

Summary
Targeted therapies have advanced the treatment options for cutaneous melanoma but many patients will progress on drug. Patient-derived xenografts (PDX) can be used to recapitulate therapy resistant tumors. Furthermore, PDX modeling can be utilized in combination with targeted sequencing and phospho-proteomic platforms providing pre-clinical basis for second-line targeted inhibitor strategies.
In this issue of Clinical Cancer Research, Krepler et al. describe the generation of patient-derived xenograft (PDX) models created from twelve BRAF V600E-harboring melanomas from individuals who have progressed on treatment with the BRAF inhibitors, vemurafenib or dabrafenib (1). Krepler and colleagues utilized these models to mimic progression on-drug by maintaining mice-bearing PDXs on BRAF inhibitor. They demonstrate that serial passaging in the PDX system faithfully recapitulates the original tumor’s histological features. By employing targeted sequencing panels and reverse phase proteomic analysis (RPPA), the group categorizes mechanisms of resistance and tests strategies for second-line combination therapies. They identified cMET amplification and phosphorylation in a subset of progressing tumors that are sensitive to cMET targeting in combination with BRAF and MEK inhibitors.
Even before President Obama’s initiative on Precision Medicine, there have been significant efforts to develop more optimal tools that will help design personalized treatment strategies. One tool at the forefront of such efforts is the development of PDX models, which reflect the heterogeneity within tumors and maintain, at least in initial passages, components of the original tumor microenvironment. Despite some limitations, PDX models have the potential to improve cancer therapies and promote precision medicine efforts. As evidenced in the current study, high-throughput screening modalities identified additional mutations/alterations in BRAF inhibitor-resistant PDX samples that when targeted in combination results in tumor shrinkage.
A likely advantage of the PDX model compared to standard cell line xenograft models is the former’s ability to better predict therapeutic response to targeted therapies and inform drug treatment scheduling to stave off the onset of resistance. One discussion within the melanoma field is the benefit of maintaining patients on continuous drug after the onset of disease progression. Whereas one study concluded that maintaining patients on BRAF inhibitors with progressive disease increases overall survival (2), there is at least one case report detailing patient benefit from discontinuing targeted therapy (3). Furthermore, multiple pre-clinical studies, as well as the present manuscript, demonstrate that a “drug holiday” delays the onset of resistance and that drug cessation following the development of resistance is detrimental to optimal growth (4, 5). Given these differences and the notion that durable resistance develops from the outgrowth of minor populations within tumors, the PDX system is ideally suited to test scheduling options to determine effects on the duration of response and whether drug tolerant cells, which are able to expand following drug removal, persist in the tumor. These represent increasingly important questions as the field moves towards combinatorial therapies and in the future may be coupled with in vivo signaling monitoring systems such as quantitative ERK1/2 reporters to assess the efficacy of scheduling across the whole tumor in a temporal and quantitative manner (6, 7).
A powerful application in the current study is the utilization of multiple platforms to highlight the example of MET amplification associated with BRAF inhibitor resistance. Notably, the authors conclude that MET amplification as determined by next generation sequencing technologies is not a sufficient indicator of its potential role in acquired resistance (1). In addition, they employ RPPA, a high throughput technique to identify alterations in signaling pathways and growth regulatory proteins. RPPA revealed elevated phospho-cMET in two of three MET amplified resistant tumors and only the tumors displaying high phospho-cMET were found to be exquisitely susceptible to cMET inhibition. In the TCGA melanoma dataset, MET is mutated or amplified in ~14% of melanoma samples (65/478), and as noted in Krepler et al. only two of nine MET amplified tumors with available RPPA data demonstrated an increase in phospho-cMET. This approach supports the notion to cross-reference next-generation sequencing data with phospho-proteomic pathway profiling to properly assess potential drug targets. Additionally, these findings highlight a role for aberrant cMET activation in drug resistant melanoma. Currently, there are multiple small molecule inhibitors to cMET in clinical trials for various cancer treatments, as well as monoclonal antibodies designed to inhibit cMET signaling either by neutralizing cMET itself or sequestering its ligand, HGF. One limitation of the NSG mouse model in this regard is that mouse HGF does not activate the human c-MET; thus, the authors are likely analyzing the effects of tumor-derived HGF or ligand-independent signaling. Utilization of human HGF knock-in mice would permit analysis of the effects of stromal-derived growth factor.
In reality the PDX system described by Krepler et al. can take several weeks to months to generate sufficient numbers of mouse “avatars” for “co-clinical trials” or drug assessments. Progressing patients may not be afforded this time frame; however, the knowledge gleaned from this system will most likely inform treatment options in other patients. An adaptation may be the ex vivo treatment of patient-derived explants, similar to experiments carried out with prostate tissue (8). These ex vivo model systems could provide rapid results more likely to guide patient treatment options in a real-time manner. Also important is the generation of models with selective tropisms. In particular, PDX models that give rise to brain metastases should be a focus given the clinical unmet need in this area. Mouse models have demonstrated spontaneous metastasis of patient derived breast cancer tissue to relevant sites of human breast cancer tropism (9). Spontaneous brain metastasis of primary melanoma has been demonstrated in mouse model (10), although this system does not utilize patient samples. Further work to establish brain metastatic model in a PDX platform needs to be explored.
