

Abstract
Objective
Lovastatin has been shown to reverse learning deficits in a mouse model of Neurofibromatosis Type 1 (NF1), a common monogenic disorder caused by a mutation in the Ras‐MAPK pathway and associated with learning disabilities. We conducted a randomized double‐blind placebo‐controlled trial to assess lovastatin's effects on cognition and behavior in patients with NF1.
Method
Forty‐four NF1 patients (mean age 25.7+/−11.6 years; 64% female) were randomly assigned to 14 weeks of lovastatin (N = 23; maximum dose of 80 mg/day for adult participants and 40 mg/day for children) or placebo (N = 21). Based on findings in the mouse model, primary outcome measures were nonverbal learning and working memory. Secondary outcome measures included verbal memory, attention, and self/parent‐reported behavioral problems, as well as tolerability of medication. Participants also underwent neuroimaging assessments at baseline and 14 weeks, to determine whether neural biomarkers were associated with treatment response. Linear mixed models assessed for differential treatment effects on outcome measures.
Results
Twelve participants dropped from the study prior to completion (8 placebo, 4 lovastatin), resulting in 32 completers (15 placebo, 17 lovastatin). Lovastatin was well‐tolerated, with no serious adverse events. Differential improvement favoring lovastatin treatment was observed for one primary (working memory; effect size f 2 = 0.70, P < 0.01) and two secondary outcome measures (verbal memory, f 2 = 0.19, P = 0.02, and adult self‐reported internalizing problems, f 2 = 0.26, P = 0.03). Exploratory moderator analyses revealed that higher baseline neural activity in frontal regions was associated with larger treatment effects.
Interpretation
These preliminary results suggest beneficial effects of lovastatin on some learning and memory functions, as well as internalizing symptoms in patients with NF1.
Introduction
Developmental learning disabilities are a highly prevalent form of nonprogressive cognitive impairment, presenting a major public health burden. However, disentangling the molecular and neural bases of these disorders is complicated by their extremely heterogeneous etiologies and to date, no effective pharmacologic treatments exist.
Neurofibromatosis type 1 (NF1) is a valuable model for understanding mechanisms of cognitive disability, as it is a common genetic disorder (incidence 1:3000) resulting from mutations in a single gene1. The disease‐causing gene, Nf1, encodes neurofibromin, a Ras GTPase‐activating protein which is highly expressed in the brain. Specific learning disabilities are among the most common neurological complications, affecting 30–65% of children with this disorder2, 3. Thirty to fifty percent of children with NF1 are diagnosed with attention deficit disorder (ADHD), a marked increase over population base rates; common symptoms, including inattention, poor working memory and executive dysfunction appear to persist into adulthood2, 4. In addition, recent studies have revealed that the NF1 behavioral phenotype greatly impacts social skills and overlaps with autism spectrum disorders5, 6. The development of a mouse model of the disorder led to the key discovery that increased Ras activity is responsible for the learning deficits in NF17, and acts by impairing long‐term potentiation (LTP) as a result of increased γ‐aminobutyric acid (GABA)‐mediated inhibition8.
Subsequent studies in the Nf1 mouse model demonstrated that the 3‐hydroxy‐3‐methyl‐glutaryl (HMG)‐CoA reductase inhibitor lovastatin, which acts as a potent inhibitor of p21Ras activity and is commonly used for the treatment of hypercholesterolemia, can reverse the biochemical, electrophysiological, and cognitive deficits observed in the mouse9. Given the high sequence homology between mouse and human neurofibromin10, this allows us to assess a pharmacologic treatment for cognitive deficits of patients with a monogenic disorder, using a medication that has been validated in preclinical studies and for which substantial clinical safety data are available.
In human subjects with NF1, two randomized trials of another statin medication, simvastatin, have been conducted; neither showed significant treatment effects on primary outcomes (intellectual function [IQ] and nonverbal learning)11, 12. However, an open‐label study of lovastatin found improvement of verbal and nonverbal memory after 3 months of treatment in children13. This could indicate a specific effect of lovastatin, or could reflect practice effects. Here we conducted the first randomized, double‐blind placebo‐controlled trial to evaluate the effect of 14 weeks of lovastatin treatment on neurobehavioral function in children with NF1, with neuroimaging measures as intermediate markers of treatment‐associated change. Based on findings in the mouse model, we hypothesized that NF1 patients treated with lovastatin would show differential improvement on measures of working memory and nonverbal learning and memory, as well as secondary measures of attention and behavioral symptoms.
Patients and Methods
Design
A prospective double‐blind, placebo‐controlled, randomized 14‐week clinical trial was conducted in individuals with NF1 between January 2010 and March 2013. The study is registered at clinicaltrials.gov, trial # NCT00352599.
Participants
Participants were recruited from three primary sources: (1) The Children's Hospital Los Angeles Neurofibromatosis Clinic, a major NF1 referral center for the greater Los Angeles region; (2) local Children's Tumor Foundation and NF Network educational symposia; and (3) websites, both NF‐specific and www.clinicaltrials.gov. Following receipt of thorough verbal and written descriptions of study requirements, and prior to initiation of any study activities, all participants provided written consent or parental permission and child/youth assent following procedures approved by the UCLA Institutional Review Board.
Participants were screened and enrolled by a pediatric neurologist (TR), a clinician with experience in caring for individuals with NF1. Inclusion criteria were as follows: (1) Meets the NIH NF1 diagnostic criteria14 and does not have segmental NF1; (2) 10–50 years of age; (3) Full‐Scale IQ of at least 70, as determined by the Wechsler Abbreviated Scale of Intelligence (WASI15); (4) Not currently taking a statin medication; (5) Not suffering from hypercholesterolemia, based on National Cholesterol Education Program (NCEP) guidelines16; (6) Normal hepatic, muscle enzyme (CPK), hematologic, renal function and cholesterol lab values on initial screening; (7) Not taking any additional medications with potential interactions with lovastatin; (8) No comorbid major neurological or psychiatric disorder (e.g., epilepsy, bipolar disorder, psychotic illness, major depression); (9) No evidence of intracranial pathology (other than asymptomatic optic pathway glioma); (10) Female participants could not be pregnant or lactating; sexually active females were required to use adequate birth control measures during the study and undergo regular urine pregnancy testing; (11) Sufficient English fluency to avoid invalidating research measures. Participants with a clinical diagnosis of ADHD were included, but those subjects on medication for ADHD treatment were required to continue the same medication dose throughout the duration of the study (Table 1). Additionally, none of the subjects included in the study had other NF1‐related gliomas.
Table 1.
