central square on the open field and allowed to move freely for 5 min. The number of peripheral and central square crossings was scored manually by the experimenter.
For the actimeter, both total locomotor activity and rearing behaviour were recorded for each mouse during two successive days using the commercially available Physiocage system (Panlab/Harvard apparatus, Cornella, Spain) and the software suite METABOLISM (V2.2.01, Panlab). Mice were singly housed in home cages placed over a sensor platform equipped with an extensiometric weight transducer and two infra‐red (IR) sensor bars including 16 IR beams spaced by 15.6 mm to allow the detection of rearing behaviour. Values of locomotor activity (arbitrary units) and rearing events were acquired by the system every second. For each animal, the total locomotor activity and the number of rearing events were calculated as the sum of the recorded values obtained for 7.5 min‐, 12 h‐ or 24 h‐interval periods.
For the fatigue tests, based either in our adjustable‐flow swimming pool or in a speed‐regulated treadmill, mice were subjected to the maximum sustainable water flow or treadmill speed and the time during which animals were able to maintain this maximum intensity was recorded as well as the value of speed. In the swimming fatigue test, the maximum water flow was 6 l min−1 and in the running fatigue test, the maximum treadmill speed was 21 m min−1.
Histological and immunohistochemical analysis
Mice were anaesthetized with 1% pentobarbital solution (6 μl g−1) and were intracardially perfused first with 20 ml of phosphate‐buffered saline (PBS) and then with 30 ml of 4% paraformaldehyde (PFA).
For motor neuron analysis, the spinal cord was dissected, post‐fixed in 4% PFA for 12 h and rinsed 2 times in PBS azide 0.01% buffer. The L1 to L5 lumbar region was isolated from spinal cord, embedded in 4% agarose type 1 and cut with a vibrating blade microtome (VT‐1000S, Leica Microsystems SAS, Nanterre, France) at 50 μm‐thick cross sections. One out of every five sections (an average of 10 sections examined corresponding to about 200 motor neurons per animal, and about 1600 motor neurons per experimental point) was processed for immunohistochemical analysis. After 30 min at room temperature in a blocking solution (with 0.5% Tween‐20, 0.5% Triton X‐100 and 4% donkey serum in Tris buffer solution (TBS)), tissues sections were incubated in primary goat choline acetyltransferase (ChAT) (polyclonal goat anti‐ChAT; 1:400; Chemicon, Inc., Temecula, CA, USA) and mouse oestrogen‐related receptor β (ERRβ; human ERRβ/NR3A2 MAb, 1:500, R&D systems Europe, France) antibody for 3 days at 4°C and diluted in the blocking solution. Sections were then washed between each subsequent step with 0.5% Tween‐20 and 0.5% Triton X‐100 in TBS. Sections were subsequently incubated with secondary Alexa Fluor® 488 donkey anti‐mouse (1:400; Jackson ImmunoResearch Europe, Newmarket, UK) and Alexa Fluor® 568 donkey anti‐goat (1:400; Jackson ImmunoResearch) for 1 h at room temperature. The sections were mounted in VectashielD® mounting medium (Vector Laboratories, Burlingame, CA, USA). The staining specificity was checked by performing the incubation in the absence of the primary antibody.
For motor end‐plate labelling, the soleus, plantaris and tibialis muscles were dissected and post‐fixed 12 h in PFA 4%. Muscle fibres were dilacerated and stained for pre‐synaptic motor nerve terminals using a mix of rabbit antibody directed against the 61 kDa isoform of neurofilament protein (NF‐L; 1:1,000; AB9568, Millipore) and rabbit anti‐synaptophysin (1:5; 80130, Life Technologies) and using Alexa Fluor 568‐conjugated‐α‐bungarotoxin (4 mg ml−1 in PBS with 4% bovine serum albumin) for post‐synaptic motor nerve terminals. The dilacerated myofibres were subsequently incubated with an Alexa Fluor 488 goat anti‐rabbit (1:400; Jackson ImmunoResearch) to reveal the pre‐synaptic staining.
For histology and typology analysis, soleus, plantaris and tibialis muscles were dissected from anaesthetized animals with 1% pentobarbital solution (6 μl g−1). All muscles were frozen in cold isopentane, maintained at −80°C on dry ice and cut in a cryostat (Leica) to give 10 μm‐thick cross sections. Slices with muscle sections were either stained by histological haematoxylin–eosine (H&E), dehydrated via an alcohol gradient and then mounted with Eukitt (VWR International, Strasbourg, France) or fixed with acetone and incubated with mouse antibodies raised against mouse myosin heavy chains (MyHC): slow type I (NCL‐MyHCs), all fast type II (NCL‐MyHCf) (Leica Microsystems, France) and intermediate type IIa (A4.74) diluted at 1:20 in 0.5% Tween 20 PBS. The secondary antibodies used were goat anti‐mouse‐specific antibodies conjugated with Cy3 (1:400, Jackson ImmunoResearch). Sections were visualized as described above. For each mouse, three sections of each muscle were studied. The percentages of fast IIx and IIb fibres were determined as the difference between the total number of type II and type IIa fibres.
All counts were performed using ImageJ software v1.47 (National Institutes of Health, Bethesda, MD, USA). Colour images were tinted using ImageJ software v1.47, and identical brightness, contrast and colour balance adjustments were applied to all groups.
In situ hybridization
Floating lumbar spinal cord sections were dehydrated in a graded series of methanol washes before storage in 100% methanol at −20°C. Sections were rehydrated by successive washes in methanol and 0.1% Tween‐20 PBS (PBT) ending up in 100% PBT. Sections were treated with 0.5% Triton X‐100 (Sigma‐Aldrich). After additional washes in PBT, sections were digested with 20 μg ml−1 proteinase K (Invitrogen). The reaction was stopped by washing in a large volume of PBT. The sections were then postfixed in 4% formaldehyde before preincubation in hybridization buffer (50% formamide, 5× SSC, pH 4.5, 1% sodium dodecyl sulphate (SDS), 50 μg ml−1 tRNA (Sigma‐Aldrich), 50 μg ml−1 heparin (Sigma‐Aldrich) in PBT). Then, chondrolectin (Chodl) probe, as previously described (Enjin et al. 2010) (1914‐2530, NM_0139134.3), at 300 ng ml−1 was heat‐denatured in hybridization buffer. Hybridization was performed overnight (14–16 h) at 65°C. Unbound probe was removed by sequential washes of buffer 1 (50% formamide, 5× SSC, pH 4.5 and 0.02% SDS in PBT) followed by buffer 2 (50% formamide, 2× SSC, pH 4.5, and PBT) at 65°C. The sections were further washed in 0.1% Tween‐20 TBS (TBST) before incubation in 1% blocking reagent (Roche Diagnostics Scandinavia, Stockholm, Sweden) together with 1:5000 diluted anti‐digoxigenin alkaline phosphatase‐conjugated antibody (Roche Diagnostics Scandinavia) overnight at 4°C. Unbound antibody was removed by sequential washes with 2 mm levamisole (GTF Fisher, Stockholm, Sweden) in TBST followed by washes with 2 mm levamisole in NTMT (100 mm NaCl, 10 mm Tris‐HCl, pH 9.5, 50 mm MgCl2 and 0.1% Tween‐20). Sections were developed in BM purple AP substrate (Roche Diagnostics Scandinavia).
Microscopy
All immunofluorescence images were collected with a CMOS camera (ORCA Flash 2.8, Hamamatsu Photonics France, Massy, France) mounted on a Zeiss AxioObserver microscope (Z1, Carl Zeiss SAS, Le Pecq, France) using the ZEN 2012 software (Carl Zeiss SAS) with 100 (×10 Zeiss Plan NeoFluar NA 0.3), 200 (×20 Zeiss EC‐Plan‐Apo NA 0.8) and 630 (×63 Zeiss Plan‐Apo Oil NA 1.4) magnifications.
Serum lactate and creatine kinase measurements
Mice were anaesthetized with 1% pentobarbital solution (6 μl g−1) diluted in 0.9% saline buffer by intraperitoneal injection. An 800 μl blood sample was taken from the right ventricle with a 1 ml syringe mounted with a 22‐gauge needle, both coated with heparin (5000 UI ml−1, Pan Pharma Luitré, France). The blood sample was centrifuged at 1500 g for 10 min at 4°C. Obtained serum was then frozen, stored at −80°C, and used within 1 month. Serum creatine kinase (CK) activity was quantified in duplicate using the EnzyChrom CK Assay Kit (ECPK‐100; BioAssay Systems, Hayward, CA, USA). For each point, a 10 μl sample of serum was incubated at 22°C in 100 μl of reactive buffer following the manufacturer's protocol in a 96‐well plate. The plate was read on a microplate spectrometer (SpectraMax 340PC384; Molecular Devices, St Grégoire, France) at 340 nm wavelength after 20 and 40 min of incubation. CK activity was evaluated following the manufacturer's protocol and given in units per litre (U l−1). Lactate levels were quantified in duplicate using a lactate assay kit (K607‐100; BioVision, Milpitas, CA, USA). For each point, 2 μl samples of serum were incubated at 22°C in 50 μl of buffer and 50 μl of reaction mix following the manufacturer's protocol in a 96‐well plate. The plate was read on the same microplate spectrometer at 570 nm wavelength after 30 min of incubation. Lactate level was given in nanomoles per microlitre (nmol μl−1).