Finally, the major obstacle to be overcome with PDX modeling is the use of a mouse with a severely compromised immune system. Immunodeficient mice are not suitable to investigate immunotherapy efficacy or the effect of targeted therapies on immune function. This hurdle is particularly relevant for cutaneous melanoma in which the advances made with targeted small molecule inhibitors have been paralleled with the remarkable clinical effects of immune checkpoint blockade agents such the anti-CTLA4 antibody, ipilimumab and the anti-PD1 agents, pembrolizumab and nivolumab. The number and type of immune cells infiltrating tumors will be important to predict the efficacy of immune checkpoint agents (11). Targeted and immunotherapy combination treatments in pre-clinical genetically engineered mouse models have demonstrated great promise (12). Furthermore, humanized mice that will serve to test immune checkpoint inhibitor therapies alone and in combination with targeted inhibitors on patient-derived samples represent the ultimate mouse avatar for personalized medicine are being established (13).
In summary, the Krepler et al. manuscript in this issue validates the power of the PDX system to define drug targets of resistant patient tumors. As outlined in Figure 1, their work suggests that proper identification of second-line therapy targets requires cross-referencing of next-generation sequencing data with phospho-proteomic pathway analysis. This study will hopefully lead the charge to add high-throughput phospho-proteomic analysis to standard clinical diagnostic practices and further expand the use of mouse avatars to bolster our understanding of drug resistance.
Figure 1.
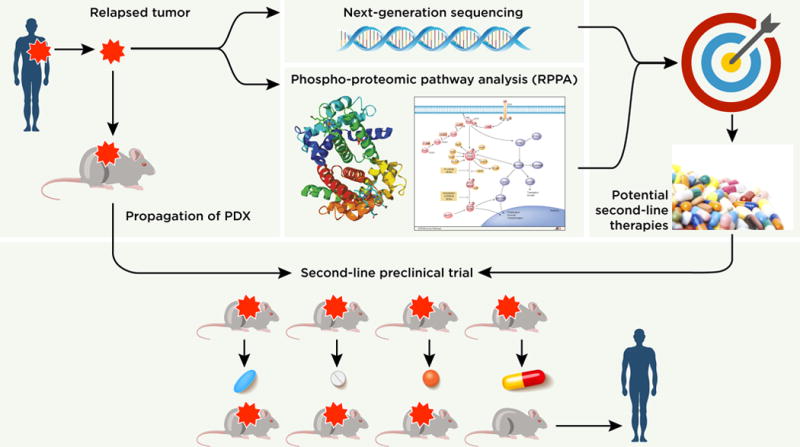
Schematic workflow depicting the identification of second-line targets from drug-resistant PDXs through next-generation sequencing and RPPA platforms. Resistant PDXs are propagated to be utilized in assays testing the efficacy of therapeutics against potential resistance mechanisms to ultimately inform second-line treatments in resistant melanoma patients.
Acknowledgments
Grant Support
E.J. Hartsough is supported by the National Cancer Center. A.E. Aplin is supported by the NIH under award numbers CA196278, CA160495, and CA182635, and grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Melanoma Research Alliance.
Footnotes
Disclosure of Potential Conflicts of Interest: A.E. Aplin reports receiving commercial research support from Pfizer. No potential conflicts of interest were disclosed by the other author.
References
- 1.Krepler C, Xiao M, Spoesser K, Brafford PA, Shannan B, Beqiri M, et al. Personalized pre-clinical trials in BRAF inhibitor resistant patient derived xenograft models identify second line combination therapies. Clin Cancer Res. 2015 Dec 16; doi: 10.1158/1078-0432.CCR-15-1762. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan MM, Haydu LE, Menzies AM, Azer MW, Klein O, Lyle M, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142–53. doi: 10.1002/cncr.28851. [DOI] [PubMed] [Google Scholar]
- 3.Seghers AC, Wilgenhof S, Lebbe C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22:466–72. doi: 10.1097/CMR.0b013e3283541541. [DOI] [PubMed] [Google Scholar]
- 4.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartsough EJ, Basile KJ, Aplin AE. Beneficial effects of RAF inhibitor in mutant BRAF splice variant-expressing melanoma. Mol Cancer Res. 2014;12:795–802. doi: 10.1158/1541-7786.MCR-13-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basile KJ, Abel EV, Dadpey N, Hartsough EJ, Fortina P, Aplin AE. In vivo MAPK reporting reveals the heterogeneity in tumoral selection of resistance to RAF inhibitors. Cancer Res. 2013;73:7101–10. doi: 10.1158/0008-5472.CAN-13-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seifert H, Hirata E, Gore M, Khabra K, Messiou C, Larkin J, et al. Extrinsic factors can mediate resistance to BRAF inhibition in central nervous system melanoma metastases. Pigment Cell Melanoma Res. 2016;29:92–100. doi: 10.1111/pcmr.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–20. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, et al. AKT1 activation promotes development of melanoma metastases. Cell Rep. 2015;13:898–905. doi: 10.1016/j.celrep.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladanyi A. Prognostic and predictive significance of immune cells infiltrating cutaneous melanoma. Pigment Cell Melanoma Res. 2015;28:490–500. doi: 10.1111/pcmr.12371. [DOI] [PubMed] [Google Scholar]
- 12.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2:643–54. doi: 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]