Baseline demographics
| NF1 Participants‐lovastatin (n = 21) | NF1 Participants‐placebo (n = 23) | P ‐value | |
|---|---|---|---|
| Age (years, ±SD) | 27.0 (12.4) | 24.4 (11.0) | 0.47 |
| Gender (N, % female) | 11 (52%) | 17 (74%) | 0.14 |
| Hispanic (N, %) | 4 (19%) | 10 (43%) | 0.08 |
| Full Scale intellectual function (IQ) (mean, ±SD) | 98.3 (13.3) | 99.8 (12.9) | 0.72 |
| Race | |||
| Caucasian | 16 (76%) | 13 (56%) | 0.28 |
| African American | 1 (5%) | 5 (21%) | |
| Asian | 4 (14%) | 4 (17%) | |
| Native American | 1 (5%) | 0 | |
| Other | 0 | 1 (4%) | |
| ADHD (attention deficit disorder diagnosis) (N, %) | 3 (14%) | 6 (26%) | 0.46 |
| Years education (SD) | 14.4 (4.0) | 13.3 (40) | 0.34 |
| Other Medication (Antidepressant/Benzodiazepine/ Psychostimulant/Mood stabilizer/ Other) | 3/0/1/0/1 | 2/1/0/1/1 | 0.60 |
Medication protocol
Subjects were randomized 1:1 to lovastatin or placebo using a computer‐generated permuted‐block, randomization list. Separate randomizations were performed for adult (n = 30) and child (n = 14) study participants to ensure that treatment assignment was balanced by age. Randomization was performed by the UCLA investigational pharmacist, who assigned participants in the order of their enrollment in the trial and dispensed the medication. Study participants, their parents, and all study investigators and personnel were blind to the treatment allocation. The randomization list was only accessible to the senior database programmer and the pharmacist. Participants' treatment allocation and test results remained de‐identified until all follow‐up was completed.
Subjects were treated once daily for 14 weeks with lovastatin or placebo. Adults receiving lovastatin were administered doses of 40 mg/day for week 1, 60 mg/day for week 2 and 80 mg/day for weeks 3–14. Children and adolescents (age 10–17) were administered 20 mg/day for week 1, 30 mg for week 2 and 40 mg for weeks 3–14. This dosing schedule was based on guidelines for adults17 and children18 with hypercholesterolemia, respectively. The capsules containing placebo or lovastatin were identical in color, shape, and size.
Medication safety monitoring and adherence
Following enrollment, participants underwent a physical examination, including height, weight, and vital signs, conducted by the study physician (TR) in order to verify the NF1 diagnosis based on NIH NF1 diagnostic criteria14 and to ensure that there were no medical contraindications to participation. Baseline laboratory studies included: complete blood count (CBC), aspartate aminotransferase (AST), alanine transaminase (ALT), bilirubin, creatinine, blood urea nitrogen (BUN), glucose, creatine phosphokinase (CPK), and lipid panels. Menstruating female participants had urine pregnancy tests. Follow‐up visits occurred at 4 weeks, 8 weeks, and 14 weeks following treatment initiation and included a comprehensive physical examination, open‐ended adverse event inquiry, documentation of concomitant medications, and repeated laboratory studies. A final study visit occurred at 18 weeks, 1 month after cessation of medication/placebo therapy, and included a final physical examination as well as adverse event monitoring. An independent study physician monitored laboratory results in parallel with study visits to insure ongoing participant safety as well as keeping the treating physician blinded to the treatment condition.
Medication compliance was assessed at the week 4, 8, and 14‐week visits. Participants were judged medication‐compliant when they took at least 80% of their study medication during the intervention period of 14 weeks, which was assessed by counting returned capsules and with a pill diary.
General lovastatin and placebo toxicity were monitored throughout the trial using standard guidelines (http://ctep.cancer.gov), and were rated as being not drug related, possibly drug related, or definitely drug related prior to unblinding.
Criteria for study discontinuation included: (1) Noncompliance with administration of lovastatin or placebo tablets, and/or with required follow‐up; (2) development of a medical condition or initiation of another medication with which lovastatin is contra‐indicated; (3) development of significant drug toxicity (defined as grade 2 or higher toxicity which persists after 10 days off lovastatin); (4) pregnancy in a female participant.
Outcome measures
Outcome measures were assessed at baseline and after 14 weeks of treatment. Parallel versions of tests were applied when available to reduce the impact of practice effects. Primary outcomes were two neuropsychological measures that were most analogous to statin‐responsive tests in Nf1 mice, specifically: the Brief Visuospatial Memory Test – Revised (BVMT, Immediate and Delayed19; nonverbal declarative memory) and the Letter‐Number Sequencing task (LNS20; working memory).
For the secondary outcomes, we selected neuropsychological and behavioral tests to assess domains that are differentially affected in NF1 patients, that is, attention, learning and memory, and social behavior2, 3: Digit Cancellation (attention/inhibitory control)21, Hopkins Verbal Learning Test (HVLT; verbal declarative memory22), WISC‐III Object Assembly (visuoconstructive ability)23, and Internalizing and Externalizing symptoms, as well as the Attention, Thought and Social Problems subscales from either the Achenbach Child Behavior Checklist (CBCL; self‐ and parent‐report) or Young Adult Self Report (YASR)24, 25.
Due to the exploratory nature of this trial, additional tertiary neurocognitive and behavioral measures (deemed less central to the characteristic NF1 cognitive profile) included: the Neuropsychological Assessment Battery (NAB) Mazes26, Delis–Kaplan Executive Function Scale (D‐KEFS) verbal fluency (category and letter fluency)27 and the Behavior Rating Inventory of Executive Function (BRIEF; General Executive Composite (GEC) score)28. All neuropsychological tests were administered and scored by trained psychometricians blind to treatment condition. Functional imaging measures were also collected at baseline and 14‐week follow‐up to explore its potential both as a predictor of eventual treatment response and as an intermediate marker of treatment‐related change.
Image acquisition
All scanning was carried out on identical Siemens 3 Tesla Tim Trio (Erlangen, Germany) MRI scanners using identical protocols at either the UCLA Brain Mapping Center or the Center for Cognitive Neuroscience (CCN). Measures of brain structure were obtained using T1‐weighted anatomical images acquired with an MPRAGE sequence with the following acquisition parameters: TR/TE/TI = 2300/2.89/900 ms; flip angle = 9 degrees; slice thickness = 1.20 mm, with a 240 × 256 acquisition matrix.
Spatial capacity working memory (SCAP) task
A spatial working memory functional MRI paradigm, designed to be analogous to the behavioral tasks used in the mouse model, was also administered. We previously found hypoactivation of fronto‐parietal neural circuitry using this task in untreated NF1 patients29. Specifically, subjects were shown a target array of 1, 3, 5, or 7 yellow circles positioned pseudo‐randomly around a central fixation, as detailed in Glahn et al30. After a variable delay, subjects were shown a single green circle and asked to indicate whether that circle was in the same position as one of the target circles. Participants took part in a behavioral training session immediately prior to the one‐hour scan. In the scanner, trial events included a 2‐sec target array, 1.5, 3, and 4.5‐sec(s) delay, 3‐sec probe array, and a jittered (average of 2 sec) intertrial interval (ITI) with a fixation. During the task, 291 functional T2*‐weighted echoplanar images (EPIs) were collected with the following parameters: slice thickness = 4 mm, 34 slices, TR = 2 sec, TE = 30 ms, flip angle = 90°, matrix 192 × 192, FOV = 192 mm.