Protein and Western blot analysis
The ventral parts of frozen spinal cords were maintained at −20°C and were selectively dissected by razor blade. A furrow still appeared in the middle of the ventral part and dorsal root ganglia were visible on the dorsal part of the spinal cord. Ventral lumbar spinal cord samples (4–10 mg) were homogenized in 100 μl per 5 mg tissues in the presence of ice‐cold RIPA buffer (50 mm Tris, 150 mm NaCl, 0.1% SDS, 1% NP40, 10 mm NaF, 1× protease inhibitor (Roche, Basel, Switzerland), 1% phosphatase inhibitor (Sigma‐Aldrich, St Louis, MO, USA)). Protein concentration of the clarified homogenates (4°C, 15 min, 17,000 g) was determined on all samples using the Bradford protein assay (Bio‐Rad Laboratories, Hercules, CA, USA). Protein samples (40 μg) were submitted to 12.5% SDS‐PAGE electrophoresis (1.5 m Tris pH 8.3, 12.5% acrylamide, 0.07% Bis, 0.1% SDS, 0.05% ammonium persulfate, 0.06% tetramethylethylenediamine). The separated proteins were transferred on PVDF membranes (Bio‐Rad Laboratories) according to the Towbin protocol (Towbin et al. 1984). Equal loading of samples was checked by Ponceau dye staining of the transferred gels. Western blot analysis was performed on membranes overnight at 4°C in 4% BSA, 0.05% Tween 20, TBS pH 7.4. The primary antibody monoclonal mouse anti‐SMN (1: 5.000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was incubated overnight at 4°C in the above blocking medium. Membranes were rinsed in 0.1% Tween 20 in TBS for 3 × 10 min at room temperature and then incubated in horseradish peroxydase‐conjugated goat secondary antibody directed against mouse immunoglobulins (1:5000; Bio‐Rad Laboratories) in 0.1 % Tween 20 in TBS for 1 h at room temperature. Bound antibody complexes were developed using the enhanced chemiluminescence (ECL) system (Amersham Biotech, Saclay, France) and exposed to hyperfilm ECL‐plus X ray film (GE Healthcare, France). Films were quantified with ImageJ v1.47.
Electrophysiological recordings from the mouse neuromuscular system in vivo
Multimodal evaluation of excitability properties of the neuromuscular system of control and SMA mice was performed in vivo by means of a minimally invasive electrophysiological method, using the Qtrac© software written by Prof. Hugh Bostock (Institute of Neurology, London, UK), as detailed previously (Boerio et al. 2009). Experiments were performed on mice anaesthetized by inhalation of 1.5% of isoflurane (AErrane®, Baxter SA, Lessines, Belgium) in air, and the experimental protocol has been approved by the French Departmental Direction of Animal Protection (no. A91‐453 to E.B.).
Briefly, an anaesthetized mouse was placed on a heating pad to maintain body temperature between 35.4 and 35.6°C throughout the experiment (measured using a rectal probe) to avoid non‐specific modifications of the measured excitability parameters. Electrical stimulation was delivered to the tibial branch of sciatic motor nerve, by means of surface electrodes, and the compound muscle action potential (CMAP) was recorded using needle electrodes inserted into the plantar muscle. The mouse was submitted to one session of excitability measurements (TRONDE protocol) which consisted of five different tests (stimulus–response relationship (C1), strength–duration relationship (C2), recovery cycle (C3), current–threshold relationship (C4) and threshold electrotonus (C5)) from which more than 30 parameters were determined (numbered 1–38) and analysed (Kiernan & Bostock, 2000; Nodera & Kaji, 2006; Vianello et al. 2014).
-
C1: the stimulus–response relationship gives information on the neuromuscular excitability state. It consisted of measuring the CMAP amplitude as a function of the intensity of a stimulation (of 1 ms duration) to evaluate notably both the CMAP maximal amplitude and the stimulation intensity that had to be applied to evoke a CMAP of 50% maximal amplitude.
-
C2: the strength–duration relationship reflects the axonal resting potential at the nodal membrane, giving information about the density and the functioning of persistent sodium channels. It was derived from the intensity–duration curve which determines the intensity in relation to the duration (0.2–1.0 ms) of a stimulus necessary to evoke a given amplitude of CMAP, in order to provide (1) the rheobase (i.e. the minimal intensity of infinitely long duration stimulation necessary to evoke a CMAP) and (2) the strength–duration time constant or chronaxie (i.e. the intensity duration of twice rheobase stimulation necessary to evoke a CMAP) given by the x‐intercept and the slope, respectively.
-
C3: the ‘recovery cycle’ corresponds to the excitability changes that occur following a CMAP. It consisted of the refractory periods (during which membrane excitability is either nil or markedly decreased and whose duration is specified by the first x‐intercept) followed by the supernormal and late subnormal periods (during which membrane excitability is increased and decreased, respectively). These excitability changes were assessed by a first supramaximal stimulation followed, at time intervals varying from 1 to 200 ms, by a test stimulation required to obtain a CMAP with 40% maximal amplitude. Then, these changes were expressed as the percentage of threshold change of the required test stimulation and plotted as a function of interstimulus intervals.
-
C4: the current–threshold relationship shows the axonal accommodation capacities to depolarization and hyperpolarization. It measured the threshold changes at the end of prolonged (200 ms) subthreshold conditioning ranging from 50 to 100% thresholds. The extent to which the slope of this relationship tended to increase in response to prolonged depolarizing and hyperpolarizing currents reflected outward and inward rectifications, respectively.
-
C5: the threshold electrotonus reflects the electrotonic changes in membrane potential. It refers to the threshold changes that occur during and after prolonged (100 ms) subthreshold conditioning depolarizing and hyperpolarizing currents applied at specified threshold percentages (20, 40 and 70%).
Statistical analysis
All data are expressed as means and standard deviation (± SD) of n different mice for each group. For the longitudinal behavioural studies, a two‐way ANOVA for repeated measures with Tukey's post hoc analysis was performed to compare all groups of mice and all ages (Systat v 8.0, SPSS Inc., Chicago, IL, USA). For other endpoint studies, a Kolmogorov–Smirnov normal distribution analysis was performed on all data followed by either Student's t test for normally distributed data or a non‐parametric Mann–Whitney U test, to verify significant differences between groups (Systat v 8.0). Statistical significance was considered when statistical power exceeds 95% (P ≤ 0.05; AnaStats.fr, France).
Results
Long‐term physical training improves resistance to muscle damages and aerobic performance in type 3 SMA‐like mice
We submitted a population of controls and type 3 SMA‐like mice to either a running or a swimming protocol for 10 months, as previously described (Grondard et al. 2008). To better characterize each exercise and to define their effects in an SMA context, we investigated the creatine kinase (CK) activity and lactate levels (indicators of potential exercise‐induced muscle lesions and energetic pathway used, respectively) in the serum of trained controls and type 3 SMA‐like mice in comparison to sedentary counterparts, at 2 and 12 months of age.
In control mice, we found that, in contrast to swimming, a single bout of running induced a significant increase in CK activity at 2 and 12 months of age (Fig. 1 A–C). Conversely and in contrast to running, a single bout of swimming exercise induced a 2‐fold increase in serum lactate levels at 2 and 12 months of age (Fig. 1 B). These data suggested that only the running‐based exercise induced muscle damage and that the swimming‐based exercise solicited more the anaerobic pathway than the running‐based training, as previously reported (Grondard et al. 2008).
Figure 1. Running‐ and swimming‐based 10‐month training programmes improve muscle resistance to exercise and aerobic capacity in type 3 SMA‐like mice .

A–D, quantification of creatine kinase activity (A and C) and lactate level (B and D) in the serum of sedentary control and type 3 SMA‐like mice (Control; SMA) at 2 months of age (A and B) after one bout of either running (One Run) or swimming (One Swim) exercise and in the serum of sedentary and 10‐month‐trained control and type 3 SMA‐like mice to running (Trained Run) or swimming (Trained Swim) at 12 months of age (C and D; n = 12 in each group). Data are represented as means ± SD (*P < 0.05).
In SMA mice, no alteration of the CK activity was detected at 2 and 12 months of age when compared to control mice, while the circulating lactate concentration levels were significantly 2‐fold higher at both ages in sedentary SMA mice (P < 0.05; Fig. 1 A and B). After a bout of exercise, independently of its type, we observed an elevation of both CK activity and lactate concentration compared to sedentary SMA mice at 2 (Fig. 1 A and B) and 12 months of age (data not shown). Interestingly, in type 3 SMA‐like mice and after 10 months of training, serum CK activity and lactate level response to a single exercise bout returned to control levels (Fig. 1 C and D). These data suggested that a 10‐month training programme in type 3 SMA‐like mice leads to (1) an improved muscle resistance to activity‐induced damage and (2) an increase in aerobic performance.
Long‐term physical training significantly improves type 3 SMA‐like mouse motor behaviour in a type‐specific manner
We next compared the impact of both physical exercises on the motor function, including motor behaviour and strength in the fore‐ and hindlimbs, of type 3 SMA‐like mice and age‐matched controls, from 2 to 12 months of age.
The grip strength test results indicated a significant decrease in maximal force from 6 months of age in the forelimbs and from 9 months of age in hindlimbs of sedentary type 3 SMA‐like mice (Fig. 2 A). Both exercise types enhanced muscular strength in type 3 SMA‐like mice reaching values comparable to sedentary controls throughout the tested age range (Fig. 2 A).
Figure 2. Running‐ and swimming‐based 10‐month training programmes differentially improve muscle strength and limit muscle fatigability in type 3 SMA‐like mice .

A and B, longitudinal measurements of the fore‐ and hindlimb muscle strength (Grip strength test; A) and the forelimb muscle fatigability to low intensity efforts (Grip test; B) of sedentary control mice (Sed Control) compared to sedentary (Sed SMA), running‐trained (Run SMA) and swimming‐trained (Swim SMA) type 3 SMA‐like mice from 2 to 12 months of age. C, longitudinal measurements of the muscle resistance to maximal intensity efforts in swimming pool of sedentary control (Sed Control) and type 3 SMA‐like (Sed SMA) mice compared to swimming‐based trained control (Swim Control) and type 3 SMA‐like (Swim SMA) mice or to running‐based trained type 3 SMA‐like (Run SMA) mice from 2 to 12 months of age (Swimming fatigability test). The minimal sustained water flow was 3 l min−1 and is indicated in the panel by ‘m’. D, measurement of the muscle resistance to maximal intensity efforts in treadmill of sedentary control (Sed Control) and type 3 SMA‐like (Sed SMA) mice compared to swimming‐based trained control (Swim Control) and type 3 SMA‐like (Swim SMA) mice or to running‐based trained type 3 SMA‐like (Run SMA) mice (Running fatigability test). The minimal sustained speed was fixed at 16 m min−1 and is indicated in the panel by ‘m’ (n = 12 in each group). Data are represented as means ± SD (*P < 0.05).
We then measured the muscular fatigability to low intensity effort (20% of maximum force) during a grip test. The resistance to fatigue for both control and type 3 SMA‐like mice increased significantly between 2 and 3 months of age, from 20 to 160 s. However, SMA‐like mice showed progressive decrease in their resistance to fatigue until 140 s, while control mice showed a significant increase reaching a plateau around 250 s at 9 months old (Fig. 2 B). After the running programme, muscle fatigability was significantly reduced in response to the low intensity effort in type 3 SMA‐like mice from 9 months of age. Interestingly, the swimming exercise improved muscle resistance to the low intensity test in both control and type 3 SMA‐like mice, with scores exceeding sedentary control levels (720 s at 6 months of age; Fig. 2 B).
When we submitted mice to the high intensity swimming fatigue test, we found a significantly higher fatigability in type 3 SMA‐like mice when compared to controls, with a decrease in the time to exhaustion from 3 months of age and in the sustained maximum water flow from 6 months of age, reaching 83 s and 3 l min−1 against 200 s and 6 l min−1, respectively (Fig. 2C). The running exercise significantly reduced fatigability in response to the high intensity effort in type 3 SMA‐like mice, while the swimming programme enhanced muscle resistance in both control and type 3 SMA‐like mice, with values exceeding control levels (500 s at 12 months of age; Fig. 2 C). These results were also confirmed in the high intensity running fatigue test at 12 months of age though the type 3 SMA‐like mice trained to swim did not exceed sedentary controls (Fig. 2 D).
Finally, no significant differences were observed in the open field test between all groups of mice from 2 to 12 months of age (Fig. 3 A). This result was confirmed at 12 months of age with the actimeter test of 48 h (Fig. 3 B). However, the number of vertical rearing events decreased significantly in type 3 SMA‐like compared to control mice at 12 months of age (Fig. 3 C). This parameter was improved by both exercise protocols in type 3 SMA‐like mice (Fig. 3 C). Taken together, these behavioural data suggested that both types of long‐term physical training were beneficial in terms of resistance to fatigue and motor behaviour in type 3 SMA‐like mice, with swimming being more effective than running.
Figure 3. Running‐ and swimming‐based 10‐month training programmes improve motor capacity despite specific adaptations in type 3 SMA‐like mice .
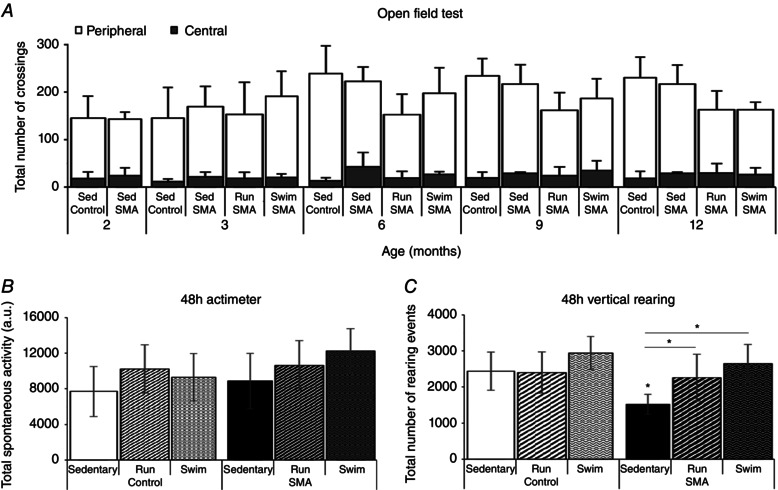
A, longitudinal measurements of the exploratory behaviour (Open field test) of sedentary control mice (Sed Control) compared to sedentary (Sed SMA), running‐trained (Run SMA) and swimming‐trained (Swim SMA) type 3 SMA‐like mice from 2 to 12 months of age. B and C, endpoint quantification of the spontaneous activity (B) and vertical rearing events (C) during 48 h (48 h actimeter and 48 h vertical rearing) in sedentary and running‐trained (Run) or swimming‐trained (swim) control and type 3 SMA‐like mice at 12 months of age (n = 12 in each group). Data are represented as means ± SD (*P < 0.05).
Specific exercise differentially protects motor neuron subpopulations in type 3 SMA‐like mice
In order to investigate the cellular mechanisms underlying the beneficial effects of physical training on the motor behaviour in type 3 SMA‐like mice, we analysed the motor neuron populations by counting the residual number of ChAT‐positive neurons in the ventral horn of the L1–L5 lumbar spinal cord in sedentary and trained control and type 3 SMA‐like mice at 12 months of age. The total number of motor neurons showed a marked decrease in the lumbar spinal cord of type 3 SMA‐like mice, with a 45% loss of ChAT‐positive neurons when compared to control mice (Fig. 4 A and B). Both exercise protocols significantly decreased motor neuron loss when compared to controls with the strongest efficiency observed with the swimming programme, reducing by half the motor neuron loss in SMA‐like mice (Fig. 4 A and B).
Figure 4. Running‐ and swimming‐based 10‐month training programmes induced neuroprotection of motor neuron subpopulations in type 3 SMA‐like mice .

A, immunodetection of ChAT‐positive motor neurons in the lumbar spinal cord (L1–L5) of sedentary control mice (left) compared to sedentary (middle left), running‐trained (middle right) and swimming‐trained (right) type 3 SMA‐like mice at 12 months of age (scale bar: 50 μm). B–D, quantitative analysis of the number of total, medial and lateral motor neurons per ventral horn (B) and of the absolute number (C) or proportion (D) of motor neurons in small (> 300 μm²), intermediate (300–600 μm²) and large (< 600 μm²) range of cell body area in the spinal cord of sedentary control mice compared to sedentary, running‐trained (Run) and swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 8 for each group). Data are represented as means ± SD (*P < 0.05).
To better characterize the neuroprotective effects of each exercise programme, we then analysed the different spinal motor neuron subpopulations, based on (1) their localization in the spinal cord (medial vs. lateral), (2) their soma size distribution (large vs. small area), and (3) their types (fast vs. slow).
Two different pools of motor neurons were distinguished on the basis of their cell body localization within the ventral spinal cord. Those with a cell body in the medial region of the ventral spinal cord are thought to specifically innervate proximal and axial muscles while those with a more laterally localized soma are thought to specifically innervate distal muscles (Bacskai et al. 2014). In the spinal cord of the sedentary type 3 SMA‐like mice, we observed a significant decrease in the number of medial motor neurons, from six to one motor neuron per slice when compared to sedentary controls, whereas the loss was limited to only 37% for lateral motor neurons (Fig. 4 B). Both exercise protocols efficiently limited medial motor neuron loss although swimming clearly provided more pronounced effects than running (Fig. 4 B).