Functional MRI analysis
fMRI data analyses were performed using tools from the FMRIB software library (www.fmrib.ox.ac.uk/fsl), version 5.0, using the same processing steps as described in our prior publications29, 31, including spatial smoothing, temporal filtering, and a three‐step registration process in which EPI images were first registered to the matched‐bandwidth high‐resolution scan, then to the structural image, and finally into standard (Montreal Neurological Institute (MNI)) space, using nonlinear transformations32.
Thirty‐seven subjects completed a baseline SCAP fMRI scan (18 statin, 19 placebo), of whom 5 were excluded due to an artifact in structural scan (n = 2) or excessive motion (translational motion >2 mm or 3 or more spikes greater than 1 mm in at least one scan; n = 3), resulting in 32 subjects (17 lovastatin, 15 placebo) in the baseline fMRI analysis. Thirty subjects (15 statin, 15 placebo) completed a follow‐up fMRI scan, of whom 10 were excluded for the following reasons: artifact in structural scan (n = 2), low signal to noise ratio (SNR; n = 2), excessive motion (n = 3), or poor registration between baseline and follow‐up scans (n = 3). Thus, 20 subjects were available for analysis at follow‐up (10 lovastatin, 10 control). Demographics of the subjects with usable MRI data did not differ from those of the overall sample. Further, there were no differences in motion (mean framewise displacement) between treatment groups (P = 0.20).
Standard model fitting was conducted for all subjects, as in Shilyansky et al.29. Briefly, for each subject, first‐level models included contrasts for each individual load and delay condition. Our primary analyses focused on the All‐Loads contrast. The six motion parameters and temporal derivatives of all regressors were included as covariates of no interest to improve statistical sensitivity. Although no differences between scanners were found in any preliminary analyses (P > 0.05 for all comparisons), we included scanner as a covariate in subsequent group‐level analyses to ensure that any subtle between‐scanner differences were controlled.
Bilateral 8 mm spherical regions of interest (ROIs) were used to extract percent signal change from the following regions at each timepoint: bilateral Brodmann's Area 10, dorsolateral prefrontal cortex (DLPFC), frontal eye fields, and parietal cortex, as well as two default mode regions which served as control regions, the ventromedial PFC and posterior cingulate cortex (PCC; Fig. 1 ). ROIs were created in MNI‐152 space and centered on peak voxel activation from the All‐Loads contrast in our previous study29. Average percent signal change values corresponding to an 8‐sec stimulus convolved with a gamma hemodynamic response function (HRF) from the All‐Loads contrast were extracted from baseline and follow‐up scans separately33.
Figure 1.
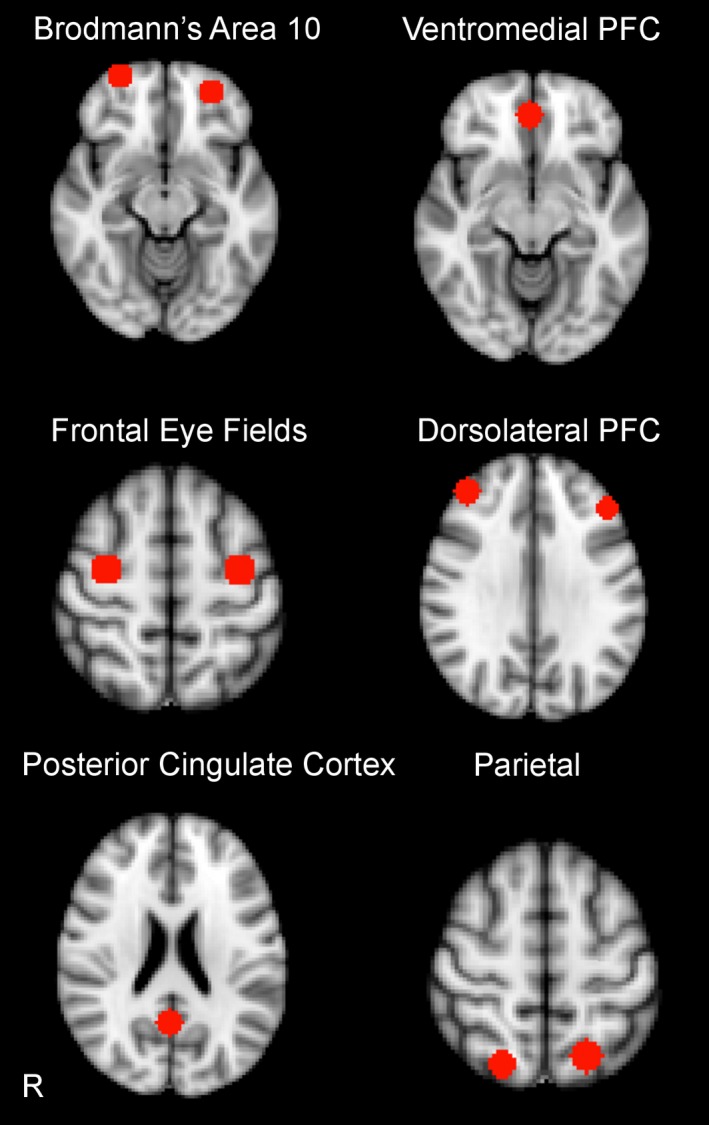
Regions of Interest for Spatial Capacity Working Memory (SCAP) Functional MRI Task.
Statistical analyses
Prior to performing the primary tests for treatment effects, t‐tests and chi‐squared tests were used to compare the lovastatin and placebo groups on baseline demographic and clinical measures to confirm the success of randomization. Similar tests were used to check for differential dropout between the treatment arms and to identify other correlates of early discontinuation. For each of the primary and secondary outcome measures, we fit a generalized linear mixed model (GLMM) with group (lovastatin, placebo), time (baseline, 14 weeks), and a group‐by‐time interaction to test for differential treatment effects (primary RCT analyses). Age‐adjusted scores were used for all cognitive and behavioral measures. For the BVMT‐R, age‐appropriate published normative data were only available for participants aged 18 and up; thus, for child participants, we applied norms from our large database of healthy control participants in a multisite project (North American Prodromal Longitudinal Study; U01MH08190234). For outcome measures where a significant treatment effect was established, follow‐up moderator analyses were conducted to determine whether baseline features of the participants influenced response to lovastatin treatment, by adding those factors and their interactions with treatment group and time to the original GLMMs. In particular, we predicted that: (1) younger participants would show stronger treatment effects due to increased malleability, and (2) participants with larger baseline deficits would experience greater treatment benefits. Parallel exploratory analyses were conducted for the neuroimaging measures, to test for differential change over time in brain structure and function in the above‐specified ROIs. For all analyses of neuroimaging measures, we controlled for age, gender and scanner location by including them as covariates in GLMMs. Finally, we examined whether greater brain volume and/or increased neural activity at baseline was associated with increased treatment response by adding them as moderators to primary models. Because child and adult participants were assessed with different versions of the CBCL (i.e., participants aged 10–17 received the parent‐report and child self‐report forms, whereas participants age 18 and up received the YASR form), these measures were analyzed separately.
We did not correct for multiple comparisons given that: (1) there were only 2 primary and 4 secondary outcome measures which were specified a priori; and (2) the neuropsychological and behavioral outcome measures are potentially intercorrelated. Multiple comparison correction would make it very difficult to detect a possible effect of treatment in this proof‐of‐concept exploratory investigation. Thus, we opted to guard against type II error.
Results
Participants
Based on known prevalence rates of NF1 (approximately 1:300035), and estimates of the number of new patients seen in local NF1 clinics annually who could conceivably enroll in the study, we estimated that it would not be feasible to recruit more than 50 participants over the 3‐year study period. We screened 53 subjects for the study; 44 were randomized to lovastatin (n = 21) or placebo (n = 23). This sample size provides 80% power to detect a moderate effect size of Cohen's f 2 = 0.05 in the mixed models for the group by time interaction, corresponding to the primary hypothesis of a differential treatment effect from baseline to 14‐week follow‐up. Participant enrollment and study flow are summarized in Figure 2.
Figure 2.
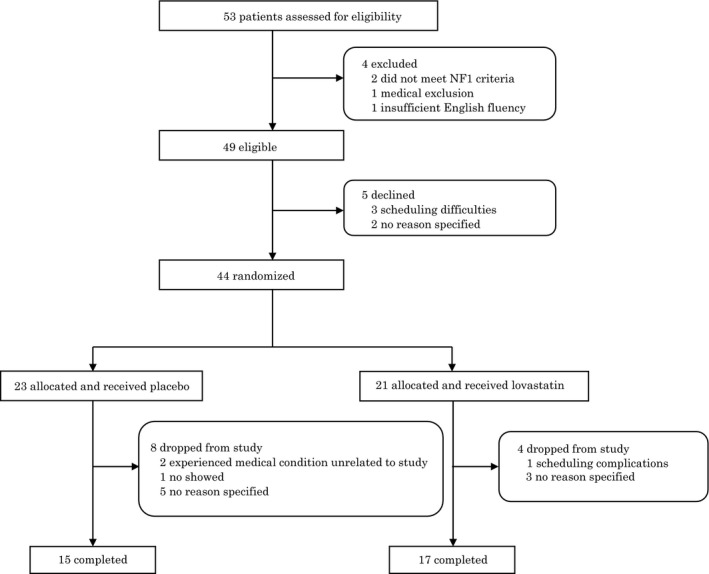
Clinical trial flow diagram of study participation. Flow chart depicts the recruitment and follow‐up of all subjects involved in the study. Of the 53 subjects originally screened, four subjects were deemed ineligible. Of the 49 eligible participants, 5 declined to participate prior to randomization. Thus, 44 were randomly assigned to the lovastatin group (n = 21) or the placebo group (n = 23). Four participants (19%) in the lovastatin group and 8 (34.8%) in the placebo group withdrew from the study before completion for personal reasons, resulting in 15 completers in the placebo group and 17 completers in the lovastatin group.
Baseline demographic characteristics did not differ between lovastatin and placebo groups (Table 1). Four participants (19%) in the lovastatin group and 8 (34.8%) in the placebo group withdrew from the study before completion, so there was no evidence of differential dropout by treatment group (χ 2 = 0.69, P = 0.41). No participants had to be withdrawn by the investigator for any reason, and no medical issues arose that resulted in unblinding prior to study completion. Study noncompleters did not differ significantly in age, gender or IQ from study completers; however, study completers on average had higher educational attainment than noncompleters (P < 0.01; 14.8 years vs. 11.7 years, respectively). Overall, medication compliance was excellent and did not differ between treatment arms (P = 0.54; mean compliance = 95.5% for the lovastatin group vs. 94.4% for the placebo group).
Effects of lovastatin on neurocognitive outcomes
There was a significant differential effect of treatment over time (i.e. significant group x time interaction), favoring the lovastatin group for one primary measure, Letter‐Number Sequencing (P < 0.01; f 2 = 0.70) and one secondary cognitive measure (HVLT; P = 0.02; f 2 =0.19; Table 2, Fig 3A–B). There was also a differential effect in favor of statin treatment for verbal category fluency (P = 0.02; f 2 = 0.19; Fig. 3C). However, there were no significant treatment effects for nonverbal memory (BVMT, Immediate and Delayed conditions) or other secondary (Object Assembly, Cancellation) or tertiary measures (Mazes, verbal letter fluency).
Table 2.
Scores on neurocognitive and behavioral outcome measures at baseline and 14 weeks in nf1 participants
| Lovastatin | Lovastatin | Placebo | Placebo | Difference in change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 14 weeks | Baseline | 14 weeks | |||||||||
| Primary measures | Mean | SD | Mean | SD | da | Mean | SD | Mean | SD | d | f 2 b | |
| Letter number sequencing* | 8.67 | 3.78 | 10.43 | 2.44 | 0.47 | 9.33 | 3.62 | 9.17 | 2.99 | −0.04 | F(1,30) = 20.9 P < 0.01 | 0.70 |
| BVMT immediate | 31.33 | 12.77 | 42.57 | 15.89 | 0.88 | 32.00 | 13.93 | 42.67 | 13.97 | 0.77 | F(1,33) = 1.3 P = 0.26 | 0.04 |
| BVMT delay | 31.33 | 10.78 | 39.00 | 14.32 | 0.71 | 33.83 | 12.97 | 42.00 | 12.68 | 0.63 | F(1,30) = 0.3 P = 0.61 | 0.01 |
| Secondary measures | ||||||||||||
| Object assembly | 7.67 | 2.81 | 8.14 | 3.34 | 0.17 | 8.83 | 1.72 | 9.83 | 2.401 | 0.58 | F(1,32) = 0.2 P = 0.69 | 0.01 |
| Cancellation | 5.17 | 2.04 | 6.14 | 1.67 | 0.48 | 4.33 | 2.66 | 6.67 | 1.37 | 0.88 | F(1,32) = 0.5 P = 0.50 | 0.02 |
| Hopkins verbal learning test* | 33.50 | 10.62 | 35.29 | 11.01 | 0.17 | 37.67 | 14.04 | 23.50 | 5.82 | −1.01 | F(1,33) = 6.4 P = 0.02 | 0.19 |
| Achenbach young adult self‐report | ||||||||||||
| Internalizing problems* | 53.67 | 9.71 | 43.5 | 9.48 | −1.05 | 54.08 | 15.20 | 53.67 | 18.26 | −0.03 | F(1,20) = 5.1 P = 0.03 | 0.26 |
| Externalizing problems | 48.93 | 9.46 | 44.25 | 7.50 | −0.49 | 52.92 | 12.41 | 53.67 | 11.31 | 0.06 | F(1,20) = 0.7 P = 0.41 | 0.04 |
| Social problems | 52.87 | 5.46 | 51.67 | 3.82 | −0.22 | 58.54 | 10.21 | 57.89 | 10.55 | −0.06 | F(1,21) = 0.2 P = 0.66 | 0.01 |
| Thought problems | 53.00 | 5.59 | 52.83 | 5.69 | −0.03 | 57.77 | 11.75 | 55.44 | 7.73 | −0.20 | F(1,22) = 1.3 P = 0.27 | 0.06 |
| Attention problems | 58.07 | 7.39 | 55.50 | 5.98 | −0.35 | 61.54 | 12.49 | 59.67 | 12.21 | −0.15 | F(1,21) = 0.4 P = 0.55 | 0.02 |
| Tertiary measures | ||||||||||||
| Mazes | 30.00 | 7.62 | 32.14 | 4.67 | 0.28 | 33.33 | 10.75 | 35.33 | 12.03 | 0.19 | F(1,32) = 0.2 P = 0.66 | 0.01 |
| Verbal letter fluency | 10.50 | 4.72 | 8.14 | 2.41 | −0.50 | 10.50 | 4.930 | 8.17 | 2.32 | −0.47 | F(1,30) = 0.1 P = 0.75 | 0.00 |
| Verbal category fluency* | 9.83 | 3.06 | 10.43 | 2.15 | 0.20 | 11.33 | 2.81 | 8.50 | 2.51 | −1.01 | F(1,31) = 5.8 P = 0.02 | 0.19 |
BVMT, the Brief Visuospatial Memory Test.