We next analysed the different subpopulations of motor neurons according to the distribution of their soma size, in lumbar spinal cord (mean of 10 slices from L1 to L5). In sedentary type 3 SMA‐like mice, we observed a significant decrease in the absolute number of large (area greater than 600 μm2) and intermediate‐area motor neurons (area ranged between 300 and 600 μm2) in comparison to sedentary controls (48 in type 3 SMA‐like mice vs. 96 in control mice and 52 in type 3 SMA‐like mice vs. 101 in control mice). In contrast, the absolute number of small‐area motor neurons (less than 300 μm2) remained stable at around 14 (Fig. 4 C). Consequently, we noted a switch in the proportion of residual motor neurons from large to small in type 3 SMA‐like mice when compared to controls, unexpectedly suggesting a motor neuron type‐dependent degeneration in type 3 SMA‐like mice (Fig. 4 D). Both exercise protocols significantly reduced the loss of intermediate‐ and large‐area motor neurons, with a maximal protection noted for large motor neurons after the swimming programme (Fig. 4 C and D).
We finally compared the impact of each training programme on the survival of fast or slow motor neurons. Since fast motor neurons are shown to express chondrolectin (Chodl) and slow motor neurons oestrogen‐related receptor β (ERRβ) (Enjin et al. 2010), we performed either in situ hybridization for Chodl mRNA or immunolocalization of ERRβ in ChAT‐positive cells in ventral horn of the lumbar L1–L5 spinal cord (Fig. 5 A–D). As expected, we found that all Chodl‐positive cells exhibited large soma surfaces, up to 600 μm2, while all of ERRβ‐ChAT‐positive neurons exhibited soma surfaces smaller than 600 μm2 (data not shown). In sedentary type 3 SMA‐like mice, the numbers of Chodl‐positive neurons (Fig. 5 A) and ERRβ‐positive motor neurons (Fig. 5 C) were 2‐fold lower compared to sedentary controls (Fig. 5 B and D). Very interestingly, the swimming‐based training significantly reduced the loss of Chodl‐positive motor neurons reaching levels comparable to controls, but failed to protect ERRβ motor neurons (Fig. 5 A–D). Conversely, we found a significant running‐induced neuroprotection of the ERRβ‐positives motor neurons but no significant effects on Chodl‐positive neurons in type 3 SMA‐like mice (Fig. 5 A–D).
Figure 5. Exercise‐induced specific neuroprotection of fast vs. slow motor neuron subpopulations in type 3 SMA‐like mice is SMN independent .
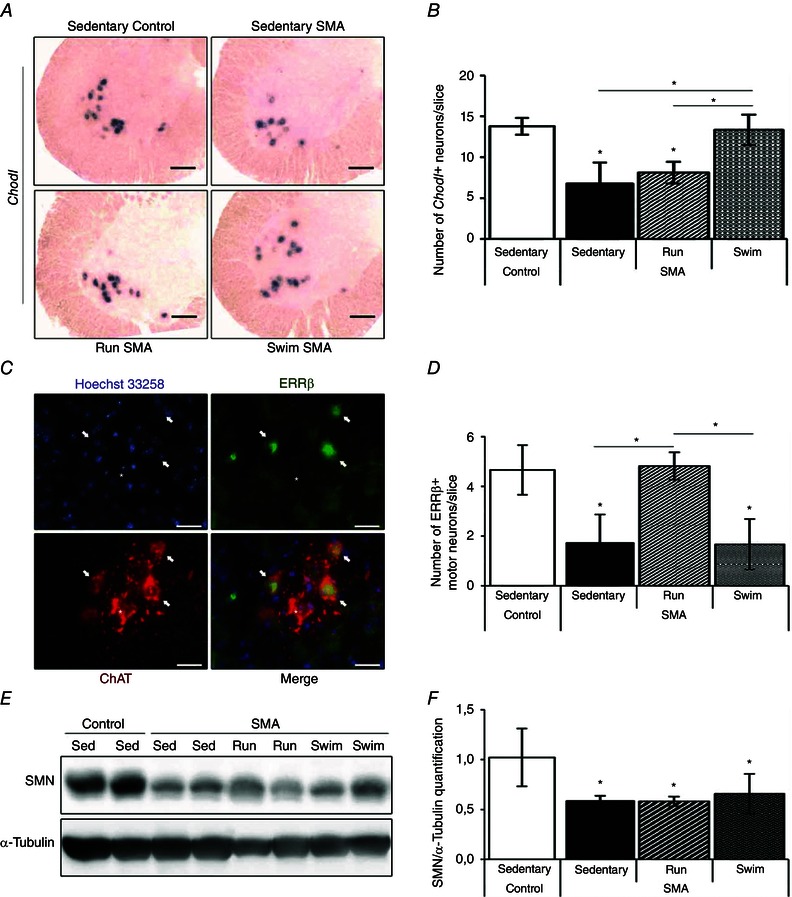
A, in situ hybridization on Chondrolectin mRNA (Chodl) in lumbar spinal cords of sedentary control (top left) and type 3 SMA‐like mice (top right) compared to running‐trained (bottom left) and swimming‐trained (bottom right) type 3 SMA‐like mice at 12 months of age (scale bar: 100 μm). B, quantitative analysis of the number of Chodl‐positive neurons in the ventral horn of lumbar spinal cords of sedentary control compared to sedentary, running‐trained (Run) and swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). C, immunodetection of the slow‐type motor neuron marker oestrogen‐related receptor β (ERRβ, green) in the nucleus (Hoechst 33258, blue) of ChAT‐positive motor neurons (ChAT, red) in the lumbar spinal cords of sedentary type 3 SMA‐like mice at 12 months of age (scale bar: 25 μm). D, quantitative analysis of the number of ERRβ‐positive motor neurons in the ventral horn of lumbar spinal cords of sedentary control mice compared to sedentary, running‐trained (Run) or swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). E and F, Western blot analysis (E) and quantification (F) of SMN protein expression in the ventral lumbar spinal cord of sedentary control and type 3 SMA‐like mice (Sed) compared to running‐trained (Run) or swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). Data are represented as means ± SD (*P < 0.05).
Finally, to determine whether such exercise‐induced neuroprotection could be linked to a modification in SMN protein expression, we performed Western blots on full‐length SMN protein in the ventral lumbar L1–L5 spinal cord of sedentary and trained controls and type 3 SMA‐like mice at 12 months of age. We found a 2‐fold decrease in SMN protein expression in type 3 SMA‐like mice when compared to controls (Fig. 5 E and F). Both physical training programmes were unable to induce SMN expression in the spinal cord, suggesting an SMN‐independent exercise‐induced benefit (Fig. 5 E and F).
Long‐term physical training protects neuromuscular junction structure in type 3 SMA‐like mice
We next analysed the shape of the neuromuscular junction (NMJ) for innervated NMJ using α‐bungarotoxin staining and neurofilament+synaptophysin immunostaining, as previously described (Biondi et al. 2008). Three different muscles of the calf, the extensor slow‐twitch soleus, the extensor fast‐twitch plantaris and the flexor fast‐twitch tibialis, were compared in control and type 3 SMA‐like mice. In adulthood, the pretzel shape organization could be pathologically affected according two main parameters: gutter fragmentation (Fig. 6 A) and total area alterations (Falk et al. 2014).
Figure 6. Running‐ and swimming‐based 10‐month training programmes reduce efficiently the neuromuscular junction defects in type 3 SMA‐like mice .

A–E, motor end‐plate labelling with α‐bungarotoxin (green) and anti‐neurofilament plus anti‐synaptophysin antibodies (NF+Synaptophysin, red), representing a typical pretzel‐shaped (A, left) and a fragmented NMJ (A, right), in the soleus of sedentary control mice (B), sedentary type 3 SMA‐like mice (C) and running‐trained (D) or swimming‐trained type 3 SMA‐like mice (E) at 12 months of age (scale bars: 5 μm for A and 10 μm for B–E). F and G, quantification of the neuromuscular junction fragmentation (F) and area (G) in the soleus, plantaris and tibialis muscles from sedentary control mice compared to sedentary, running‐trained (Run) or swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). Data are represented as means ± SD (*P < 0.05).
At 12 months of age, a significant increase in fragmented NMJs was only recorded in the two extensor soleus (Fig. 6 B and C) and plantaris muscles of sedentary type 3 SMA‐like mice compared to control mice (Fig. 6 F). Interestingly, both exercise protocols were able to significantly reduce SMA‐induced NMJ fragmentation, with a complete or a partial decrease in the plantaris and the soleus (Fig. 6 D and E) muscles, respectively, when compared to controls (Fig. 6 F).
Concerning the total gutter area at 12 months of age, only the plantaris NMJs appeared significantly affected by the disease, with a significant increase in the proportion of small and intermediate plantaris NMJ area, associated with a decrease in the proportion of large plantaris NMJ area in type 3 SMA‐like mice compared to controls (Fig. 6 G). Both exercise protocols slightly reduced the SMA‐induced decrease in NMJ area. In addition, only the running exercise induced an enlargement of the NMJ area in the soleus muscle (Fig. 6 G).