d = effect size for within‐group change from baseline to follow‐up.
f 2 = effect size for group x time interaction.
Figure 3.
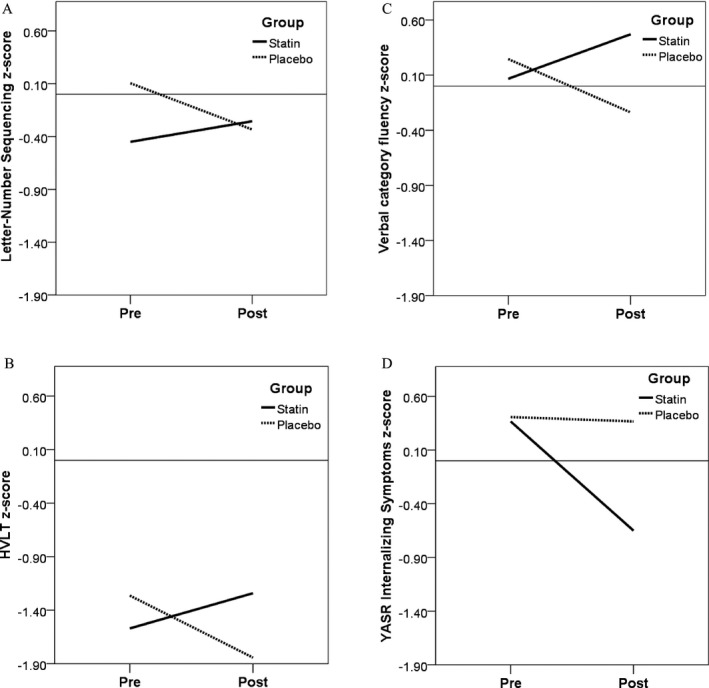
Group x Time Interactions for Measures showing significant treatment‐associated change. Scores at pre‐ and post‐treatment timepoints (baseline and 14‐week follow‐up) for each measure are shown on the Y‐axis. Scores are standardized to z‐scores, for comparability across measures. (A). Letter‐Number Sequencing showed a significant difference in trajectories between the two treatment groups (f 2 = 0.70; P < 0.01), with greater improvement seen in the statin group. (B). Hopkins Verbal Learning Test showed a significant difference in trajectories between the two treatment groups (f 2 = 0.19; P = 0.02), with greater improvement seen in the statin group. (C). Verbal category fluency showed a significant difference in trajectories between the two treatment groups (f 2= 0.19; P = 0.02), with greater improvement seen in the statin group. (D). YASR Internalizing Problems showed a significant difference in trajectories between the two treatment groups (f 2= 0.26; P = 0.03), with greater improvement seen in the statin group.
Effects of lovastatin on behavioral outcomes
The YASR Internalizing Problems showed a differential treatment effect (P = 0.03; f 2 = 0.26), with greater improvement seen in the statin group (Fig. 3D). However, no significant effects of treatment were observed for YASR Externalizing Problems, nor the Thought, Attention, or Social Problems subscales (Table 2).
For the parent‐report and child self‐report versions of the CBCL, there was no evidence of a differential treatment effect for Internalizing or Externalizing Problems (Table 3), or for the Thought, Attention, or Social Problems subscales. Similarly, no significant treatment effect was observed for the BRIEF GEC score.
Table 3.
Child Self‐report and Parent–report Measures (Child Behavior Checklist and BRIEF) at Baseline and 14 Weeks
| Lovastatin | Lovastatin | Placebo | Placebo | f 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 14 weeks | Baseline | 14 weeks | Difference in change | ||||||||
| Mean | SD | Mean | SD | d | Mean | SD | Mean | SD | d | |||
| Parent reporta | ||||||||||||
| Internalizing problems | 53.80 | 14.27 | 45.75 | 13.60 | −0.56 | 54.86 | 8.57 | 47.17 | 11.05 | −0.90 | F(1,8) = 0.2 P = 0.67 | 0.03 |
| Externalizing problems | 48.80 | 16.90 | 51.25 | 14.73 | 0.14 | 50.57 | 8.44 | 49.33 | 10.07 | −0.15 | F(1,8) = 1.4 P = 0.26 | 0.18 |
| Social problems | 61.20 | 13.16 | 61.75 | 6.24 | 0.04 | 60.71 | 12.71 | 59.17 | 11.92 | −0.12 | F(1,8) = 0.0 P = 0.82 | 0.00 |
| Thought problems | 57.60 | 9.92 | 60.00 | 7.44 | 0.24 | 57.57 | 8.81 | 56.50 | 9.71 | −0.12 | F(1,8) = 2.6 P = 0.15 | 0.33 |
| Attention problems | 65.20 | 13.42 | 67.75 | 16.68 | 0.19 | 68.29 | 9.93 | 62.67 | 9.77 | −0.57 | F(1,8) = 1.8 P = 0.21 | 0.23 |
| BRIEF GEC (General Executive Composite) | 64.00 | 9.508 | 62.43 | 14.57 | −0.17 | 59.83 | 11.79 | 57.50 | 16.11 | −0.20 | F(1,11) = 0.0 P = 0.92 | 0.00 |
| Child reporta | ||||||||||||
| Internalizing problems | 48.80 | 13.83 | 44.20 | 6.42 | −0.33 | 53.29 | 6.18 | 48.17 | 5.38 | −0.83 | F(1,9) = 0.0 P = 0.82 | 0.00 |
| Externalizing problems | 44.80 | 9.26 | 44.20 | 10.50 | −0.06 | 48.71 | 6.82 | 46.33 | 5.96 | −0.35 | F(1,9) = 0.7 P = 0.41 | 0.08 |
| Social problems | 57.20 | 9.86 | 58.00 | 10.79 | 0.08 | 61.71 | 7.23 | 58.67 | 8.19 | −0.42 | F(1.9) = 2.8 P = 0.12 | 0.31 |
| Thought problems | 59.60 | 10.46 | 56.40 | 7.70 | −0.31 | 57.71 | 5.85 | 54.67 | 7.47 | −0.52 | F(1,10) = 0.0 P = 0.98 | 0.00 |
| Attention problems | 59.20 | 11.90 | 60.40 | 13.69 | 0.10 | 58.29 | 9.01 | 59.67 | 7.82 | 0.15 | F(1,9) = 0.0 P = 0.90 | 0.00 |
scores only available on participants under 18 (n = 14 at baseline, n = 13 at follow‐up).