Specific exercise differentially limited muscle atrophy and fast‐to‐slow myofibre transitions in type 3 SMA‐like mice
Based on previous studies of SMA‐induced muscle alterations in diverse mice models (Biondi et al. 2008; Grondard et al. 2008; Boyer et al. 2013; Cobb et al. 2013), we analysed muscles alterations, in soleus, plantaris and tibialis of controls and type 3 SMA‐like mice at 12 months of age. Haematoxylin–eosin staining showed an atrophy in all the studied SMA muscles, with a significant decrease in the proportion of the largest myofibres (cross‐sectional area > 1500 μm² for both extensors and > 2500 μm² for the tibialis) in favour of the smallest myofibres (cross‐sectional area < 1500 μm² for soleus and tibialis and < 1000 μm² for plantaris; Fig. 7 A–D). Interestingly, both exercise types significantly reduced muscle atrophy and, in addition, induced an exercise‐specific hypertrophy when compared to sedentary control mice (Fig. 7 A–D). Accordingly, the running‐based exercise preferentially induced a hypertrophy of intermediate‐area myofibres for the two extensor muscles and of large‐area myofibres for the flexor tibialis, whereas the swimming‐based exercise preferentially induced a hypertrophy of the largest myofibres in the three muscles (Fig. 7 B–D).
Figure 7. Exercise‐specific adaptations of skeletal muscles in type 3 SMA‐like mice .

A, haematoxylin–eosin staining of plantaris of sedentary control mice (top left) compared to sedentary (top right), running‐trained (bottom left) and swimming‐trained (bottom right) type 3 SMA‐like mice at 12 months of age (scale bar: 25 μm). B–D, quantification of the cross‐sectional area of myofibres from soleus (B), plantaris (C) and tibialis (D) muscles of sedentary control mice compared to sedentary, running‐trained (Run) and swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). E, immunodetection of type IIa myosin heavy chain (MyHC) in the plantaris of sedentary control mice (top left) compared to sedentary (top right), running‐trained (bottom left) and swimming‐trained (bottom right) type 3 SMA‐like mice at 12 months of age (scale bar: 50 μm). F, analysis of adult (i.e. I, II, IIa and IIx/IIb) MyHC isoforms typology of the soleus, plantaris and tibialis of sedentary control mice compared to sedentary, running‐trained (Run) and swimming‐trained (Swim) type 3 SMA‐like mice at 12 months of age (n = 4 in each group). Data are represented as means ± SD (*P < 0.05).
Concerning the muscle typology, we next determined the percentage of myofibres expressing slow (type I), intermediate (type IIa) or fast (IIx+IIb) myosin heavy chain isoforms, a signature of the myofibre metabolic profile (Schiaffino & Reggiani, 2011), as previously described (Biondi et al. 2008, 2010). At 12 months of age, we noted a transition to a slower, more oxidative, phenotype in the three SMA muscles compared to controls, as revealed by a gain in type I fibres for the soleus muscle and in type IIa fibres for the two fast‐twitch muscles. Thus, the two extensor muscles were more affected than the flexor muscles in type 3 SMA‐like mice (Fig. 7 E and F). The swimming exercise reduced SMA‐induced myofibre transitions in type 3 SMA‐like mice (Fig. 7 F), restoring the type I proportion and favouring the more glycolytic intermediate and fast‐twitch myofibres (Fig. 7 F). In contrast, the running‐based training led to a gain in type I myofibres compared to sedentary SMA‐like mice (Fig. 7 F).
Long‐term physical training significantly improved alterations of neuromuscular excitability in type 3 SMA‐like mice
Finally, to ascertain that the exercise‐induced protection of the motor unit structure resulted in retained functional properties, we evaluated, in vivo, the neuromuscular excitability properties of hindlimb plantar muscle in all groups of mice. Profound significant alterations of neuromuscular excitability waveforms (Fig. 8 A) and associated analysed parameters (Table 1) were detected in 12‐month‐old sedentary type 3 SMA‐like mice when compared to age‐matched controls. In particular, for the stimulus–response relationship analysis (label C1, Table 1), a significant 11% enhanced latency (parameter 19 in Table 1) between the nerve stimulation onset and the CMAP peak amplitude was recorded in sedentary SMA‐like mice compared to sedentary controls. In addition, as revealed by the strength–duration relationship analysis (label C2, Fig. 8 A and Table 1), the chronaxie (parameter 03 in Table 1) was 31% lower in sedentary SMA‐like mice compared to control mice. Furthermore, as derived from the recovery cycle analysis (label C3, Fig. 8 A and Table 1), the sedentary SMA‐like mice displayed a 42% higher and 15% longer refractory period than controls, as determined by (1) the percentage of threshold change at 2 ms interstimulus interval (parameter 32 in Table 1) and (2) the relative refractory period (parameter 10, i.e. the first x‐intercept in Table 1), respectively. The SMA‐like mice also displayed a 60–76% lower supernormal period, as determined by the percentage of threshold change between 5 and 7 ms interstimulus intervals (parameters 13, 34 and 33 in Table 1). The current–threshold relationship analysis (label C4, Fig. 8 A and Table 1) showed that the most prominent abnormality in sedentary SMA mice was a 23% reduction of the hyperpolarizing slope (parameter 28 in Table 1). Finally, abnormalities in threshold electrotonus analysis (label C5, Fig. 8A and Table 1) were a 14–18% lower threshold increase in response to the hyperpolarizing current (parameters 22, 30, 11 and 31 in Table 1) in sedentary SMA‐like mice compared to controls. Taken together, these results suggest a hyperexcitability of the neuromuscular system in sedentary SMA‐like mice due to axonal membrane depolarization.
Figure 8. Running‐ and swimming‐based 10‐month training programmes counteract the neuromuscular excitability defect in a type 3 SMA‐like mice .
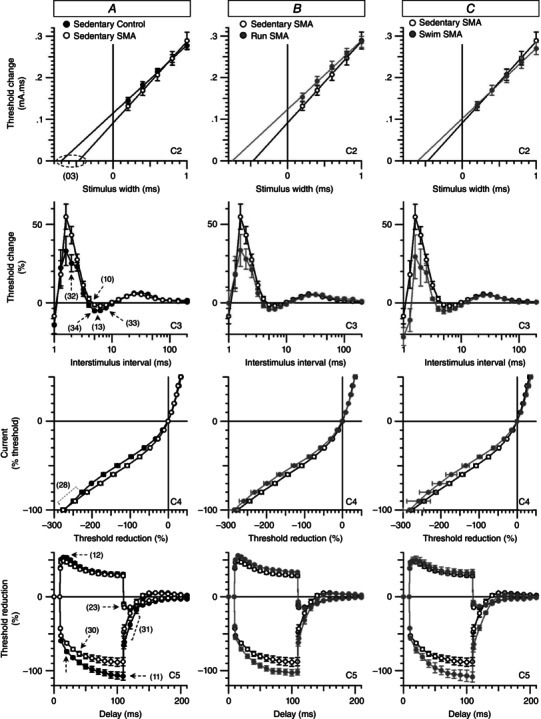
A–C, superimposed excitability waveforms obtained in vivo by stimulating the tibial branch of the sciatic nerve and recording the CMAP from plantar muscle in sedentary control mice compared to sedentary type 3 SMA‐like mice (A) and in sedentary type 3 SMA‐like mice compared to either running‐trained (B) or swimming‐trained (C) type 3 SMA‐like mice. The significant differences in excitability parameters between sedentary control and SMA mice are indicated by numbers and dashed lines or arrows. C2, strength–duration relationship. C3, recovery cycle. C4, current–threshold relationship. C5, threshold electrotonus (subthreshold conditioning depolarizing and hyperpolarizing current set to 40%). Data are represented as means ± SD of 4–8 animals per group (*P < 0.05).
Table 1.
Comparison of neuromuscular excitability parameters
| Curve a | Derived excitability parameter b | Sedentary control | Sedentary SMA | Run SMA | Swim SMA |
|---|---|---|---|---|---|
| C1 | 06: Peak response (mV) | 5.2 ± 1.2 | 4.4 ± 1.21 | 5 ± 1.2 | 4.6 ± 1.2 |
| C1 | 19: Latency (ms) | 2.9 ± 0.08 | 3.3 ± 0.1* | 3 ± 0.1 | 2.9 ± 0.2 |
| C1 | 01: Stimulus (mA) for 50% max response | 0.2 ± 1.08 | 0.2 ± 1.1 | 0.2 ± 1 | 0.27 ± 1.2 |
| C1 | 05: Stimulus–response slope | 4.4 ± 1.17 | 3.7 ± 1.1 | 4.9 ± 1.2 | 4.3 ± 1.2 |
| C2 | 03: Strength–duration time constant (ms) | 0.7 ± 0.05 | 0.5 ± 0.04* | 0.7 ± 0.04 | 0.6 ± 0.02 |
| C2 | 04: Rheobase (mA) | 0.16 ± 0,01 | 0.2 ± 0.01 | 0.17 ± 1.03 | 0.16 ± 1.05 |
| C3 | 10: Relative refractory period (ms) | 3.6 ± 0.1 | 4.3 ± 0.2* | 3.6 ± 1.0 | 3.84 ± 1.1 |
| C3 | 32: Refractoriness at 2 ms (%) | 25.1 ± 5.3 | 43.1 ± 5.5* | 27.8 ± 6.4 | 22.6 ± 11.7 |
| C3 | 13: Superexcitability (%) | −7.9 ± 2.1 | −2.4 ± 0.8* | −5.6 ± 1.7 | −6.5 ± 1.9 |
| C3 | 34: Superexcitability at 5 ms (%) | −5 ± 0.7 | −1.2 ± 1.3* | −4.2 ± 0.4 | −5.1 ± 1.6 |
| C3 | 33: Superexcitability at 7 ms (%) | −4 ± 0.5 | −1.6 ± 1* | −3.2 ± 0.6 | −4 ± 0.9 |
| C3 | 14: Subexcitability (%) | 6 ± 0.4 | 5.2 ± 0.3* | 4.7 ± 0.8 | 4.6 ± 0.4 |
| C4 | 07: Resting I–V slope | 0.8 ± 0.05 | 1.1 ± 0.1 | 0.9 ± 0.04 | 0.8 ± 0.07 |
| C4 | 08: Minimum I–V slope | 0.2 ± 0,01 | 0.25 ± 0.01 | 0.25 ± 0.01 | 0.26 ± 0.02 |
| C4 | 28: Hyperpolarization I–V slope | 0.4 ± 0.01 | 0.32 ± 0.01* | 0.41 ± 0.05 | 0.5 ± 0.1 |
| C5 | 12: TEd (10–20 ms) | 50.2 ± 1.9 | 44.6 ± 2.2 | 51.4 ± 1.3 | 48.3 ± 5 |
| C5 | 20: TEd (40–60 ms) | 35.3 ± 1.9 | 32.9 ± 2.1 | 38.3 ± 1.2 | 35.8 ± 4.6 |
| C5 | 21: TEd (90–100 ms) | 30.6 ± 2.2 | 28.5 ± 2.1 | 33.5 ± 1 | 32.5 ± 4.1 |
| C5 | 22: TEh (10–20 ms) | −78.2 ± 1.9 | −66.8 ± 2.2* | −75.7 ± 2.7 | −76.5 ± 4.2 |
| C5 | 30: TEh (20–40 ms) | −88.5 ± 2.7 | −76.3 ± 3.1* | −87.3 ± 3.1 | −89.5 ± 5.3 |
| C5 | 11: TEh (90–100 ms) | −106.9 ± 5 | −88.1 ± 5.6* | −102.4 ± 4.2 | −107.7 ± 7.3 |
| C5 | 31: TEh (slope 101–140 ms) | 1.62 ± 0.1 | 1.2 ± 0.1* | 1.4 ± 0.09 | 1.7 ± 0.1 |
aC1: stimulus–response relationship; C2: strength–duration relationship; C3: recovery cycle; C4: current–threshold relationship; and C5: threshold electrotonus. bTEd: threshold electrotonus from depolarizing currents (40%); TEh: threshold electrotonus from hyperpolarizing currents (40%). Derived excitability parameters (means ± SD) from plantar muscle recordings from sedentary controls (n = 8), sedentary SMA (n = 8), running‐trained SMA (Run SMA; n = 7) and swimming‐trained SMA (Swim SMA; n = 4) mice. Each group of SMA mice were compared to sedentary control animals and differences were considered significant when P ≤ 0.05 ( * and highlighted in grey).
Importantly, all these alterations were counteracted by both exercise protocols in type 3 SMA‐like mice, as revealed by the analysis of the neuromuscular excitability waveforms (Fig. 8 B and C). In addition, excitability parameters of 12‐month‐old running and swimming type 3 SMA‐like mice reached levels comparable to age‐matched controls (Table 1).
Discussion
Admittedly, submitting SMA patients to an exercise programme may be considered hazardous due to motor weakness, muscular fatigue of patients and lack of studies investigating physical training effects in SMA (Preisler et al. 2009; Lewelt et al. 2015). The present study provides the first lines of evidence indicating that two different long‐term exercises could be beneficial for mild SMA. While the running and swimming protocols used here were based on different velocity, amplitude of movement, energetic metabolic involvement and motor unit recruitment (Grondard et al. 2008), they both resulted in significant improvements in motor performance (grip strength), reduction in muscle fatigability (grip test and fatigability test), motor neuron neuroprotection, and prevention of NMJ shape and muscle phenotype alterations in a type 3 SMA‐like mouse model (Table 2). However, each exercise protocol exhibited specificities, notably regarding the effects on different motor neuron populations and on specific muscle fibres. The swimming protocol efficiently protected fast motor neurons, while the running protocol only protected slow motor neurons. Furthermore, the swimming protocol significantly increased muscle resistance to fatigability to reach higher values compared to sedentary control mice, while running slightly prevented the appearance of muscle fatigability. Finally, swimming exercise preferentially enhanced the cross‐sectional area of large myofibres while running enhanced the cross‐sectional area of intermediate myofibres (Table 2). In addition, the present study strongly suggests that the forced and repetitive use of motor units sufficiently protects them from SMA‐induced alterations, independently of SMN gene expression.
Table 2.
Comparison of running and swimming benefits in type 3 SMA‐like mice
| Physical exercise improvements | Run SMA | Swim SMA |
|---|---|---|
| Grip strength preservation | +++ | +++ |
| Muscle resistance to low intensity test | + | +++ |
| Muscle resistance to high intensity test | + | +++ |
| Motor neuron protection | ++ | +++ |
| Medial motor neurons | ++ | +++ |
| Large motor neurons | + | +++ |
| Chodl + neurons | − | +++ |
| ERRβ+ motor neurons | +++ | − |
| Preservation of NMJ shape | +++ | +++ |
| Muscle hypertrophy (increased CSA) | +++ | +++ |
| Intermediate myofibres | +++ | ++ |
| Large myofibres | + | +++ |
| Neuromuscular excitability | +++ | +++ |
Comparison of the 10‐month running and swimming exercises efficiency of the analysed parameters (motor capacity and motor unit) which have been improved by physical exercise in the type 3 SMA‐like mice. The level of efficiency to improve each parameter is represented by ‘–’ for no efficiency, + for low efficiency (under sedentary control levels), ++ for moderate efficiency and +++ for high efficiency (equal or superior to sedentary control levels). The grey highlighting indicate which exercise induced the better effect on a considered parameter.
Importantly, both exercise protocols efficiently protected the subpopulation of motor neurons that are particularly sensitive to SMA (Mentis et al. 2011). In type 3 SMA‐like mice, the main part of the medial motor neuron population, innervating the proximal and axial muscles, had disappeared as already observed in a severe SMA mouse model (Mentis et al. 2011), while the lateral motor neurons, innervating distal muscles, remained present at 62%. Whatever the cause of the preferential loss of medial motor neurons, a significant protection was shown with both exercise paradigms, with the highest efficiency obtained with swimming exercise. This swimming efficiency could be explained by the higher solicitation of proximal muscles to maintain the body afloat compared to running. This hypothesis, linking the activation levels of a specific motor unit to the efficacy of its motor neuron protection, is further supported by the preferential neuroprotection of motor neurons subpopulations by specific exercise. Yet, the low intensity and amplitude running exercise was shown to preferentially recruit the slow and intermediate motor units (Grondard et al. 2008) and we observed that this type of exercise protected essentially small cell body ERRβ+ motor neurons, referred to as slow motor neurons (Enjin et al. 2010). In contrast, the high intensity and amplitude swimming exercise was shown to recruit intermediate and fast motor units (Grondard et al. 2008) and we observed that this type of exercise protected preferentially large cell body Chodl + motor neurons, referred to as fast motor neurons (Enjin et al. 2010).
The fact that the exercise‐induced neuroprotection in type 3 SMA‐like mice occurred independently of SMN protein expression is puzzling and unexpected when compared with the effects of a 5‐day training programme performed in type 2 SMA‐like mice (Grondard et al. 2005). In contrast to the former study (Grondard et al. 2005), performed in immature 8‐day‐old mice (Biondi et al. 2008), the present study used an adult type 3 SMA mouse model characterized by low expression levels of SMN compared to more precocious ages. Undoubtedly, the molecular mechanisms underlying the beneficial effects of exercise in each case would be specifically linked to the structure of the responding system. Furthermore, several compounds such as Rho‐associated protein kinase (ROCK) inhibitors (Bowerman et al. 2010) or the Histone deacetylases (HDAC) inhibitor trichostatin A (Liu et al. 2014) were also shown to significantly improve the motor behaviour and motor unit state of an intermediate SMA‐like mouse model independently of SMN expression.
Interestingly, several previous studies have highlighted the importance of sensory–motor circuits in the physiopathology of SMA (Mentis et al. 2011; Imlach et al. 2012). Yet, alterations of proprioceptive neurons and cholinergic interneurons have been postulated to represent the primary cause of motor neuron and muscle dysfunctions in SMA, based on cellular and electrophysiological studies in the spinal cord of severe type SMA mice and in Drosophila (Mentis et al. 2011; Imlach et al. 2012). Since physical exercise is known to systematically involve myotatic reflexes to control the muscle contraction and the antagonist muscle coordination, it is tempting to speculate that both exercises exerted their beneficial effects on SMA motor neurons through an improvement of sensory–motor circuits, and de facto increasing the number of synaptic inputs on motor neurons, including proprioceptive inputs.