Functional neuroimaging measures: changes over time
There were no differential treatment effects for changes in neural activity during spatial working memory (SCAP performance; all P ≥ 0.05).
Moderators of treatment effects
Baseline neuroimaging indices
For each of the four unique outcome measures that showed significant differential treatment effects, we tested whether baseline neural activity (i.e., percent signal change within any of the 10 ROIs) was a potential treatment moderator. For Letter‐Number Sequencing (LNS), baseline percent signal change in the right frontal eyefields significantly modulated the magnitude of treatment response (P = 0.01). For verbal category fluency, baseline percent signal change in the right DLPFC was a significant moderator of treatment effect (P = 0.05). For YASR Internalizing Problems, baseline percent signal change in both the left frontal eyefields (P = 0.05) and the right DLPFC were significant moderators of treatment effect (P = 0.03). For the HVLT, however, there was no effect on the magnitude of treatment response for baseline functional activation in any ROI.
Age and baseline cognition/symptoms
Baseline performance was not a significant moderator of the observed treatment effect for any of the cognitive measures (LNS [P = 0.26], HVLT [P = 0.42], or verbal category fluency [P = 0.71]). For YASR Internalizing Symptoms there was a trend toward baseline symptom severity modulating the magnitude of the treatment effect (P = 0.06), with NF1 patients with higher baseline levels of internalizing symptoms showing greater relative benefits from lovastatin versus placebo (i.e., the relative benefit of lovastatin is greater with increasing baseline symptoms). Age was not a significant moderator of the treatment effect for any cognitive or behavioral measures.
Safety and effects on laboratory measures
Lovastatin and placebo were well tolerated throughout the study. All reported adverse events and abnormal lab values were determined to be CTCAE Grade 1 (mild) and clinically insignificant. There were no CTCAE Grade 3–4 adverse events and no subjects required a dosage adjustment or removal from the study due an adverse event. At least one Grade 1 side effect was reported in 12 of 17 (70.5%) subjects taking Lovastatin and 14 of 15 (93%) subjects taking placebo. These side effects were deemed unlikely to be related to study medication. There was no significant difference in the percentage of subjects reporting one or more side effects between treatment groups. No subjects on lovastatin complained of myalgias (musculoskeletal system) or muscle weakness (musculoskeletal system) and none had an elevation in CPK. Four subjects on placebo complained of muscle weakness and 5 complained of myalgia. Fatigue (general body system) was the most common side effect reported in the placebo group, followed by headaches (neurologic system). Headache was the most common side effect reported in the lovastatin group, followed by insomnia (Table 4).
Table 4.
Adverse events summary
| Adverse events by organ system | Lovastatin (n = 17) | Placebo (n = 15) |
|---|---|---|
| Gastrointestinal system disorders | 7 (6) | 37 (10) |
| General/Whole body system disorders | 9 (7) | 23 (9) |
| Neurologic system disorders | 11 (9) | 21 (8) |
| Musculoskeletal system disorders | 0 (0) | 12 (6) |
| Respiratory system disorders | 3 (2) | 8 (5) |
| Cardiovascular system disorders | 1 (1) | 7 (5) |
| Psychiatric disorders | 1 (1) | 6 (4) |
| Visual system disorders | 0 (0) | 3 (2) |
| Dermatologic system disorders | 2 (2) | 7 (4) |
| Hematologic system disorders | 1 (1) | 6 (4) |
Data indicate the number of events reported, for study completers (number of patients who reported an event). All adverse events were determined to be CTCAE Grade 1 (mild).
After 14 weeks, total and LDL cholesterol levels were significantly reduced in the lovastatin‐treated relative to the placebo group (P < 0.001; Table 5). There was no differential change in levels of HDL cholesterol or triglycerides between groups (P > 0.3 for both comparisons).
Table 5.
Treatment‐Associated Changes in Cholesterol Levels
| Percent change from baseline | NF1 participants‐ statin | NF1 participants‐ placebo | P ‐Value |
|---|---|---|---|
| Total Cholesterol (±SD) | −17.30 (18.99) | 5.8 (11.49) | <0.001 |
| LDL Cholesterol (±SD) | −25.51 (16.3) | 9.05 (14.51) | <0.001 |
| HDL Cholesterol (±SD) | −0.55 (8.12) | 2.80 (11.36) | 0.33 |
| Triglycerides (±SD) | 17.4 (44.4) | 7.8 (35.92) | 0.67 |
Discussion
We conducted the first randomized double‐blind placebo‐controlled trial to assess the effects of lovastatin, an HMG‐CoA reductase inhibitor, on neurobehavioral function in patients with NF1. Although exploratory, we saw differential improvement favoring lovastatin treatment for one primary outcome measure (working memory), analogous to a statin‐responsive measure in the Nf1 mouse model, as well as declarative memory and verbal fluency, and in self‐reported internalizing (but not externalizing) symptoms. We also found lovastatin to be safe and well‐tolerated, with only mild side effects reported, all of which were determined unlikely to be related to study medication.
While our results indicated effects of lovastatin on neurocognition and behavior, we were unable to detect effects of the drug on underlying neural activity. The neurocognitive measures that showed significant treatment‐associated change (LNS, verbal declarative memory and verbal fluency) have verbal function in common, suggesting that neural activation during spatial working memory might not have been the optimal choice for a neuroimaging biomarker. Thus, two possible explanations for this pattern of findings are that: (1) we did not have power to detect a true effect, given the reduced sample size for those with usable neuroimaging measures (n = 37); and/or (2) the neuroimaging variables we studied were not sensitive indices of the behavioral changes. While this may be a function of the specific measures we selected, it is also worth noting that the psychometric properties of the neurocognitive and behavioral measures we used are very well established. In contrast, for the neuroimaging measures very little data exist on repeatability over a three‐month time period36.
Although NF1 is a monogenic disorder there is substantial variability across patients in the mutation characteristics, which may be related to the variability we observed within groups both in baseline performance and in the amount of change over time. Moderator analyses revealed that participants with greater baseline percent signal change in frontal regions (frontal eyefields and right DLPFC) during a working memory task showed greater differential improvement on lovastatin vs. placebo, suggesting that frontal ‘engagement’ during effortful cognitive processing may be associated with better treatment response.