In addition to protecting motor neurons in SMA mice, both exercise protocols improved neuromuscular excitability properties. Several specific perturbations of the neuromuscular functions have already been reported in SMA, including (1) a CMAP decrease (Kissel et al. 2011), (2) motor neuron hyperexcitability (Mentis et al. 2011) and (3) an increase in latency (Yonekawa et al. 2013). However, if we confirmed some of these electrophysiology ‘markers’ of SMA such as increased latency (parameter 19, C1), we described here for the first time higher and longer refractory periods (parameters 32 and 10, C3), lower supernormal period (parameters 13, 34 and 33, C3) and smaller threshold changes in sedentary type 3 SMA‐like mice, suggesting axonal motor neuron membrane depolarization through increased excitability of the SMA neuromuscular system (Kiernan & Bostock, 2000; Kiernan et al. 2002). These alterations could be linked to a dysfunction either in Na+ and K+ channels along the motor neuron axons or in the oxygen‐dependent Na+/K+‐ATPase (Kiernan & Bostock, 2000). Importantly, both physical exercises were able to counteract these neuromuscular defects, returning to control electrophysiological values. Therefore, our results concur with an exercise‐induced compensatory hyperactivity of the Na+/K+‐ATPase or a recovery of ion channel functionality, leading to membrane hyperpolarization.
Taken together, our results support the concept of a double adaptive effect in type 3 SMA‐like mice, involving both an intrinsic muscle adaptation and an exercise‐induced specific neuroprotection. Indeed, the running exercise induced a transition from type II to type I myofibres in the context of preferential neuroprotection of slow motor neurons, which could be attributable to exercise‐induced neuroprotection, but also a hypertrophy of intermediate fibres or large fibres, which may depend on intrinsic muscle adaptations. The swimming exercise induced a transition from type IIx/IIb to type IIa myofibres in both fast‐twitch muscles with a hypertrophy of large fibres, which may result from intrinsic muscle adaptations, in a context of maintenance of IIx/IIb myofibres, likely to be attributable to the neuroprotection of fast motor neurons.
From a mechanistic point of view, the chemo‐communications between motor neurons and muscles are likely to be involved in the specific effects of each exercise protocol on SMA motor unit adaptations. These communications occur through growth/neurotrophic factor release in an autocrine/paracrine mode and through axonal transport (de la Cruz et al. 1996; Wright et al. 1997; Chowdary et al. 2012; Glat et al. 2016). Interestingly, the axonal transport at the levels of the sciatic nerve has been shown to be differentially modulated by the type of physical solicitation, i.e. swim or run (Jasmin et al. 1987), thus potentially representing a likely mechanism to explain, at least partly, the specific effects of each exercise on SMA motor units.
Furthermore, based on structural (NMJ and myofibre phenotypes) and on behavioural data (vertical rearing events), we confirmed the preferential alteration of extensor muscles in the type 3 SMA‐like mouse model (Biondi et al. 2008). In both exercise types, the extensors muscles are solicited, notably during the pushing phase of the movement (Gruner et al. 1980). The fact that these muscles were significantly improved by both exercises further substantiated the hypothesis of a direct link between activity and neuroprotection leading to beneficial motor unit adaptations.
Interestingly, several exercise‐induced adaptations observed in SMA, such as increased muscle resistance to high and low intensity efforts, myofibre hypertrophy and muscle typology adaptations, were also found in controls. These observations suggest that SMA‐like mice respond adequately to different physical exercise workloads. Thus, because each exercise type enhances specific motor unit adaptations, the exercise prescription should take into account the disease context and the expected motor unit adaptations.
Finally and most importantly, the present study provides several clues for designing rehabilitation protocols for mild SMA patients. The swimming‐based training clearly provided the most profound benefits in terms of neuroprotection, muscle phenotype maintenance and motor behaviour, including a significant increase in muscle resistance to fatigue. Nevertheless, the running exercise also had specific beneficial effects on SMA motor units, i.e. protection of slow motor neurons and hypertrophy of intermediate myofibres, suggesting that the design of active physiotherapy protocols based on a mixture of running and swimming exercises could synergize the specific benefits induced by each exercise. The only limitation of such application could be the muscle weakness of type 2 and some type 3 SMA patients, especially on hindlimbs, due to high gravity strains. Therefore, short periods of high intensity swimming exercise essentially targeting the recruitment of extensor muscles should be clinically relevant for moderately severe SMA patients.
Additional information
Competing interests
The authors declare no competing financial interest. A patent on active physiotherapy for SMA patients is pending.
Author contributions
F.C. conducted and analysed the majority of experiments. C.D. and L.H. conducted the Western blot experiments and participated in the behavioural testing experiments. E.B. conducted and analysed the electrophysiological experiments on neuromuscular excitability. T.R. and B.B. conducted and analysed the spontaneous activity and vertical rearing experiments. B.D.G. conducted the in situ hybridization experiments. J.B. conducted NMJ experiments and developmental analysis of NMJ and muscle. P.L. conducted the behaviour analysis and helped in writing the manuscript. C.P. assisted in the majority of experiments and helped in writing the manuscript. C.C. helped in the analysis of muscle data. F.C. helped in supervising the project and experiments and helped in writing the manuscript. O.B. supervised the project and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
F.C. is the recipient of a PhD fellowship from the AXA Research fund and Fondation Garches. This work is supported by AFM (Association Française contre les Myopathies).
Translational perspective
Growing evidence from the last decade indicates the positive impact of physical exercise on neuromuscular disorders, including Parkinson's disease and spinal muscular atrophy (SMA). However, in some cases like amyotrophic lateral sclerosis and Duchenne muscular dystrophy, the potential role of exercise remains under debate. ‘Physical exercise’ is a term that covers a wide range of conditions, including voluntary vs. forced and long‐term vs. acute exercise, concentric vs. eccentric contractions and anaerobic vs. oxidative energetic pathways. The present study evaluated whether two different forced long‐term exercises (10 consecutive months), based either on a low intensity running exercise, favouring the use of oxidative energetic pathways, or on a high intensity swimming exercise, favouring the use of anaerobic energetic pathways, could be beneficial for mild type SMA‐like mice. We reported that while both types of exercise were beneficial, the motor unit structure and function did not equally respond: swimming‐based exercise revealed further beneficial effects compared to running, especially on the protection of specific motor neuron subpopulations. Only the slow motor neurons have been protected after running, while both intermediate and fast motor neurons have been protected after swimming, leading to specific adaptations of other components of the motor unit, notably muscle fibres. Therefore, the present study reinforces the idea that a better understanding of the effects of ‘well controlled’ physical exercise on motor units could provide several clues when designing rehabilitation programmes for SMA patients. This work has been used to design an innovative clinical trial for SMA patients, which is currently ongoing in France.
Acknowledgements
All the authors wish to thank C. Mader and J. Coppet for animal care and Klas Kullander and its laboratory for delivery of in situ hybridization probes.