There are inherent challenges in conducting a ‘bench to bedside’ exploratory clinical trial, particularly the selection of outcome measures that are both clinically meaningful and analogous to the probes used in animal models, dosing schedule, and trial duration. Notably, we found significant treatment‐associated changes in measures of working and declarative memory; in the mouse model, lovastatin improved spatial learning and normalized phosphorylated MAPK in the cortex and hippocampus9. Thus, in both the mouse and human studies, improvements were observed in frontal‐ and hippocampal‐dependent learning systems. In contrast to the findings in the mouse model, however, we did not find improvements in measures of attention. This may be attributable to the way attention was measured in the mouse versus human studies (lateralized reaction time test. versus self‐ and parent‐report measures), or could be due to the low rates of overt ADHD in our sample (20% overall). Further, the observed reduction in blood cholesterol levels over the course of the trial indicates that target engagement (i.e., inhibition of the HMG‐CoA reductase pathway12) was achieved, at least in the liver, in NF1 patients randomized to lovastatin. While it cannot be determined whether similar inhibition of this pathway was attained in the brain, the significant effects observed on some measures of cognition and behavior suggest that this is the case.
Previously, a small, open‐label, single‐arm study of lovastatin in children with NF1 suggested that lovastatin improved memory and attention, and ‘normalized’ resting functional connectivity within the default mode network13, 37. A larger, multisite randomized trial of lovastatin is currently underway (NCT00853580), although this study included only children and no biomarker assessments. One additional small study of 10 NF1 patients – which involved a randomized placebo‐controlled crossover design –examined the effects of 4 days of a high dose of lovastatin (200 mg) on electrophysiological outcome measures using transcranial magnetic stimulation (TMS)38. In this study, 4 days of lovastatin treatment was found to increase motor evoked potential amplitude after paired associative stimulation, reduce short‐interval cortical inhibition, and to improve phasic alertness in NF1 patients. The relevance of these changes to cognitive and behavioral outcome measures is not known, but suggests that TMS measures may provide a valuable intermediate link between the animal model and human studies with regard to “LTP‐like” cortical plasticity.
In contrast, a 12‐week randomized trial of another statin drug, simvastatin, did not find significant improvement in primary and secondary outcome measures, with the exception of a 1.5 standard deviation improvement on a secondary measure of visuoconstructive ability (Object Assembly)11. In a subsequent trial, Van der Vaart and colleagues12 found that 12 months of treatment with 20–40 mg daily of simvastatin did not result in differential improvement (relative to placebo) on measures of IQ, attention, or internalizing behavioral problems in 8–16‐year‐old children with NF1. In a related commentary, Acosta39 suggests possible reasons that this trial was negative, namely that outcome measures such as IQ are overly broad in terms of the cognitive domain being targeted, and the use of subjective measurements (i.e., parent‐ and self‐report measures). The additional suggestion is made that “to improve the design of future trials …efforts should be made to ensure that the study design for the human trial is more similar to the mouse‐model study design, while acknowledging the difficulties in behavioral comparison between species”. In contrast to that recommendation, we found treatment‐associated changes in verbally mediated cognitive domains, which cannot be measured in a mouse.
It is also possible that differences in the chemical composition and blood‐brain permeability of simvastatin versus lovastatin40 may be relevant to the different sets of results across studies. A recent review concluded that monacolin J derivatives (natural and semi‐synthetic statins), of which lovastatin is one, have the best neuroprotective potential, due to their higher capacity for blood–brain barrier penetration, cholesterol lowering effect on neurons with a satisfactory safety profile, and in vitro protection against cell death40.
Although we used age‐adjusted measures, given the wide age range in our study we also assessed age as a moderating variable. However, we did not find a moderating effect of age on measures showing a differential treatment effect, nor were any results present in the individual age groups that did not appear in the overall group. In contrast, internalizing symptoms showed differential treatment‐associated change only within the adult patients. Whether this is a result of larger baseline variability of the parent‐report measures (as shown in Table 3), and/or lower reliability of childrens' self‐report, is not known.
There is increasing evidence that statins may play a neuroprotective role in several disorders, including neurodegenerative conditions like Alzheimer's disease40 as well as neuroinflammatory disorders41. In animal models statins have been shown to have cell protective properties and to inhibit leukocyte migration through the blood–brain barrier40. Notably, high doses of simvastatin (80 mg/daily) were found to reduce the annualized rate of whole‐brain atrophy in patients with multiple sclerosis compared with placebo, and were well‐tolerated and safe41.
Several limitations of this study should be noted, particularly the small sample size and wide age range. This was unavoidable, given the challenges of recruiting patients with a disorder with a prevalence of 1:3000 in a single location. These limitations are, at least in theory, offset by the known, homogeneous etiology of this disorder, in which the molecular pathways leading to cognitive dysfunction are well understood7, 42. Although we found a consistent pattern of results across verbally mediated cognitive measures, indicating differential improvement in the statin‐treated group, the estimate of the magnitude of treatment effect sizes may be inflated due to the sample size43. In addition, we elected not to control for multiple comparisons given the exploratory nature of this trial. Thus, while results are promising, further investigation and replication in larger samples is clearly warranted.
Collectively, these findings offer preliminary evidence for lovastatin's efficacy in improving some aspects of cognition and internalizing symptomatology in patients with the monogenic disorder NF1. These findings have potential clinical relevance not only for NF1, but also for other ‘Ras‐opathies’ caused by disruptions of the RAS/ERK signaling pathway, such as Noonan and Costello syndrome, and Tuberous Sclerosis Complex (TSC)44. The genes affected in these disorders belong to a pathway that is coregulated by RAS but also critically dependent on RHEB, another farnesylated protein of the RAS family. Given the substantial association between genetic disruptions in these pathways and cognitive dysfunction, agents shown to reverse synaptic plasticity deficits by targeting these pathways offer hope for enhancing cognition in patients suffering from these highly disabling disorders.
Author Contributions
CEB designed the study and drafted the manuscript. GH conducted the statistical analyses and contributed to drafting the manuscript. AS and JJM contributed to the study conceptualization and protocol development, and edited the manuscript. CAS advised on the study design and statistical analyses. TR and SAH served as the study physicians and contributed to drafting of the manuscript. JYW reviewed laboratory results and provided critical feedback on the manuscript. NE and JH recruited and assessed study participants, analyzed neurocognitive data, and edited the manuscript. LP, CM, and RJ conducted the preprocessing, quality control and analysis of neuroimaging data, and edited the manuscript.
Conflicts of Interest
The authors have no relevant conflicts of interest to report.
Acknowledgment
We thank the individuals who participated in this study. This work was supported by grants from the National Institute of Mental Health (R34 MH089299; to C.E.B. and RO1 MH084315 to A.S.)
References
- 1. Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor‐prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 2010;152A:327–332. [DOI] [PubMed] [Google Scholar]
- 2. Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 2005;65:1037–1044. [DOI] [PubMed] [Google Scholar]
- 3. North KN, Riccardi V, Samango‐Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology 1997;48:1121–1127. [DOI] [PubMed] [Google Scholar]
- 4. Mautner VF, Granstrom S, Leark RA. Impact of ADHD in adults with neurofibromatosis type 1: associated psychological and social problems. J Attention Disord 2015;19:35–43. [DOI] [PubMed] [Google Scholar]
- 5. Garg S, Lehtonen A, Huson SM, et al. Autism and other psychiatric comorbidity in neurofibromatosis type 1: evidence from a population‐based study. Dev Med Child Neurol 2013;55:139–145. [DOI] [PubMed] [Google Scholar]
- 6. Adviento B, Corbin IL, Widjaja F, et al. Autism traits in the RASopathies. J Med Genet 2014;51:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Costa RM, Federov NB, Kogan JH, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002;415:526–530. [DOI] [PubMed] [Google Scholar]
- 8. Cui Y, Costa RM, Murphy GG, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008;135:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li W, Cui Y, Kushner SA, et al. The HMG‐CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 2005;15:1961–1967. [DOI] [PubMed] [Google Scholar]
- 10. Bernards A, Snijders AJ, Hannigan GE, et al. Mouse neurofibromatosis type 1 cDNA sequence reveals high degree of conservation of both coding and non‐coding mRNA segments. Hum Mol Genet 1993;2:645–650. [DOI] [PubMed] [Google Scholar]
- 11. Krab LC, de Goede‐Bolder A, Aarsen FK, et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA 2008;300:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Vaart T, Plasschaert E, Rietman AB, et al. Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1‐SIMCODA): a randomised, placebo‐controlled trial. Lancet Neurol 2013;12:1076–1083. [DOI] [PubMed] [Google Scholar]
- 13. Acosta MT, Kardel PG, Walsh KS, et al. Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. Pediatr Neurol 2011;45:241–245. [DOI] [PubMed] [Google Scholar]
- 14. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis . Bethesda, Md., USA, July 13‐15, 1987. Neurofibromatosis 1988;1:172–178. [PubMed] [Google Scholar]
- 15. Wechsler D. Wechsler abbreviated scale of intelligence.. New York, NY: The Psychological Corporation: Harcourt Brace & Company, 1999. [Google Scholar]
- 16. Expert Panel on Detection E ,. Treatment of High Blood Cholesterol in A. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486–2497. [DOI] [PubMed] [Google Scholar]
- 17. Bradford RH, Shear CL, Chremos AN, et al. Expanded Clinical Evaluation of Lovastatin (EXCEL) study results. I. Efficacy in modifying plasma lipoproteins and adverse event profile in 8245 patients with moderate hypercholesterolemia. Arch Intern Med 1991;151:43–49. [DOI] [PubMed] [Google Scholar]
- 18. Stein EA, Illingworth DR, Kwiterovich PO Jr, et al. Efficacy and safety of lovastatin in adolescent males with heterozygous familial hypercholesterolemia: a randomized controlled trial. JAMA 1999;281:137–144. [DOI] [PubMed] [Google Scholar]
- 19. Benedict HRB. Brief Visuospatial Memory Test–Revised professional manual. Odessa: FL Psychological Assessment Resources Inc,1997. [Google Scholar]
- 20. Wechsler D. The Wechsler intelligence scale for children—fourth edition. London: Pearson Assessment, 2004. [Google Scholar]
- 21. Wechsler D. Wechsler adult intelligence scale‐fourth edition (WAIS‐IV). London: Pearson Assessment, 2008. [Google Scholar]
- 22. Brandt J, Benedict RHopkins. Verbal learning test‐revised: professional manual. Florida: PAR, 2001. [Google Scholar]
- 23. Wechsler D. The wechsler intelligence scale for children—third edition. San Antonio, Texas: The Psychological Corporation, 1991. [Google Scholar]
- 24. Achenbach T. Manual for the child behavior checklist/4–18. Burlington: VT University of Vermont Department of Psychiatry, 1991. [Google Scholar]
- 25. Achenbach T. Manual for the young adult behavior checklist and young adult self‐report. Burlington,.VT: University of Vermont Department of Psychiatry, 1997. [Google Scholar]
- 26. White T, Stern RA. Neuropsychological assessment battery (nab): demographically corrected norms manual. Lutz, FL: Psychological Assessment Resources, Inc., 2003. [Google Scholar]
- 27. Delis D, Kaplan E, Kramer J. Delis‐Kaplan executive function system (D‐KEFS™). London: Pearson Asssessment, 2001. [Google Scholar]
- 28. Gioia GA, Isquith PK, Guy SC, Kenworthy L. Behavior rating inventory of executive function. Child Neuropsychol 2000;6:235–238. [DOI] [PubMed] [Google Scholar]
- 29. Shilyansky C, Karlsgodt KH, Cummings DM, et al. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc Natl Acad Sci USA 2010;107:13141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glahn DC, Kim J, Cohen MS, et al. Maintenance and manipulation in spatial working memory: dissociations in the prefrontal cortex. NeuroImage 2002;17:201–213. [DOI] [PubMed] [Google Scholar]
- 31. Montojo CA, Ibrahim A, Karlsgodt KH, et al. Disrupted working memory circuitry and psychotic symptoms in 22q11.2 deletion syndrome. Neuroimage Clin. 2014;4:392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andersson JLR, Jenkinson M, Smith S. Non‐linear registration aka spatial normalization FMRIB technical report TR07JA2. Oxford, United Kingdom: FMIRB Analysis Group of the University of Oxford, 2007. [Google Scholar]
- 33. Mumford JA, Poldrack RA. Modeling group fMRI data. Soc Cogn Affect Neurosci. 2007;2:251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Addington J, Cadenhead KS, Cannon TD, et al. North American Prodrome Longitudinal Study: a collaborative multisite approach to prodromal schizophrenia research. Schizophr Bull 2007;33:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radtke HB, Sebold CD, Allison C, et al. Neurofibromatosis type 1 in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns 2007;16:387–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carter CS, Barch DM, Bullmore E, et al. Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia II: developing imaging biomarkers to enhance treatment development for schizophrenia and related disorders. Biol Psychiatry 2011;70:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chabernaud C, Mennes M, Kardel PG, et al. Lovastatin regulates brain spontaneous low‐frequency brain activity in neurofibromatosis type 1. Neurosci Lett 2012;515:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mainberger F, Jung NH, Zenker M, et al. Lovastatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol 2013;13:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Acosta MT. Challenges of cognitive research in neurofibromatosis type 1. Lancet Neurol 2013;12:1040–1041. [DOI] [PubMed] [Google Scholar]
- 40. Sierra S, Ramos M, Molina P, et al. Statins as neuroprotectants: a comparative in vitro study of lipophilicity, blood‐brain barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J Alzheimer's Dis 2011; 23(iss:2):307–318. [DOI] [PubMed] [Google Scholar]
- 41. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high‐dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS‐STAT): a randomised, placebo‐controlled, phase 2 trial. Lancet 2014;383:2213–2221. [DOI] [PubMed] [Google Scholar]
- 42. Costa RM, Yang T, Huynh DP, et al. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat Genet 2001;27:399–405. [DOI] [PubMed] [Google Scholar]
- 43. Button KS, Ioannidis JP, Mokrysz C, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci 2013;14:365–376. [DOI] [PubMed] [Google Scholar]
- 44. Krab LC, Goorden SM, Elgersma Y. Oncogenes on my mind: ERK and MTOR signaling in cognitive diseases. Trends Genet 2008;24:498–510. [DOI] [PubMed] [Google Scholar]