References
- Bacskai T, Rusznak Z, Paxinos G & Watson C (2014). Musculotopic organization of the motor neurons supplying the mouse hindlimb muscles: a quantitative study using Fluoro‐Gold retrograde tracing. Brain Struct Funct 219, 303–321. [DOI] [PubMed] [Google Scholar]
- Biondi O, Branchu J, Sanchez G, Lancelin C, Deforges S, Lopes P, Pariset C, Lecolle S, Cote J, Chanoine C & Charbonnier F (2010). In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J Neurosci 30, 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O, Grondard C, Lecolle S, Deforges S, Pariset C, Lopes P, Cifuentes‐Diaz C, Li H, della Gaspera B, Chanoine C & Charbonnier F (2008). Exercise‐induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 28, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladen CL, Thompson R, Jackson JM, Garland C, Wegel C, Ambrosini A, Pisano P, Walter MC, Schreiber O, Lusakowska A, Jedrzejowska M, Kostera‐Pruszczyk A, van der Pol L, Wadman RI, Gredal O, Karaduman A, Topaloglu H, Yilmaz O, Matyushenko V, Rasic VM, Kosac A, Karcagi V, Garami M, Herczegfalvi A, Monges S, Moresco A, Chertkoff L, Chamova T, Guergueltcheva V, Butoianu N, Craiu D, Korngut L, Campbell C, Haberlova J, Strenkova J, Alejandro M, Jimenez A, Ortiz GG, Enriquez GV, Rodrigues M, Roxburgh R, Dawkins H, Youngs L, Lahdetie J, Angelkova N, Saugier‐Veber P, Cuisset JM, Bloetzer C, Jeannet PY, Klein A, Nascimento A, Tizzano E, Salgado D, Mercuri E, Sejersen T, Kirschner J, Rafferty K, Straub V, Bushby K, Verschuuren J, Beroud C & Lochmuller H (2014). Mapping the differences in care for 5,000 spinal muscular atrophy patients, a survey of 24 national registries in North America, Australasia and Europe. J Neurol 261, 152–163. [DOI] [PubMed] [Google Scholar]
- Boerio D, Greensmith L & Bostock H (2009). Excitability properties of motor axons in the maturing mouse. J Peripher Nerv Syst 14, 45–53. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Beauvais A, Anderson CL & Kothary R (2010). Rho‐kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet 19, 1468–1478. [DOI] [PubMed] [Google Scholar]
- Boyer JG, Murray LM, Scott K, De Repentigny Y, Renaud JM & Kothary R (2013). Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet Muscle 3, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdary PD, Che DL & Cui B (2012). Neurotrophin signaling via long‐distance axonal transport. Annu Rev Phys Chem 63, 571–594. [DOI] [PubMed] [Google Scholar]
- Cobb MS, Rose FF, Rindt H, Glascock JJ, Shababi M, Miller MR, Osman EY, Yen PF, Garcia ML, Martin BR, Wetz MJ, Mazzasette C, Feng Z, Ko CP & Lorson CL (2013). Development and characterization of an SMN2‐based intermediate mouse model of Spinal Muscular Atrophy. Hum Mol Genet 22, 1843–1855. [DOI] [PubMed] [Google Scholar]
- Deforges S, Branchu J, Biondi O, Grondard C, Pariset C, Lecolle S, Lopes P, Vidal PP, Chanoine C & Charbonnier F (2009). Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J Physiol 587, 3561–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz RR, Pastor AM & Delgado‐Garcia JM (1996). Influence of the postsynaptic target on the functional properties of neurons in the adult mammalian central nervous system. Rev Neurosci 7, 115–149. [DOI] [PubMed] [Google Scholar]
- Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Memic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ & Kullander K (2010). Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J Comp Neurol 518, 2284–2304. [DOI] [PubMed] [Google Scholar]
- Falk DJ, Todd AG, Lee S, Soustek MS, ElMallah MK, Fuller DD, Notterpek L & Byrne BJ (2014). Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet 24, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glat MJ, Benninger F, Barhum Y, Ben‐Zur T, Kogan E, Steiner I, Yaffe D & Offen D (2016). Ectopic muscle expression of neurotrophic factors improves recovery after nerve injury. J Mol Neurosci 58, 39–45. [DOI] [PubMed] [Google Scholar]
- Goes AT, Souza LC, Filho CB, Del Fabbro L, De Gomes MG, Boeira SP & Jesse CR (2014). Neuroprotective effects of swimming training in a mouse model of Parkinson's disease induced by 6‐hydroxydopamine. Neuroscience 256, 61–71. [DOI] [PubMed] [Google Scholar]
- Grondard C, Biondi O, Armand AS, Lecolle S, Della Gaspera B, Pariset C, Li H, Gallien CL, Vidal PP, Chanoine C & Charbonnier F (2005). Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 25, 7615–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondard C, Biondi O, Pariset C, Lopes P, Deforges S, Lecolle S, Gaspera BD, Gallien CL, Chanoine C & Charbonnier F (2008). Exercise‐induced modulation of calcineurin activity parallels the time course of myofibre transitions. J Cell Physiol 214, 126–135. [DOI] [PubMed] [Google Scholar]
- Gruner JA, Altman J & Spivack N (1980). Effects of arrested cerebellar development on locomotion in the rat. Cinematographic and electromyographic analysis. Exp Brain Res 40, 361–373. [DOI] [PubMed] [Google Scholar]
- Harding AE & Thomas PK (1980). The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103, 259–280. [DOI] [PubMed] [Google Scholar]
- Hsieh‐Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH & Li H (2000). A mouse model for spinal muscular atrophy. Nat Genet 24, 66–70. [DOI] [PubMed] [Google Scholar]
- Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L & McCabe BD (2012). SMN is required for sensory‐motor circuit function in Drosophila. Cell 151, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin BJ, Lavoie PA & Gardiner PF (1987). Fast axonal transport of acetylcholinesterase in rat sciatic motoneurons is enhanced following prolonged daily running, but not following swimming. Neurosci Lett 78, 156–160. [DOI] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno‐Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT & Monani UR (2008). Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17, 2552–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan MC & Bostock H (2000). Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 123, 2542–2551. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Walters RJ, Andersen KV, Taube D, Murray NM & Bostock H (2002). Nerve excitability changes in chronic renal failure indicate membrane depolarization due to hyperkalaemia. Brain 125, 1366–1378. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Hernandez D, Bradley WG & Moraes CT (2003). Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann Neurol 53, 804–807. [DOI] [PubMed] [Google Scholar]
- Kissel JT, Scott CB, Reyna SP, Crawford TO, Simard LR, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D'Anjou G, LaSalle B, Prior TW, Sorenson S, Maczulski JA, Bromberg MB, Chan GM & Swoboda KJ (2011). SMA CARNIVAL TRIAL PART II: a prospective, single‐armed trial of L‐carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One 6, e21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch‐Marce M, Griffin JW, Rich MM & Sumner CJ (2009). Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M et al (1995). Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 80, 155–165. [DOI] [PubMed] [Google Scholar]
- Lewelt A, Krosschell KJ, Stoddard GJ, Weng C, Xue M, Marcus RL, Gappmaier E, Viollet L, Johnson BA, White AT, Viazzo‐Trussell D, Lopes P, Lane RH, Carey JC & Swoboda KJ (2015). Resistance strength training exercise in children with spinal muscular atrophy. Muscle Nerve 52, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebetanz D, Hagemann K, von Lewinski F, Kahler E & Paulus W (2004). Extensive exercise is not harmful in amyotrophic lateral sclerosis. Eur J Neurosci 20, 3115–3120. [DOI] [PubMed] [Google Scholar]
- Liu H, Yazdani A, Murray LM, Beauvais A & Kothary R (2014). The Smn‐independent beneficial effects of trichostatin A on an intermediate mouse model of spinal muscular atrophy. PLoS One 9, e101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HL, Zhao G, Zhang H & Shi LD (2013). Long‐term treadmill exercise inhibits the progression of Alzheimer's disease‐like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res 256, 261–272. [DOI] [PubMed] [Google Scholar]
- Lorson CL & Androphy EJ (2000). An exonic enhancer is required for inclusion of an essential exon in the SMA‐determining gene SMN. Hum Mol Genet 9, 259–265. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH & Androphy EJ (1998). SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 19, 63–66. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ & O'Donovan MJ (2011). Early functional impairment of sensory‐motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodera H & Kaji R (2006). Nerve excitability testing and its clinical application to neuromuscular diseases. Clin Neurophysiol 117, 1902–1916. [DOI] [PubMed] [Google Scholar]
- Preisler N, Andersen G, Thogersen F, Crone C, Jeppesen TD, Wibrand F & Vissing J (2009). Effect of aerobic training in patients with spinal and bulbar muscular atrophy (Kennedy disease). Neurology 72, 317–323. [DOI] [PubMed] [Google Scholar]
- Russman BS, Melchreit R & Drennan JC (1983). Spinal muscular atrophy: the natural course of disease. Muscle Nerve 6, 179–181. [DOI] [PubMed] [Google Scholar]
- Schiaffino S & Reggiani C (2011). Fiber types in mammalian skeletal muscles. Physiol Rev 91, 1447–1531. [DOI] [PubMed] [Google Scholar]
- Towbin H, Schoenenberger C, Ball R, Braun DG & Rosenfelder G (1984). Glycosphingolipid‐blotting: an immunological detection procedure after separation by thin layer chromatography. J Immunol Methods 72, 471–479. [DOI] [PubMed] [Google Scholar]
- Tsai LK, Tsai MS, Lin TB, Hwu WL & Li H (2006). Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol Dis 24, 286–295. [DOI] [PubMed] [Google Scholar]
- Tuon T, Valvassori SS, Lopes‐Borges J, Luciano T, Trom CB, Silva LA, Quevedo J, Souza CT, Lira FS & Pinho RA (2012). Physical training exerts neuroprotective effects in the regulation of neurochemical factors in an animal model of Parkinson's disease. Neuroscience 227, 305–312. [DOI] [PubMed] [Google Scholar]
- Veldink JH, Bar PR, Joosten EA, Otten M, Wokke JH & van den Berg LH (2003). Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord 13, 737–743. [DOI] [PubMed] [Google Scholar]
- Vianello S, Bouyon S, Benoit E, Sebrie C, Boerio D, Herbin M, Roulot M, Fromes Y & de la Porte S (2014). Arginine butyrate per os protects mdx mice against cardiomyopathy, kyphosis and changes in axonal excitability. Neurobiol Dis 71, 325–333. [DOI] [PubMed] [Google Scholar]
- Wright DE, Zhou L, Kucera J & Snider WD (1997). Introduction of a neurotrophin‐3 transgene into muscle selectively rescues proprioceptive neurons in mice lacking endogenous neurotrophin‐3. Neuron 19, 503–517. [DOI] [PubMed] [Google Scholar]
- Xu ZQ, Zhang LQ, Wang Q, Marshall C, Xiao N, Gao JY, Wu T, Ding J, Hu G & Xiao M (2013). Aerobic exercise combined with antioxidative treatment does not counteract moderate‐ or mid‐stage Alzheimer‐like pathophysiology of APP/PS1 mice. CNS Neurosci Ther 19, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekawa T, Komaki H, Saito Y, Sugai K & Sasaki M (2013). Peripheral nerve abnormalities in pediatric patients with spinal muscular atrophy. Brain Dev 35, 165–171. [DOI] [PubMed] [Google Scholar]